

Abstract
Zinc (Zn) deficiency during early development can result in multiple brain abnormalities and altered neuronal functions. In rats, a gestational deficit of Zn can affect the fetal brain cytoskeleton, and signaling cascades involved in cellular processes that are central to brain development. In the current paper, we tested the hypothesis that oxidative stress is involved in Zn deficiency-induced altered tubulin dynamics and the associated dysregulation of transcription factor NF-κB. For this purpose, we used two cell culture models (rat cortical neurons, human IMR-32 neuroblastoma cells) and an animal model of Zn deficiency. A low rate of in vitro tubulin polymerization, an increase in tubulin oligomers and a higher protein cysteine oxidation were observed in the Zn deficient neuronal cells, and in gestation day 19 fetal brains obtained from dams fed marginal Zn diets throughout pregnancy. These alterations could be prevented by treating the Zn deficient cells with the reducing agent tris(2-carboxyethyl)phosphine, or the presence of N-acetyl cysteine (NAC) and α-lipoic acid (LA). Consistent with the above, Zn deficiency-induced tubulin-mediated alterations in transcription factor NF-κB nuclear translocation were prevented by treating IMR-32 cells with LA and NAC. Binding of the NF-κB protein p50, dynein and karyopherin alpha (components of the NF-κB transport complex) to β-tubulin as well as the expression of NF-κB dependent genes (bcl-2, cyclin D1 and c-myc) were also restored by the addition of LA and NAC to Zn deficient cells. In conclusion, a deficit in Zn viability could affect early brain development through: 1) an induction of oxidative stress; 2) tubulin oxidation; 3) altered tubulin dynamics, and 4) deregulation of signals (e.q. NF-κB) involved in critical developmental events.
Keywords: zinc, brain, redox state, tubulin, NF-κB
Introduction
Microtubules are key components of the neuronal cytoskeleton and are critical for proper neuronal function [1, 2]. They are formed through the reversible polymerization of tubulin, a heterodimer composed of 50 kDa α and β subunits. Oxidation and modification of protein cysteines can have profound effects on protein structure and function [3]. In this regard, tubulin with its 20 free sulfhydryl groups is highly susceptible to oxidation. For instance, oxidants that react with cysteines, such as peroxynitrite, H2O2, and thiol-reactive agents, can inhibit tubulin polymerization. This inhibition can be reversed by disulfide reducing agents [3-6].
Zinc (Zn) is an essential nutrient that has multiple roles in the nervous system [7]. Deficits of this nutrient result in adverse effects on brain structure and function [8-10]. In humans, developmental Zn deficiency can affect infant behavior, cognitive and motor performance [11-13], and it has been associated with attention-deficit/hyperactivity disorders [14]. Illustrative of the potential positive effects of Zn on the nervous system, are reports that supplementation of undernourished children with Zn can improve developmental quotients, activity patterns and neuropsychological functions [11-13, 15]. Consistent with the human literature, in experimental animals severe Zn deficiency during early development can result in multiple congenital anomalies, including brain malformations [16-18]. Marginal Zn deficiency during early development does not necessary result in gross teratogenicity. In rats, it has been shown to result in altered brain signaling and gene expression which can adversely impact behavioral and cognitive abilities latter in life [19].
Neurons have been shown to be highly sensitive to a deficit of Zn. In rats, postnatal Zn deficiency impairs the normal development of the cerebellum, affecting the number and differentiation of Purkinje, basket, stellate, and granule cells [10]. Zn deficiency in adult rats decreases proliferation of neural precursor cells in the subgranular zone and granular cell layer of the dentate gyrus [20]. We have recently shown using primary cultures of rat cortical neurons and human neuroblastoma IMR-32 cells, that Zn deficiency reduces cell viability, decreases cell proliferation and increases neuronal apoptotic cell death [21]. A decreased Zn availability causes several alterations in neuronal cells physiology which have been in part attributed to increased oxidative stress. In IMR-32 cells with Zn deficiency there can be a rapid increase in reactive oxygen and nitrogen species as a consequence of the activation of the NMDA receptor [22] leading to NADPH oxidase and nitric oxide synthase activation [23], and the triggering the activation of multiple oxidant-sensitive signalling cascades [24, 25]. Importantly, several of these cascades have also been shown to be activated in rat fetal brain obtained from dams fed marginal Zn diets throughout gestation [22].
The structure and dynamics of the neuronal cytoskeleton, particularly microtubules, is altered by a deficit of Zn. This was previously observed in fetal and adult rat brain as well as in IMR-32 cells [17, 26-30]. Although evidence clearly demonstrates that Zn deficiency affects the microtubule network in the nervous system, the mechanisms underlying these negative effects and their functional consequences are not well understood. We have provided evidence that a major functional consequence of low Zn concentrations can be altered tubulin polymerization that results in an altered transport of activated transcription factors NF-κB and NFAT into the nucleus [26, 28]. Such alterations could affect affect key developmental processes including neurogenesis and neuronal apoptosis [20, 31]. Given the participation of microtubules in axonal transport and in the communication of signals from the synapse into the nucleus [32], it is reasonable to suggest that developmental Zn deficiency can result in widespread alterations in fetal and neonatal brain signaling, with consequential behavioral, cognitive and locomotor alterations in the offspring [26, 28]. In the current study we tested the hypothesis that Zn deficiency-induced oxidative modifications of tubulin contribute to the altered tubulin polymerization that has been associated with decreased neuronal/brain Zn. The microtubule-associated impaired nuclear transport of NF-κB was examined as a potential functional consequence of neuronal Zn deficiency. The capacity of LA and NAC and oxidized thiol reductants to prevent the alterations in tubulin dynamics, and NF-κB nuclear transport were investigated. A combination of cell culture and whole animal models were used in these studies.
Materials and methods
Materials
IMR-32 neuroblastoma cells were obtained from the American Type Culture Collection (Rockville, MA). Cell culture media and reagents were purchased from Invitrogen Life Technologies (Carlbad, CA). The oligonucleotides containing the consensus sequence for NF-κB (5’-AGTTGAGGGGACTTTCCCAGGC-3’) and OCT-1, and the reagents for the EMSA assay were obtained from Promega (Madison, WI). Antibodies for p50 (sc-7178), dynein (sc-7526), karyopherin α (sc-6918), c-myc (sc-764), cyclin D1 (sc-718), hRNP (sc-32301), and β-tubulin (sc-9104) were from Santa Cruz Biotechnology (Santa Cruz, CA). Bcl-2 antibody was from Cell Signaling Technology. PVDF membranes were obtained from BIO-RAD (Hercules, CA). The ECL Plus Western blotting detection reagents were from GE Healthcare (Piscataway, NJ). Chroma Spin-10 columns were obtained from Clontech (Mount View, CA). (±)-α-lipoic acid (LA), N-acetyl cysteine, (NAC), tris(2-carboxyethyl)phosphine (TCEP), and all other reagents were from the highest quality available and were purchased from Sigma (St. Louis, MO).
Cell cultures and incubation
Zn deficient fetal bovine serum (FBS) was prepared by chelation with diethylenetriamine pentaacetic acid (DTPA) as previously described [26, 33]. The chelated FBS was subsequently diluted with complex medium (55% (v/v) DMEM high glucose, 30% (v/v) Ham F-12, 5% (v/v) α-MEM) to a final concentration of 3 mg protein/ml to match the protein concentration of the control non-chelated medium (10% (v/v) FBS). The concentration of Zn in the deficient medium was 1.5 μM. Aliquots of the Zn deficient media were supplemented with ZnSO4 to a final Zn concentration of 15 μM.
IMR-32 cells were cultured at 37 °C in complex medium supplemented with 10 % (v/v) FBS and antibiotics-antimicotic. Cells were grown in complex medium containing 10% (v/v) non-chelated FBS) until 90% confluence. IMR-32 cells were incubated in control non-chelated medium or in chelated media containing 1.5 μM Zn. Cells in control non-chelated medium or in chelated media were incubated in the absence or the presence of 0.5 mM LA or 1 mM NAC. LA and NAC were added at the time that the medium was changed to Zn deficient or control medium.
