

Abstract
Transcription factors AP-1, NF- κB and NFAT are central to brain development by regulating the expression of genes that modulate cell proliferation, differentiation, apoptosis, and synaptic plasticity. This work investigated the consequences of feeding zinc deficient and marginal zinc diets to rat dams during gestation on the modulation of AP-1, NF- κB and NFAT in fetal brain. Sprague-Dawley rats were fed from gestation day 0 a control diet ad libitum (25 μg zinc/g diet, C), a zinc deficient diet ad libitum (0.5 μg zinc/g diet, ZD), the control diet in the amounts eaten by the ZD rats (restrict fed, RF), or a diet containing a marginal zinc concentration ad libitum (10 μg zinc/g diet, MZD) until gestation day 19. AP-1-DNA binding was higher (50-190%) in nuclear fraction isolated from ZD, RF and MZD fetal brains compared to controls. In MZD fetal brain high levels of activation of the upstream mitogen-activated protein kinases JNK and p38 and low levels of ERK phosphorylation were observed. Total levels of NF- κB and NFAT activation were higher or similar in the ZD and MZD groups than in controls, respectively. However, NF- κB- and NFAT-DNA binding in nuclear fractions was markedly lower in ZD and MZD fetal brain than in controls (50-80%). The latter could be related to zinc deficiency-associated alterations of the cytoskeleton which is required for NF- κB and NFAT nuclear transport. In summary, suboptimal zinc nutrition during gestation could cause long term effects on brain function, partially through a deregulation of transcription factors AP-1, NF- κB and NFAT.
Keywords: zinc, brain, gestation, AP-1, NF-κB, NFAT
Introduction
Gestational zinc deficiency in humans has adverse effects on neurologic and behavioral development [for a review see (1) and references therein]. Current estimates of dietary and total zinc intake collected in the third National Health and Nutrition Examination Survey (NHANES III) were used to assess zinc intake in the U.S. population and to identify population groups which zinc status may be of concern. Based on the NHANES III data (1988-1994), Briefel et al show that more than 40 % of pregnant women have inadequate intakes of zinc and that young children are at greatest risk for inadequate zinc intake (2). In addition, zinc deficiency may arise from low bioavailability and/or interactions with other nutrients and losses of the mineral in different disease states (3, 4). Therefore, a condition of marginal zinc deficiency prevails among the population worldwide.
Zinc is critical for the physiology of the nervous system and zinc deficiency has been associated with altered neurodevelopment [(5) and references therein]. In humans, developmental zinc deficiency affects infant behavior, cognitive and motor performance (6-8), and could be associated with attention-deficit/hyperactivity disorders (9). As further evidence of the relevance of zinc for the developing nervous system, the supplementation of undernourished children with zinc improves developmental quotients, activity patterns and neuropsychological functions (6-8, 10). In animal models, gestational severe zinc deficiency is highly teratogenic affecting the nervous system as well as other organs (11). In rats, zinc deficiency causes neural tube defects, hydrocephaly, exencephaly, and anencephaly (12). Developmental marginal zinc deficiency does not cause teratogenicity or growth retardation in rats, but affects brain gene expression (13). In adolescent monkeys, and before signs of growth retardation, a moderate zinc deprivation alters daytime activity levels and attention performance (14). Given the potential adverse effects of developmental zinc deficiency on the onset of behavioral alterations and neurological disorders later in life, the understanding of the potential mechanisms involved is of critical relevance.
We previously observed in neuronal cells in culture, that zinc deficiency causes alterations in select cell signaling pathways. Zinc deficiency was associated with the activation of transcription factor AP-1 and an increase transcription of AP-1-driven genes (15). The activation of AP-1 and of the upstream mitogen-activated protein kinases (MAPKs) JNK and p38 occurs as a consequence of the increased oxidant production associated with neuronal zinc deficiency (15). Zinc deficiency also affects the modulation of transcription factors NF-κB and NFAT in neuronal cells (16-18). NF-κB and NFAT are activated in the cytosol by degradation of the IκB subunit or calcineurin-mediated dephosphorylation, respectively. Upon activation, a nuclear localization signal is unmasked which drives the translocation of NF-κB and NFAT into the nucleus. In IMR-32 cells, a decrease in intracellular zinc levels is associated with the activation of the initial events of the NF-κB and NFAT signaling cascades (17-19) that is triggered by a rise in cellular oxidants (17, 19). However, the active NFAT and NF-κB accumulate in the cytosol secondary to impairment in the translocation of the active transcription factors from the cytosol to the nucleus (16-18). Consistently, this was associated with a decreased transactivation of NF-κB- and NFAT-driven genes (16-18).