Rat cortical neurons were isolated as previously described [18]. Isolated cells were suspended in Neurobasal culture medium supplemented with 2% (v/v) serum-free additive B27, and plated on poly-L-lysine-coated dishes. Cell cultures were incubated at 37°C in an atmosphere of 95% (v/v) air, 5% (v/v) CO2 with 90–95% (v/v) humidity. After 24 h in culture, the media was replaced by DMEM high glucose supplemented with 10% (v/v) FBS. After 72 h, cell cultures were supplemented with 40 μM cytosine-β-D-arabinoside for 24 h to inhibit non-neuronal cell proliferation. After 7-9 d in culture, the media was replaced by control non-chelated medium, or chelated medium containing 1.5 μM Zn, and cells were incubated for an additional 24 h.
Animals and animal care
All procedures were in agreement with standards for the care of laboratory animals as outlined in the NIH Guide for the Care and Use of Laboratory Animals. All procedures were administered under the auspices of the Animal Resource Services of the University of California, Davis, which is accredited by the American Association for the Accreditation of Laboratory Animal Care. Experimental protocols were approved before implementation by the University of California, Davis Animal Use and Care Administrative Advisory Committee, and were administered through the Office of the Campus Veterinarian.
Adult Sprague-Dawley rats (Charles River, Wilmington, MA) (200-225 g) were housed individually in suspended stainless steel cages in a temperature (22–23°C)-and photoperiod (12 h l/d)-controlled room. An egg white protein based diet with adequate Zn (25 μg Zn/g) was the standard control diet [34]. Animals were fed the control diet for one week before breeding. Males and females were caged together overnight and the following morning the presence of a sperm plug confirmed a successful breeding. On gestation day 0 (GD 0), rats (7 animals/group) were divided into two groups and fed ad libitum: a control diet (25 μg Zn/g diet, C), or a diet containing a marginal concentration of Zn (10 μg Zn/g diet, MZ) until GD19. Food intake was recorded daily, and body weight was measured at 5-d intervals. On gestation day 19, the dams were anesthetized with isofluorane (2mg/kg body weight), and laparatomies were performed. The gravid uterus was removed, and fetuses collected. Fetal brains were excised, rinsed in ice-cold PBS, the meninges removed, weighed and a pool of 4-5 brains was homogenized for tubulin polymerization assays.
In Vitro Microtubule Assembly
Cells (35 × 106) were rinsed three times with 0.1 m Pipes buffer, pH 7.0 containing 0.1 mM GTP (Pipes). Cell pellets were manually homogenized using a glass homogenizer to a final volume of 0.4 ml. Fetal brains were rinsed twice with Pipes and homogenized in 3 volumes of the same buffer with a motor-driven glass homogenizer with a Teflon pestle. Both cellular and brain homogenates were incubated for 30 min at 4 °C to allow microtubule depolymerization, and then centrifuged at 100,000 × g for 30 min at 4 °C. The supernates were decanted, 200-μl aliquots were placed in a 96-well plate, and tubulin assembly was followed as the increase in absorbance at 340 nm for 90 min. The polymerization was started when the samples were placed at 37 °C in a PerkinElmer HTS 7000 Plus Bio Assay Reader (PerkinElmer Life Sciences) and was followed for 90 min.
Determination of Zn concentrations
Zn concentrations in the diets, in fetal brains and IMR-32 cell 100,000 × g supernates, were measured following procedures previously described [35]. Briefly, diet samples were wet ashed with 16 m nitric acid (Baker’s Instra-analyzed: J.T. Baker, Philipsburg, NJ). Concentrations of Zn were determined by ICP-AES (Trace Scan; Thermo Elemental, Franklin, MA). Certified reference solutions (QC 21, Spec CentriPrep, Metuchen, NJ) were used to generate standard curves for each element. A sample of a National Bureau of Standards Bovine Liver (SRM 1577; United States Department of Commerce, National Bureau of Standards, Washington, DC) was included with the samples to ensure accuracy and reproducibility.
Determination of total protein thiols and tubulin cysteine oxidation
Total protein thiols were measured in brain and IMR-32 100,000 × g supernates, obtained as described above. Thiol quantitation was done using a commercial kit based in the Ellman′s reaction (Thiol and Sulfide quantitation kit T-6060, Molecular Probes), and following the manufacturer’s instructions. The determination of tubulin cysteine oxidation was done following a previously published protocol [36]. Briefly, aliquots of IMR32 cells, cortical neurons and fetal brain 100,000 × g supernates were first treated with 100 mM N-ethyl maleimide to block free thiols, and excess reagent was removed by passage through a Micro Bio-Spin® 6 chromatography column. To reduce oxidized thiols, samples were subsequently incubated with 1 mM dithiothreitol for 10 min at room temperature. Iodoacetamidefluorescein (IAF) in N,N-dimethylformamide was added to a final concentration of 1.5 mm, samples were incubated at 37 °C for 30 min, and passed through a second column to remove excess IAF. Proteins were resolved by SDS-PAGE on a 7.5% gel under reducing conditions, and gel images were captured in a phosphoimager 840 (GE HealthCare, Piscataway, NJ).
Electrophoretic mobility shift assay (EMSA)
Nuclear and cytosolic fractions were isolated as previously described [37, 38], with minor modifications [26]. For the EMSA, the oligonucleotides containing the consensus sequence for NF-κB or OCT-1 were end labelled with [γ-32P] ATP using T4 polynucleotide kinase and purified using Chroma Spin-10 columns. Samples were incubated with the labeled oligonucleotide (20,000-30,000 cpm) for 20 min at room temperature in 1X binding buffer [5X binding buffer: 50 mM Tris-HCl buffer, pH 7.5, containing 20% (v/v) glycerol, 5 mM MgCl2, 2.5 mM EDTA, 2.5 mM DTT, 250 mM NaCl and 0.25 mg/ml poly(dI-dC)]. The products were separated by electrophoresis in a 6% (w/v) non-denaturing polyacrilamide gel using 0.5 × TBE (Tris/borate 45 mM, EDTA 1mM) as the running buffer. The gels were dried and the radioactivity quantitated in a Phosphoimager 840 (GE HealthCare, Piscataway, NJ).
Western blot analysis
For the preparation of total tissue extracts, fetal brains were homogenized [50 mg of tissue/400 μl of lysis buffer: 50 mmol/L Tris (pH 7.5), 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 50 mM NaF, 2 mM NaVaO4 containing inhibitors of proteases and phosphatases and 1 % (v/v) Igepal]. The homogenates were incubated at 4°C for 30 min and centrifuged at 10,000 × g for 20 min. The supernates were collected and protein concentration was measured [39].
Aliquots of total or nuclear fractions containing 25-50 μg protein were separated by reducing 10% (w/v) polyacrylamide gel electrophoresis and electroblotted to PVDF membranes. Colored molecular weight standards (Amersham, Piscataway, NJ) were ran simultaneously. Membranes were blotted overnight in 5% (w/v) non-fat milk and incubated in the presence of the corresponding primary antibodies (see Materials section) for 90 min at 37°C. After incubation with the secondary antibody (HRP-conjugated) (1:10,000 dilution), 90 min at room temperature, the conjugates were visualized by chemiluminescence detection in a Phosphoimager 840.
Immunoprecipitation
Total cell lysates were prepared as described above. 350 μg of total cell proteins were pre-cleared for 30 min using 0.25 μg of the appropriate control IgG from the host specie together with 20 μl of Protein A/G-Agarose. The precleared-supernatant fractions were immunoprecipitated with an anti β-tubulin antibody (1 μg) together with 25 μl Protein A/G-Agarose overnight at 4 °C. After centrifugation, the beads were washed twice with PBS, and the final pellets were resuspended in 30 μl of 1X sample buffer. Samples were boiled for 5 min, 15 μl of each sample were loaded for the electrophoresis, and Western blots were immediately done as described above. To evaluate the specificity of the immunoprecipitation, a control total cell fraction was immunoprecipitated with control IgG together with Protein A/G-Agarose, instead of the corresponding β-tubulin antibody. There was no evidence of non-specific immunoprecipitation when control samples were immunoprecipitated with control IgG, instead of the corresponding primary β-tubulin.
Results
Zn deficiency impairs tubulin polymerization in IMR-32 cells: LA and NAC can correct the inhibition
We first investigated the possible involvement of tubulin oxidative modifications in the altered tubulin polymerization associated with neuronal Zn deficiency by incubating the Zn deficient cells with LA and NAC. We have previously shown that under similar experimental conditions, LA and NAC prevented the increase in IMR-32 cell oxidants and the decrease in glutathione levels associated with the decreased cellular Zn [24]. Figure 1A depicts the typical kinetics of tubulin polymerization in 100,000 × g supernates isolated from IMR-32 cells incubated in control or Zn deficient (1.5 μM Zn, ZD) media for 24 h. The rates of tubulin assembly were lower in the Zn deficient (74 %, p < 0.001) cells than in controls. The addition of either 0.5 mM LA or -1 mM NAC- to the cells incubated in Zn deficient media restored tubulin polymerization rates to control values (Fig.1 A). Zn concentrations in the 100,000 × g supernatant fractions from the Zn deficient cells were 39 % (p<0.05) lower than in controls. Zn concentrations were not affected by LA or NAC treatment (Fig.1 B). The concentration of thiols in the supernatant proteins was reduced by 22% (p<0.05) in the Zn deficient cells (Fig.1C). The co-incubation with NAC or LA restored the protein thiol levels in the Zn deficient cells to control values (Fig. 1C).
Figure 1. Zinc deficiency-induced inhibition of tubulin polymerization in IMR-32 cellsis reverted by NAC and LA.
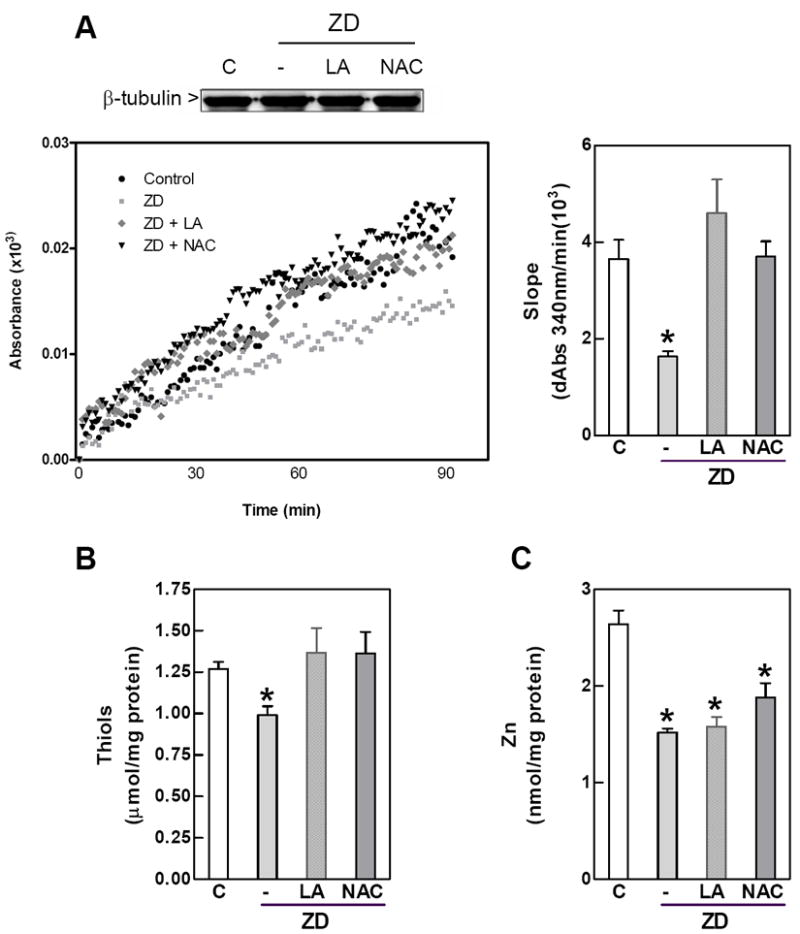
IMR-32 cells were incubated for 24 h in control non-chelated media (C) or in chelated media containing 1.5 μM Zn (ZD) without or with the addition of 0.5 mM LA (LA) or 1mM NAC (NAC). 100,000 × g supernates were prepared as described under Materials and Methods. A- In vitro tubulin polymerization. Left panel shows a representative kinetics. Tubulin polymerization rates were calculated as the slope in the initial linear phase of the polymerization (right panel).; B- Zinc concentration and C- protein thiol concentration in 100,000 × g supernatants Results are shown as means ± SEM of at least 3 independent experiments. *Significantly different compared with the other groups (p<0.05, one-way ANOVA test).
The disulfide reductant TCEP prevents Zn deficiency-associated impaired tubulin polymerization in fetal brain and IMR-32 cells
We previously reported that a severe gestational Zn deficiency can affect tubulin polymerization in the fetal brain [29]. We now evaluated if feeding rats marginal Zn (MZ) diets during gestation could also affect tubulin assembly in the brain of GD19 fetuses. Using this experimental model we previously reported that this level of marginal Zn deficiency does not affect food intake, maternal body weight and it does not result in gross fetal abnormalities [40]. The rate of in vitro tubulin polymerization was 50% (p<0.05) lower in fetal brain 100,000 × g supernates isolated from MZ fetal brain compared to controls (Fig. 2 A). The concentration of Zn in MZ supernates was 21% (p<0.05) lower than in controls (Fig. 2 B). The concentration of protein thiols was significantly lower in brain supernates from MZ fetuses than in controls. (Fig. 2 B). To evaluate if the decrease in the rate of tubulin polymerization was due to alterations in the redox state of tubulin thiols, supernatants were incubated with 1.5 mM TCEP. Treatment of MZ supernates with TCEP restored the tubulin polymerization rates to levels similar to those of controls (Fig. 2 A). TCEP did not affect tubulin polymerization rates in control supernatants. Accordingly, the treatment of 100,000 × g supernatants isolated from IMR-32 cells incubated for 24 h in Zn deficient media with TCEP reverted the decrease in tubulin assembly rates (Fig. 2 C). Prior to the polymerization reaction, total protein (data not shown) and β-tubulin (Fig. 1A, 2A) concentrations in the 100,000 × g supernates were not affected by Zn deficiency in either the fetal brains or the IMR-32 cells.
Figure 2. Zinc deficiency inhibits tubulin polymerization in rat fetal brain and IMR-32 cells: effects of a thiol reductant.