Transcription factors AP-1, NF-κB and NFAT play critical roles in brain development and function through the regulation of multiple events including cell proliferation, differentiation, apoptosis, and synaptic plasticity (20-26). A deregulation of these cell signals during early brain development could explain the teratogenic effects of severe zinc deficiency. Of higher relevance for human populations, marginal zinc nutrition during gestation could lead to irreversible and long term effects in brain function, in part, by altering the modulation of AP-1, NF-κB and NFAT. To test this hypothesis we investigated, in vivo, if a severe and a marginal zinc nutrition imposed to rats during gestation can affect the modulation of transcription factors AP-1, NF-κB, and NFAT in the brain from gestation day (GD) 19 fetuses. The activation of upstream events and NFAT and NF-κB nuclear transport were investigated.
Materials and Methods
Materials
The oligonucleotide containing the consensus sequence for NFAT, primary antibodies for β-tubulin (sc-9104), IκB (sc-371), ERK1/2 (sc-93), p- ERK1/2 (sc-7383), heterogeneous nuclear ribonucleoprotein (hnRNP) (sc-32301), NFATc4 (sc-1153), p38 (sc-7149), p50 (sc-7178), and secondary anti rabbit IgG-HRP (sc-2970), anti mouse IgG2a-HRP (sc-2970),and anti goat IgG-HRP (sc-2056) antibodes were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Primary antibodies for p-IκB (# 2859), JNK (# 9252), p-JNK1/2 (# 9251), and p-p38 (# 9211), and secondary anti mouse IgG-HRP (# 7074) antibody were obtained from Cell Signaling Technology (Beverly, MA). The oligonucleotides containing the consensus sequence for NF- κB, AP-1 and OCT-1, and the reagents for the EMSA assay were obtained from Promega (Madison, WI). PVDF membranes were obtained from BIO-RAD (Hercules, CA) and Chroma Spin-10 columns were obtained from Clontech (Palo Alto, CA). The ECL plus western blotting system was from Amersham Pharmacia Biotech Inc. (Piscataway, NJ). All other reagents were from the highest quality available and were purchased from Sigma (St. Louis, MO).
Animals and animal care
All procedures were in agreement with standards for the care of laboratory animals as outlined in the NIH Guide for the Care and Use of Laboratory Animals. All procedures were administered under the auspices of the Animal Resource Services of the University of California, Davis, which is accredited by the American Association for the Accreditation of Laboratory Animal Care. Experimental protocols were approved before implementation by the University of California, Davis Animal Use and Care Administrative Advisory Committee, and were administered through the Office of the Campus Veterinarian.
Adult Sprague-Dawley rats (Charles River, Wilmington, MA) (200-225 g) were housed individually in suspended stainless steel cages in a temperature (22–23°C)- and photoperiod (12 h/d)-controlled room. An egg white protein based diet with adequate Zn (25 μg Zn/g) was the standard control diet (27). Animals were fed the control diet for one week before breeding. Males and females were caged together overnight and the following morning the presence of a sperm plug confirmed a successful breeding. On gestation day 0 (GD 0), rats (7 animals/group) were divided into four groups and fed one of the following diets: a control diet ad libitum (25 μg zinc/g diet, C), a zinc deficient diet ad libitum (0.5 μg zinc/g diet, ZD), the control diet in the amounts eaten by the zinc deficient rats (the average intake of the ZD group was used to calculate the amount fed to the restrict fed group, RF), or a diet containing a marginal zinc concentration ad libitum (10 μg zinc/g diet, MZD) until gestation day 19 (GD 19). Initial maternal body weights were similar among the groups. Zinc concentration of the diets was verified by ICP-AES following procedures previously described (28). Food intake was recorded daily, and body weight was measured at 5-d intervals.
On GD 19, the dams were anesthetized with isofluorane (2mg/kg body weight), and laparatomies were performed. Maternal blood was collected by cardiac puncture into heparinized syringes (Sarstead, Princeton, NJ) and centrifuged at 800 × g for 10 min. The plasma was removed and stored at −80°C until analyzed. The gravid uterus was removed, its weight recorded, and individual placentas and fetuses were subsequently collected. Fetuses were examined for gross structural malformations and weighed. Fetal brains were excised, rinsed in ice-cold PBS, the meninges removed, and after weighing, the tissue was processed according to the corresponding assays.