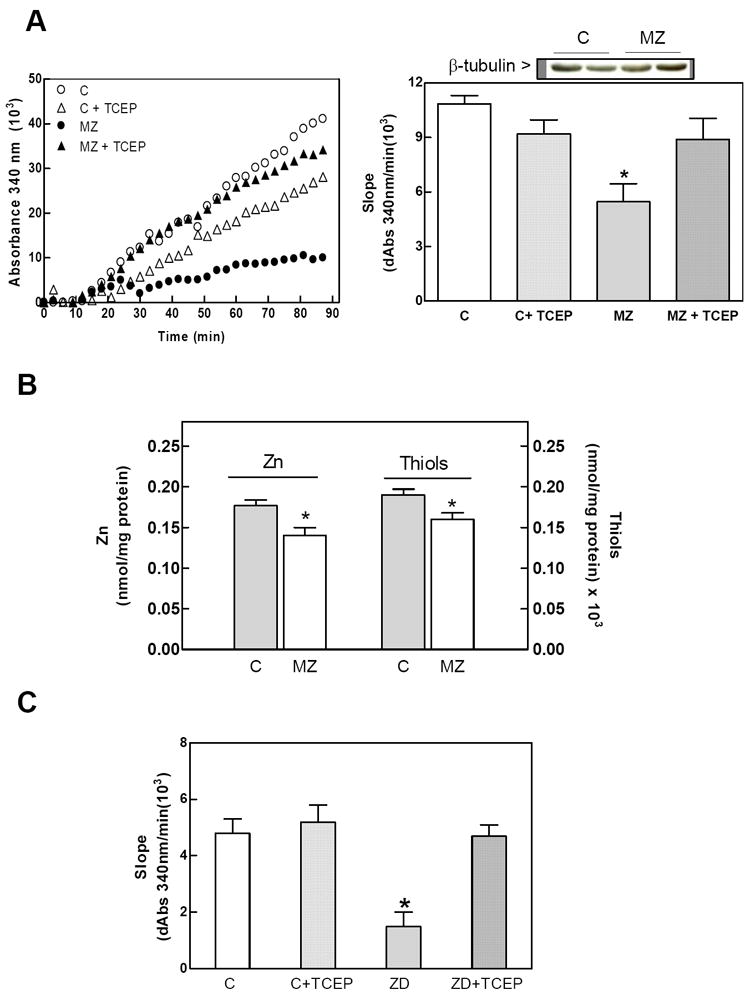
GD19 rat brain and IMR-32 100,000 × g supernates were prepared as described under Materials and Methods. Supernates were incubated without or with 50 mM TCEP. A- In vitro tubulin polymerization in GD19 fetal brain from dams fed control (C) or marginal Zn diets (MZ) throughout gestation. Left panel shows a representative kinetics for supernates incubated without (C,MZ) or with (C + TCEP, MZ + TCEP) 2.5 mM TCEP. Tubulin polymerization rates were calculated as the slope in the initial linear phase of the polymerization (right panel). B- Zinc and thiol concentrations in 100,000 × g supernates from GD19 fetal brain. C- Tubulin polymerization rates in 100,000 × g supernates from IMR-32 cells incubated for 24 h in control (C) or Zn deficient media (ZD). Portions of the supernates were incubated with 5 mM TCEP (C + TCEP, ZD + TCEP) for 30 min prior to evaluating tubulin polymerization. Results are shown as means ± SEM of at least 3 independent experiments for IMR-32 cells or 5-7 litters for GD19 fetal brain. *Significantly different compared with the other groups (p<0.05, one-way ANOVA test).
Zinc deficiency causes tubulin thiol oxidation and the formation of tubulin oligomers in IMR-32 cells, cortical neurons, and fetal brain
We next investigated the potential thiol oxidation and formation of inter-subunit disulphide bonds. Proteins in neuronal cell and fetal brain supernatants were separated using non-reducing SDS-PAGE, and β-tubulin was identified by Western blot. As shown in Fig. 3, in neuronal cells and fetal brain, Zn deficiency was associated with an increase in tubulin oligomers of molecular weight higher than 100 kDa. Treatment of cells with LA and NAC, or the addition of TCEP to supernates obtained from IMR-32 cells, cortical neurons and fetal brain resulted in the disappearance of most of the high molecular weight bands (Fig. 3). These findings suggest that tubulin oligomers could are due to thiol oxidation and to the formation of disulfide bridges between monomers.
Figure 3. Zinc deficiency induces an increase in tubulin oligomers in rat fetal brain, rat cortical neurons and IMR-32 cells.
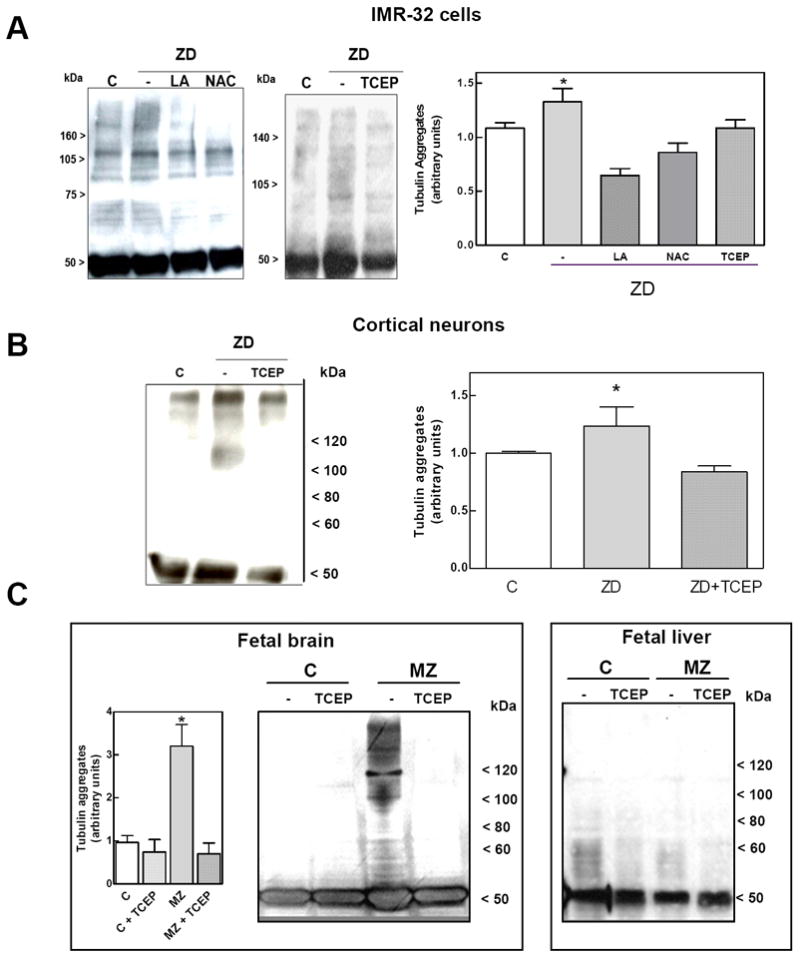
Tubulin oligomers in 100,000 × g supernates were evaluated under the following experimental conditions. A-Tubulin oligomers in IMR-32 cells incubated for 24 h in IMR-32 cells in control non-chelated media (C) or in chelated media containing 1.5 μM Zn (ZD) without or with the addition of 0.5 mM LA (LA) or 1mM NAC (NAC) (left panel). Zn deficient supernates were treated 1.5 mM TCEP (center panel) for 30 min. B, C- 100,000 × g supernates isolated from (B) cortical neurons, and (C) fetal brain and liver, were incubated in the absence or the presence of 1.5 mM TCEP for 30 minutes. Tubulin oligomers in the supernates were assessed by separating proteins by SDS-PAGE under non-reducing conditions, and subsequent Western blot for β-tubulin. Tubulin oligomers of MW >100 kDA were quantitated. Results are shown as means ± SEM of 4 independent experiments for IMR-32 cells or 5-7 litters for GD19 brain. *Significantly different compared with the other groups (p<0.05, one-way ANOVA test).
Cysteine oxidation is a classic redox modification of proteins. To evaluate if tubulin oxidation and formation of oligomers in Zn deficiency could be due to thiol oxidation, we next measured the relative content of thiol groups using the fluorescent probe IAF. Thiol groups were initially blocked with iodoacetamide, disulfides were then reduced with DTT, and subsequently derivatized with IAF. An increase in the intensity of fluorescence in the tubulin band is due to higher levels of oxidized thiols. Zn deficiency led to an increase (38 %, 75 %, and 93%, respectively) in thiol oxidation in IMR-32 cells, rat cortical neurons and fetal brain homogenates, respectively (Fig. 4). Thiol oxidation in the Zn deficient IMR-32 cell and cortical neurons was prevented when cultures were supplemented with LA during the 24 h incubation in Zn deficient media (Fig.4).
Figure 4. Zinc deficiency induces tubulin cysteine oxidation.
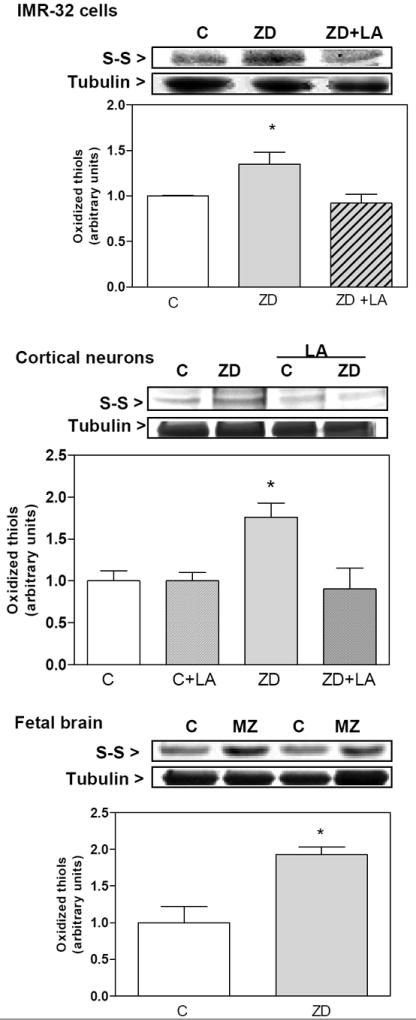
Tubulin thiols were measured in 100,000 × g supernates isolated from A- IMR-32 cells, B-cortical neurons and C- E19 brain. To measure tubulin thiols, supernates were first treated with N-ethyl maleimide to block free thiols, followed by reduction with dithiothreitol, and reaction with IAF as described in Material and Methods. After SDS-PAGE, the fluorescence intensity of the tubulin bands was quantitated. Results are shown as means ± SEM of 4 independent experiments for IMR-32 cells and cortical neurons, or 5-7 litters for GD19 brain. *Significantly different compared with the other groups (p<0.05, one-way ANOVA test).
We next evaluated whether protein carbonylation and nitration could occur as a consequence of low Zn in neuronal cells and brain. For this purpose, we investigated the presence of chemical modifications in tubulin mediated by the increase in reactive oxygen and nitrogen species. Under these experimental conditions, Zn deficiency was not associated with increases in carbonylation or nitration of tubulin (data not shown).
LA and NAC restore NF-κB nuclear translocation in Zn deficient cells
We investigated the capacity of NAC and LA to restore NF-κB nuclear translocation in Zn deficient IMR-32 cells. A disruption in the transport of the active NF-κB into the nucleus and a decreased expression of NF-κB-driven genes is a major consequence of the impaired tubulin assembly in Zn deficiency neuronal cells [28]. NF-κB-DNA binding was measured in nuclear and cytosolic fractions by EMSA (Fig. 5A). NF-κB-DNA binding was 61% lower in nuclear fractions isolated from the IMR-32 cells incubated for 24 h in 1.5 μM Zn medium. NF-κB-DNA binding in cytosolic fractions was higher (2.2-fold) in Zn deficient cells than in controls (Fig. 5A). The treatment of the Zn-deficient cells with LA or NAC restored the nuclear transport of NF-κB (Fig. 5 A). EMSA results were confirmed by Western Blot assays that measured the p50 protein distribution in nuclear and cytosolic fractions While p50 levels were lower in nuclear fractions, it accumulated in the cytosolic fractions isolated from Zn deficient cells compared to controls. In addition, and supporting the altered nuclear transport of the active NF-κB protein complex in Zn deficiency cells, the decrease in p50 nuclear content from Zn deficient IMR-32 cells, was also restored by the addition of LA and NAC (Fig. 5 A,B).
Figure 5. LA and NAC restore NF-κB nuclear translocation in zinc deficient IMR-32 cells.
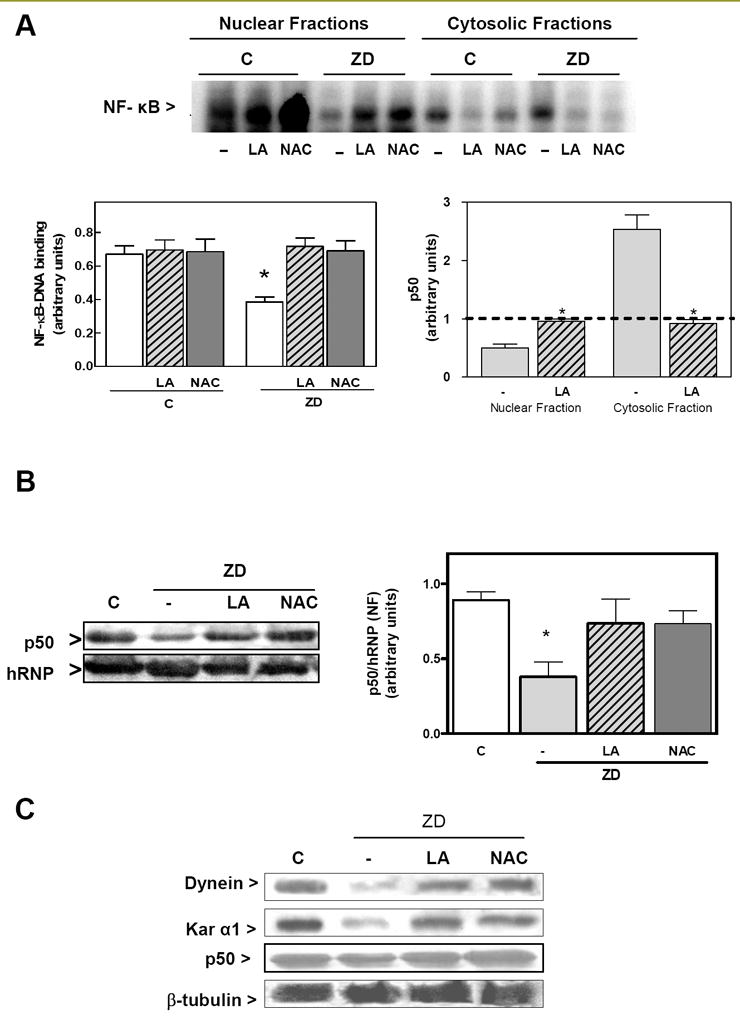
IMR-32 cells were incubated for 24 h either in control non-chelated media (C) or in chelated media containing 1.5 μM Zn (ZD); without or with the addition of 0.5 mM LA or 1 mM NAC. A- Upper panel: representative EMSA image for NF-κB in nuclear fractions; Lower panel right: quantification of EMSA bands corresponding to NF κB-DNA complexes; Lower panel left: p50 levels in nuclear and cytosolic fractions measured by Western blot. B- Nuclear content of the NF-κB protein p50 was measured by Western blot and normalized for the hRNP protein content, C- After immunoprecipitation of whole cell extracts with a β-tubulin antibody adsorbed to agarose pellet beads, Western blots for dynein, p50, karyopherin and β-tubulin were done. One representative Western blot out of three independent experiments is shown. A,B- Bands were quantitated and results are shown as means ± SEM of at least 3 independent experiments. *Significantly different compared with the other groups (p<0.05, one-way ANOVA test).
The altered nuclear transport of NF-κB in the Zn deficient cells can be in part a consequence of an impaired formation of the transport complex tubulin-dynein-karyopherin-p50 [28]. Immunoprecipitation of β-tubulin, shows its interaction with dynein, karyopherin, and p50 (Fig. 5 C), which is impaired in IMR-32 cells incubated for 24 h in Zn deficient media. Consistent with the recovered NF-κB nuclear transport, in Zn deficient cells treated with NAC and LA the interaction tubulin-dynein-karyopherin-p50 was restored (Fig. 5 C).
The consequence of Zn deficiency-induced impaired NF-κB nuclear transport is a decreased transactivation of NF-κB-regulated genes. As previously observed, the incubation of IMR-32 cells for 24 h in Zn deficient media led to a decreased expression of the NF-κB-regulated proteins Bcl-2, cyclin-D1, and c-myc (Fig. 6). Supplementation of the Zn deficient media with NAC and LA restored the expression of these proteins to control values.