Electrophoretic mobility shift assay (EMSA)
Nuclear and cytosolic fractions were isolated as previously described (29, 30), with minor modifications (18). Briefly, fetal brains were manually homogenized in ice-cold hypotonic buffer [50 mg of tissue/ 200 μl of buffer A: 10 mM HEPES (pH 8.0), 1.5 mM MgCl2, 5 mM KCl, 0.5 mM dithiothreitol, 0.4 mM NaVaO4, 0.5 mM PMSF and inhibitors of proteases and phosphatases]. Once the homogenates were obtained, another 200 μl of buffer A containing 0.2% (v/v) of Igepal were added and the samples were incubated on ice for 10 min. After 15 min of centrifugation at 850 × g, the supernatant (cytosolic fraction) was separated and stored at − 80 °C. The nuclear pellet was gently resuspended in 100 μl of buffer B [20 mM Tris (pH 8.0), 25% (v/v) glycerol, 1.5 mM MgCl2, 0.4 M NaCl, 0.2 mM EDTA, 0.5 mM dithiothreitol, 0.4 mM NaVaO4, 0.5 mM PMSF and inhibitors of proteases and phosphatases] and incubated for 15 min on ice. After centrifuging for 30 min at 14,000 × g, the supernatant (nuclear fraction) was transferred into new tubes and stored at −80 °C. Total tissue fractions were prepared as described below (Western blot analysis). For the EMSA, the oligonucleotides containing the consensus sequence for AP-1, NFAT, NF-κB or OCT-1 were end labelled with [γ −32P] ATP using T4 polynucleotide kinase and purified using Chroma Spin-10 columns. Samples were incubated with the labelled oligonucleotide (20,000-30,000 cpm) for 20 min at room temperature in 1× binding buffer [5× binding buffer: 50 mM Tris-HCl buffer, pH 7.5, containing 20% (v/v) glycerol, 5 mM MgCl2, 2.5 mM EDTA, 2.5 mM DTT, 250 mM NaCl and 0.25 mg/ml poly(dI-dC)]. The products were separated by electrophoresis in a 6% (w/v) non-denaturing polyacrilamide gel using 0.5 × TBE (Tris/borate 45 mM, EDTA 1mM) as the running buffer. The gels were dried and the radioactivity quantitated in a Phosphoimager 840 (Amersham Pharmacia Biotech. Inc., Piscataway, NJ).
Western blot analysis
For the preparation of total tissue extracts, fetal brains were homogenized [50 mg of tissue/ 400 μl of lysis buffer: 50 mmol/L Tris (pH 7.5), 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 50 mM NaF, 2 mM NaVaO4 containing inhibitors of proteases and phosphatases and 1 % (v/v) Igepal]. The homogenates were incubated at 4°C for 30 min and centrifuged at 10,000 × g for 20 min. The supernatants were collected and protein concentration was measured (31).
Aliquots of total or nuclear fractions containing 25-50 μg protein were separated by reducing 10% (w/v) polyacrylamide gel electrophoresis and electroblotted to PVDF membranes. Colored molecular weight standards (Amersham, Piscataway, NJ) were ran simultaneously. Membranes were blotted overnight in 5% (w/v) non-fat milk and incubated in the presence of the corresponding primary antibodies (see Materials section) for 90 min at 37°C. After incubation with the secondary antibody (HRP-conjugated) (1:10,000 dilution), 90 min at room temperature, the conjugates were visualized by chemiluminescence detection in a Phosphoimager 840.