Figure 6. LA and NAC restore NF-κB-driven transactivating activity.
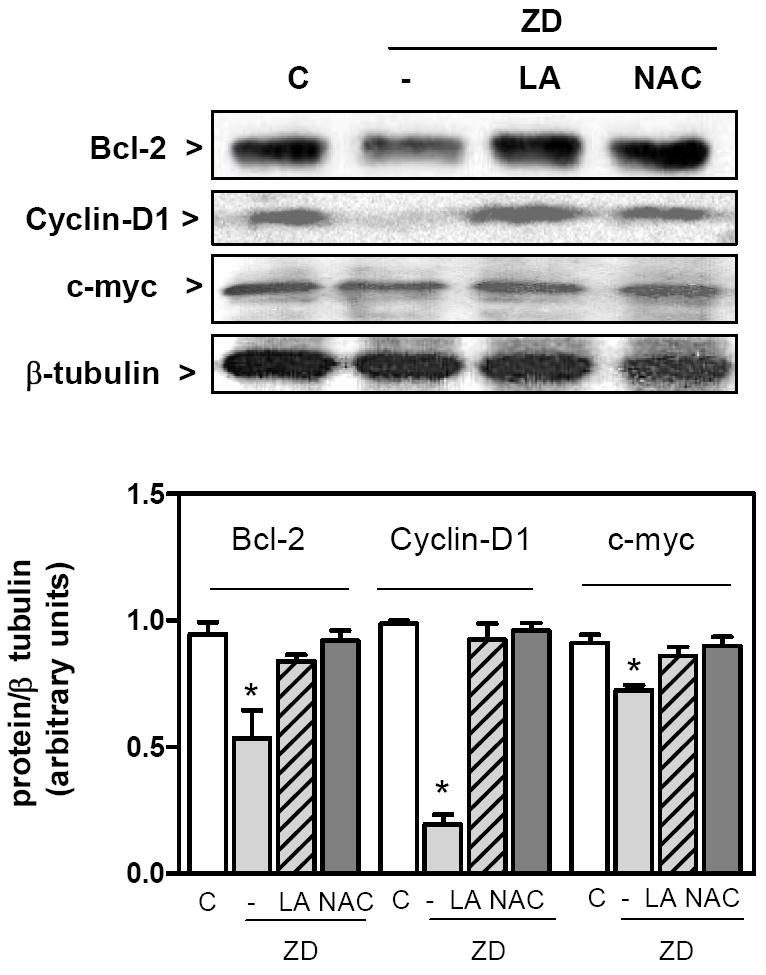
IMR-32 cells were incubated for 24 h in control non-chelated media (C) or in chelated media containing 1.5 μM Zn (ZD); without or with the addition of 0.5 mM LA or 1 mM NAC. Upper panel: typical Western blot images for bcl-2, cyclin-D1, cMyc, and β-tubulin in total cell fractions isolated from IMR-32 cells incubated for 24 h in the different experimental conditions. Lower: after quantitation of Western blots, results for bcl-2, cyclin-D1, c-myc were normalized for the β -tubulin. Results are shown as means ± SEM of 3 independent experiments. *Significantly lower compared to the other groups (p < 0.03, one way ANOVA test).
DISCUSSION
Oxidative modifications of tubulin can significantly affect the dynamics of its assembly into microtubules. Tubulin posttranslational modifications mark subpopulations of microtubules and selectively affect downstream microtubule-based functions [41]. A decreased Zn availability affects tubulin polymerization kinetics and structure in rat brain and neuronal cells, with significant consequences for cellular signaling [26, 28, 42]. However, the underlying mechanisms of Zn deficiency-induced microtubule alterations are unknown. In this study we show that a decrease in Zn can be associated with increased tubulin oxidation in neuronal cells and rat fetal brain. Tubulin oxidative modifications mostly involve the oxidation of thiols groups that in part causes tubulin crosslinking. These oxidative modifications led to major functional consequences for the neuronal cell, given that the prevention by LA or NAC of tubulin oxidation and of the associated alterations in tubulin dynamics, resulted in an improved nuclear transport of NF-κB.
Zn is critical for the physiology of the nervous system, and Zn deficiency has been associated with altered neurodevelopment [43]. However the underlying mechanisms are still speculative. Oxidative stress, altered redox signaling, and altered patterns of cell proliferation and apoptosis could in part underlie the adverse effects of developmental Zn deficiency. Microtubules, common to those processes, are highly susceptible to oxidation; they participate in signaling events, and directly or indirectly fect cell fate. Brain and neuronal microtubule assembly is significantly affected by Zn deficiency in animal and cell models. Control and Zn deficient cells show very distinct differences in the kinetics of microtubule polymerization. An extended lag phase and a decrease in the maximal rate of polymerization are observed in Zn deficiency. Significantly, similar alterations have been observed in cells treated with peroxinitrite [5]. The finding that LA and NAC prevented the altered tubulin kinetics in IMR-32 cells, while it did not prevent the decrease in cellular Zn, strongly support the concept that a Zn deficiency-induced increase in cellular oxidative stress contributes to the occurrence of altered tubulin polymerization and function. Zn deficiency decreased protein thiol concentrations in IMR-32 cell, which were restored to control values by LA and NAC. Consistently, we previously demonstrated that in Zn deficient IMR-32 cells, LA and NAC prevent the increase in H2O2 production [24], restore reduced glutathione levels [24], and prevent the activation of the oxidant-sensitive mitogen activated signaling kinases JNK and p-38 [25]
Another key observation was that the extended tubulin polymerization lag and reduced rate caused by Zn deficiency in IMR-32 cells and GD19 brain supernates is fully reversed by TCEP, a disulfide reducing agent. Oxidation and modification of protein cysteines can have profound effects on protein structure and function. In this line, tubulin with its 20 free sulfhydryl groups is an ideal candidate for oxidation. The presence of disulfide links between tubulin subunits was evidenced by the presence of tubulin oligomers of a molecular weight higher than 100 kDa in Zn deficient cortical neurons, IMR-32 cells and GD19 brain that disappear upon reduction with TCEP. Further evidence of this type of modifications was the direct measurement of higher levels of oxidized thiols under Zn deficiency conditions. Interestingly, two other commons oxidative modifications, carbonylation and nitration, were not detected [44]. It should be considered that not only cysteine oxidation of tubulin, but that of other related proteins, such as tau and microtubule associated protein 2 [5], can affect the assembly of microtubules.
Upon exposure to a Zn deficient media, a rapid increase in oxidant and H2O2 levels are observed in differentiated PC-12 and IMR-32 cells [22]. This is in part triggered by the activation of the NMDA receptor, the influx of calcium, and the associated activation of NADPH oxidase and nitric oxide synthase [22]. This is accompanied by a reduction in cellular reduced glutathione, and as currently observed, by a reduction in protein thiols both in vitro (IMR-32 cells) and in vivo (GD19 fetal rat brain) conditions of Zn deficiency. Accordingly, an altered tubulin polymerization was also observed in both models. These findings strongly supports the concept that even marginal Zn deficiencies, similar to those found in human populations, can have a major impact on brain physiology through oxidative stress and cytoskeletal alterations.
Transcription factor NF-κB is widely distributed in the nervous system. Although their target genes are still not fully defined, NF-κB regulates central processes to neurodevelopment [45-48]. NF-κB is highly expressed during neurodevelopment and in the mature mouse brain in areas of active neurogenesis which suggests its involvement in cell proliferation [49]. NF-κB protects neurons from different pro-apoptotic stimuli (reviewed in [46]). The antiapoptotic action of NF-κB can be related to its role in the regulation of several pro-survival genes. Furthermore, a large body of evidence indicates that NF-κB modulates synaptic plasticity, and memory (reviewed in [50]). The nuclear transport of the active NF-κB is impaired secondary to alterations in tubulin polymerization [28], and also when in the dynein and dynactin complex is affected [51]. As a consequence of a decreased NF-κB nuclear translocation, a lower expression of NFκB-regulated genes occurs in Zn deficient IMR-32 cells [28]. Similarly, we also observed an altered nuclear translocation of NF-κB, in GD19 fetal brain from rats fed zinc deficient or marginal zinc diets, compared to controls [22]. The fact that NAC and LA can restore the translocation of NF-κB and the expression of NF-κB-regulated genes (bcl-2, cyclin D1 and c-myc) supports the concept that the NF-κB nuclear translocation mechanism in neurons is in part dependent on tubulin redox-state. Of important physiological implications, Chowanadisai et al. [16] demonstrated that a suboptimal Zn nutrition during development can alter the expression of NF-κB-dependent genes, including the NMDA receptor subunits NR1, NR2A, and NR2B. Therefore, gestational suboptimal Zn nutrition, with its associated oxidative stress condition, can have a major impact on fetal brain NF-κB modulation.
In summary, in the current paper we advance a potential mechanism by which tubulin polymerization dynamics in the nervous system can be affected by a deficit of Zn. Oxidative modifications of tubulin, and possibly of other associated proteins, contribute to the defective tubulin polymerization. Prevention of tubulin oxidation by LA and NAC can help to restore tubulin dynamics and NF-κB nuclear translocation and transactivating activity, a key signal for proper cell growth and differentiation in the developing brain.
Acknowledgments
This work was supported by grants from the University of California and NIH (grant # HD 01743), USA.
Abbreviations
- LA
(±)-α-lipoic acid
- EMSA
electrophoretic mobility shift assay
- FBS
fetal bovine serum
- GD
gestation day
- hRNP
heterogeneous nuclear ribonucleoprotein
- MAPKs
mitogen-activated protein kinases
- MZ
marginal zinc
- NAC
N-acetyl cysteine
- TCEP
tris(2-carboxyethyl)phosphine
- Zn
zinc
- ZD
zinc deficient
References
- 1.Schnapp BJ, Vale RD, Sheetz MP, Reese TS. Single microtubules from squid axoplasm support bidirectional movement of organelles. Cell. 1985;40:455–462. doi: 10.1016/0092-8674(85)90160-6. [DOI] [PubMed] [Google Scholar]
- 2.Schnapp BJ, Vale RD, Sheetz MP, Reese TS. Microtubules and the mechanism of directed organelle movement. Ann N Y Acad Sci. 1986;466:909–918. doi: 10.1111/j.1749-6632.1986.tb38476.x. [DOI] [PubMed] [Google Scholar]
- 3.Banan A, Choudhary S, Zhang Y, Fields JZ, Keshavarzian A. Oxidant-induced intestinal barrier disruption and its prevention by growth factors in a human colonic cell line: role of the microtubule cytoskeleton. Free Radic Biol Med. 2000;28:727–738. doi: 10.1016/s0891-5849(00)00160-x. [DOI] [PubMed] [Google Scholar]
- 4.Landino LM, Moynihan KL, Todd JV, Kennett KL. Modulation of the redox state of tubulin by the glutathione/glutaredoxin reductase system. Biochem Biophys Res Commun. 2004;314:555–560. doi: 10.1016/j.bbrc.2003.12.126. [DOI] [PubMed] [Google Scholar]
- 5.Landino LM, Robinson SH, Skreslet TE, Cabral DM. Redox modulation of tau and microtubule-associated protein-2 by the glutathione/glutaredoxin reductase system. Biochem Biophys Res Commun. 2004;323:112–117. doi: 10.1016/j.bbrc.2004.08.065. [DOI] [PubMed] [Google Scholar]
- 6.Bernhard D, Csordas A, Henderson B, Rossmann A, Kind M, Wick G. Cigarette smoke metal-catalyzed protein oxidation leads to vascular endothelial cell contraction by depolymerization of microtubules. Faseb J. 2005;19:1096–1107. doi: 10.1096/fj.04-3192com. [DOI] [PubMed] [Google Scholar]
- 7.Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci. 2005;6:449–462. doi: 10.1038/nrn1671. [DOI] [PubMed] [Google Scholar]
- 8.Keen CL, Taubeneck MW, Daston GP, Gershwin ME, Ansari A, Rogers J. Primary and secondary Zn deficiency as factors contributing to abnormal central nervous system development. Dev Brain Dysfunct. 1995;8:79–89. [Google Scholar]
- 9.Rogers JM, Oteiza PI, Keen CL. Zn and Manganese Deficiencies in Prenatal and Neonatal Development with Special Reference to the Central Nervous System. New York: Wiley-Liss; 1990. [Google Scholar]
- 10.Sandstead HH. Zinc is essential for brain development and function. J Trace Elem Exp Med. 2003;16:165–173. [Google Scholar]
- 11.Bentley ME, Caulfield LE, Ram M, Santizo MC, Hurtado E, Rivera JA, Ruel MT, Brown KH. Zinc supplementation affects the activity patterns of rural Guatemalan infants. J Nutr. 1997;127:1333–1338. doi: 10.1093/jn/127.7.1333. [DOI] [PubMed] [Google Scholar]
- 12.Gardner JM, Powell CA, Baker-Henningham H, Walker SP, Cole TJ, Grantham-McGregor SM. Zinc supplementation and psychosocial stimulation: effects on the development of undernourished Jamaican children. Am J Clin Nutr. 2005;82:399–405. doi: 10.1093/ajcn.82.2.399. [DOI] [PubMed] [Google Scholar]
- 13.Penland JG, Sandstead HH, Alcock NW, Dayal HH, Chen XC, Li JS, Zhao F, Yang JJ. A preliminary report: effects of zinc and micronutrient repletion on growth and neuropsychological function of urban Chinese children. J Am Coll Nutr. 1997;16:268–272. doi: 10.1080/07315724.1997.10718684. [DOI] [PubMed] [Google Scholar]
- 14.Arnold LE, DiSilvestro RA. Zinc in attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2005;15:619–627. doi: 10.1089/cap.2005.15.619. [DOI] [PubMed] [Google Scholar]
- 15.Brown KH, Peerson JM, Rivera J, Allen LH. Effect of supplemental zinc on the growth and serum zinc concentrations of prepubertal children: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2002;75:1062–1071. doi: 10.1093/ajcn/75.6.1062. [DOI] [PubMed] [Google Scholar]
- 16.Rudolf E, Cervinka M. Responses of human gingival and periodontal fibroblasts to a low-zinc environment. Altern Lab Anim. 2010;38:119–138. doi: 10.1177/026119291003800213. [DOI] [PubMed] [Google Scholar]
- 17.Hesketh JE. Impaired microtubule assembly in brain from zinc-deficient pigs and rats. Int J Biochem. 1981;13:921–926. doi: 10.1016/0020-711x(81)90019-7. [DOI] [PubMed] [Google Scholar]
- 18.Wang F, Zhao F, Guo J, Jing N. Mechanism of impairment to microtubule polymerization resulting from zinc deficiency during pregnancy and lactation in mice. Wei Sheng Yan Jiu. 2000;29:156–158. [PubMed] [Google Scholar]
- 19.Chowanadisai W, Kelleher SL, Lonnerdal B. Maternal zinc deficiency reduces NMDA receptor expression in neonatal rat brain, which persists into early adulthood. J Neurochem. 2005;94:510–519. doi: 10.1111/j.1471-4159.2005.03246.x. [DOI] [PubMed] [Google Scholar]
- 20.Corniola RS, Tassabehji NM, Hare J, Sharma G, Levenson CW. Zinc deficiency impairs neuronal precursor cell proliferation and induces apoptosis via p53-mediated mechanisms. Brain Res. 2008;1237:52–61. doi: 10.1016/j.brainres.2008.08.040. [DOI] [PubMed] [Google Scholar]
- 21.Adamo AM, Zago MP, Mackenzie GG, Aimo L, Keen CL, Keenan A, Oteiza PI. The role of zinc in the modulation of neuronal proliferation and apoptosis. Neurotox Res. 17:1–14. doi: 10.1007/s12640-009-9067-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aimo L, Cherr GN, Oteiza PI. Low extracellular zinc increases neuronal oxidant production through nadph oxidase and nitric oxide synthase activation. Free Radic Biol Med. 2010;48:1577–1587. doi: 10.1016/j.freeradbiomed.2010.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bananis E, Murray JW, Stockert RJ, Satir P, Wolkoff AW. Microtubule and motor-dependent endocytic vesicle sorting in vitro. J Cell Biol. 2000;151:179–186. doi: 10.1083/jcb.151.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mackenzie GG, Zago MP, Erlejman AG, Aimo L, Keen CL, Oteiza PI. alpha-Lipoic acid and N-acetyl cysteine prevent zinc deficiency-induced activation of NF-kappaB and AP-1 transcription factors in human neuroblastoma IMR-32 cells. Free Radic Res. 2006;40:75–84. doi: 10.1080/10715760500312305. [DOI] [PubMed] [Google Scholar]
- 25.Zago MP, Mackenzie GG, Adamo AM, Keen CL, Oteiza PI. Differential Modulation of MAP Kinases by Zinc Deficiency in IMR-32 Cells: Role of H(2)O(2) Antioxid Redox Signal. 2005;7:1773–1782. doi: 10.1089/ars.2005.7.1773. [DOI] [PubMed] [Google Scholar]
- 26.Mackenzie GG, Zago MP, Keen CL, Oteiza PI. Low intracellular zinc impairs the translocation of activated NF-kappa B to the nuclei in human neuroblastoma IMR-32 cells. J Biol Chem. 2002;277:34610–34617. doi: 10.1074/jbc.M203616200. [DOI] [PubMed] [Google Scholar]
- 27.Mackenzie GG, Keen CL, Oteiza PI. Zinc status of human IMR-32 neuroblastoma cells influences their susceptibility to iron-induced oxidative stress. Dev Neurosci. 2002;24:125–133. doi: 10.1159/000065691. [DOI] [PubMed] [Google Scholar]
- 28.Mackenzie GG, Keen CL, Oteiza PI. Microtubules are required for NF-kappaB nuclear translocation in neuroblastoma IMR-32 cells: modulation by zinc. J Neurochem. 2006;99:402–415. doi: 10.1111/j.1471-4159.2006.04005.x. [DOI] [PubMed] [Google Scholar]
- 29.Oteiza PI, Cuellar S, Lonnerdal B, Hurley LS, Keen CL. Influence of maternal dietary zinc intake on in vitro tubulin polymerization in fetal rat brain. Teratology. 1990;41:97–104. doi: 10.1002/tera.1420410110. [DOI] [PubMed] [Google Scholar]
- 30.Oteiza PI, Hurley LS, Lonnerdal B, Keen CL. Marginal zinc deficiency affects maternal brain microtubule assembly in rats. J Nutr. 1988;118:735–738. doi: 10.1093/jn/118.6.735. [DOI] [PubMed] [Google Scholar]
- 31.Peters JM, Wiley LM, Zidenberg-Cherr S, Keen CL. Influence of short-term maternal zinc deficiency on the in vitro development of preimplantation mouse embryos. Proc Soc Exp Biol Med. 1991;198:561–568. doi: 10.3181/00379727-198-43289. [DOI] [PubMed] [Google Scholar]
- 32.Hanz S, Perlson E, Willis D, Zheng JQ, Massarwa R, Huerta JJ, Koltzenburg M, Kohler M, van-Minnen J, Twiss JL, Fainzilber M. Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron. 2003;40:1095–1104. doi: 10.1016/s0896-6273(03)00770-0. [DOI] [PubMed] [Google Scholar]
- 33.Oteiza PI, Clegg MS, Zago MP, Keen CL. Zinc deficiency induces oxidative stress and AP-1 activation in 3T3 cells. Free Radic Biol Med. 2000;28:1091–1099. doi: 10.1016/s0891-5849(00)00200-8. [DOI] [PubMed] [Google Scholar]
- 34.Keen CL, Peters JM, Hurley LS. The effect of valproic acid on 65Zn distribution in the pregnant rat. J Nutr. 1989;119:607–611. doi: 10.1093/jn/119.4.607. [DOI] [PubMed] [Google Scholar]
- 35.Clegg MS, Keen CL, Lonnerdal B, Hurley LS. Influence of ashing techniques in the analysis of trace elements in animal tissue. I. Wet ashing. Biol Trace Element Res. 1981;3:107–115. doi: 10.1007/BF02990451. [DOI] [PubMed] [Google Scholar]
- 36.Baty JW, Hampton MB, Winterbourn CC. Detection of oxidant sensitive thiol proteins by fluorescence labeling and two-dimensional electrophoresis. Proteomics. 2002;2:1261–1266. doi: 10.1002/1615-9861(200209)2:9<1261::AID-PROT1261>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 37.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osborn L, Kunkel S, Nabel GJ. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc Natl Acad Sci U S A. 1989;86:2336–2340. doi: 10.1073/pnas.86.7.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 40.Aimo L, Mackenzie GG, Keenan AH, Oteiza PI. Gestational zinc deficiency affects the regulation of transcription factors AP-1, NF-kappaB and NFAT in fetal brain. J Nutr Biochem. 2010 doi: 10.1016/j.jnutbio.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verhey KJ, Gaertig J. The tubulin code. Cell Cycle. 2007;6:2152–2160. doi: 10.4161/cc.6.17.4633. [DOI] [PubMed] [Google Scholar]
- 42.Mackenzie GG, Oteiza PI. Zinc and the cytoskeleton in the neuronal modulation of transcription factor NFAT. J Cell Physiol. 2007;210:246–256. doi: 10.1002/jcp.20861. [DOI] [PubMed] [Google Scholar]
- 43.Caulfield LE, Zavaleta N, Shankar AH, Merialdi M. Potential contribution of maternal zinc supplementation during pregnancy to maternal and child survival. Am J Clin Nutr. 1998;68:499S–508S. doi: 10.1093/ajcn/68.2.499S. [DOI] [PubMed] [Google Scholar]
- 44.Landino LM, Hagedorn TD, Kim SB, Hogan KM. Inhibition of tubulin polymerization by hypochlorous acid and chloramines. Free Radic Biol Med. 2011;50:1000–1008. doi: 10.1016/j.freeradbiomed.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Albensi BC, Mattson MP. Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse. 2000;35:151–159. doi: 10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 46.Mattson MP, Culmsee C, Yu Z, Camandola S. Roles of nuclear factor kappaB in neuronal survival and plasticity. J Neurochem. 2000;74:443–456. doi: 10.1046/j.1471-4159.2000.740443.x. [DOI] [PubMed] [Google Scholar]
- 47.Mattson MP. NF-kappaB in the survival and plasticity of neurons. Neurochem Res. 2005;30:883–893. doi: 10.1007/s11064-005-6961-x. [DOI] [PubMed] [Google Scholar]
- 48.Mattson MP, Meffert MK. Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006;13:852–860. doi: 10.1038/sj.cdd.4401837. [DOI] [PubMed] [Google Scholar]
- 49.Denis-Donini S, Caprini A, Frassoni C, Grilli M. Members of the NF-kappaB family expressed in zones of active neurogenesis in the postnatal and adult mouse brain. Brain Res Dev Brain Res. 2005;154:81–89. doi: 10.1016/j.devbrainres.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 50.Romano A, Freudenthal R, Merlo E, Routtenberg A. Evolutionarily-conserved role of the NF-kappaB transcription factor in neural plasticity and memory. Eur J Neurosci. 2006;24:1507–1516. doi: 10.1111/j.1460-9568.2006.05022.x. [DOI] [PubMed] [Google Scholar]
- 51.Shrum CK, Defrancisco D, Meffert MK. Stimulated nuclear translocation of NF-kappaB and shuttling differentially depend on dynein and the dynactin complex. Proc Natl Acad Sci U S A. 2009;106:2647–2652. doi: 10.1073/pnas.0806677106. [DOI] [PMC free article] [PubMed] [Google Scholar]