Results
Animal outcome
As reported previously (32), the food intake of the dams in the ZD group was significantly lower (p < 0.05) since the fifth day of gestation compared to the C group (data not shown). The food intake of the MZD and C rats was similar throughout the studied period. At GD 19, the cumulative food intake was 401 ± 43, 305 ± 13, 302 ± 40, and 423 ± 53 g for the C, RF, ZD and MZD groups, respectively. Maternal weight gain at GD 19 was significantly lower (p < 0.01) in the RF and ZD groups (66 ± 4 and 18 ± 4 g, respectively), compared to the C and MZD groups (121 ± 5 and 123 ± 5 g, respectively). Even though the ZD and RF groups were fed the same amount of food, the maternal weight gain was significantly lower in the ZD group compared to the RF group (p<0.05). As previously observed (32), the zinc deficient diet caused a marked decrease in maternal zinc plasma levels (66 %) compared to the control diet and adversely affected different gestational parameters (Table 1). Significantly, in the mentioned study (32) the concentration of zinc in GD 19 fetal brain supernatants was 35% lower than in controls. A lower litter weight, live fetuses per litter, placental and fetal weight were observed in the ZD animals compared to all other groups. As expected, a high percentage of resorptions, gross fetal structural malformations, and sites affected, characterized the ZD group. Although the RF dams gained significantly less weight during gestation than the C dams, neither the maternal plasma zinc levels nor the measured gestational parameters were affected by the low food intake. Maternal plasma zinc concentration was significantly lower (54 %) in the MZD group at GD 19 than in the C group. However, the gestational parameters in the MZD group were similar to those of the C group, except for a significant higher fetal weight (30 %, p<0.05). Given the well known deregulation of glucose homeostasis in zinc deficiency, the occurrence of maternal hyperglycemia during gestation could explain the high fetal weight in the MZD group. Brain weight was lower in ZD and higher in MZD fetuses compared to controls (p < 0.05). Although no significant differences were observed in the brain/body weight ratio, future studies should characterize the different brain structures for size and cellularity, in order to determine if brain weight differences are simply related to fetal size or if they are accompanied by structural brain abnormalities.
Table 1.
Reproductive parameters in dams fed diets containing different zinc concentrations.
| Parameters | Control | ZD | RF | MZD |
|---|---|---|---|---|
| Litter weight (g) | 66.0 ± 2.1a | 17.6 ± 6.3 b | 57.4 ± 2.2 a | 65.9 ± 2.8 a |
| Live fetuses/litter | 15.7 ± 0.3 a | 6.8 ± 2.3 b | 14.5 ± 0.9 a | 14.7 ± 0.7 a |
| Fetal weight (g) | 2.20 ± 0.05 a | 1.35 ± 0.12 b | 2.09 ± 0.07 a | 2.51 ± 0.15 c |
| Placental weight (g) | 0.43 ± 0.01 a | 0.29 ± 0.02 b | 0.39 ± 0.01 a | 0.48 ± 0.02 a |
| Brain weight (mg) | 76.2 ± 1.4 a | 52.8 ± 5.9 b | 76.4 ± 0.8 a | 83.6 ± 2.2 c |
| Malformed fetuses (%)1 | 0.5 ± 0.5 a | 60.0 ± 13.9 b | 0.0 ± 0.0 a | 1.0 ± 1.0 a |
| Resorptions (%)2 | 0.5 ± 0.5 a | 51.7 ± 14.9 b | 3.5 ± 2.2 a | 1.7 ± 1.1 a |
| Sites affected (%)3 | 1.0 ± 0.7 a | 81.2 ± 8.0 b | 3.5 ± 2.2 a | 2.6 ± 1.9 a |
| Maternal plasma Zn (μM) | 15.4 ± 0.6 a | 5.2 ± 0.8 b | 14.4 ± 1.0 a | 7.1 ± 0.7 c |
Values are shown as ± S.E.M., n: 6-8. Data with different superscripts are statistically different by one way ANOVA .(p< 0.05)
(Malformed/Total live fetuses) . 100
(Resorptions/Total sites) . 100
(Resorptions + malformed fetuses/Total sites) . 100
Zinc deficiency affects the modulation of transcription factors AP-1, NF-κB and NFAT in fetal brain
The overall activation of AP-1, NF-κB and NFAT was investigated by measuring in fetal brain total fractions the AP-1, NF-κB and NFAT binding to their respective consensus oligonucleotides by EMSA (Fig.1). The OCT-1-DNA binding was also evaluated as a loading control, since this transcription factor was not found to be affected by zinc deficiency in cells in culture (16). NF-κB-DNA binding in fetal brain total fractions from the ZD animals was significantly higher (34 and 63%, respectively) compared to the C and RF groups. There was a trend (p < 0.065) for higher brain AP-1- DNA binding in the ZD group than in the C and RF groups. NFAT- and OCT-1-DNA binding in total fetal brain fractions was similar among the evaluated groups (Fig.1). The transcription factor-DNA binding was subsequently evaluated in cytosolic and nuclear fractions isolated from fetal brain (Fig. 2). The NF-κB- and NFAT-DNA binding in nuclear fractions was markedly lower in the ZD compared to the C and RF groups. In ZD fetal brain, the nuclear/cytosolic ratio for NF-κB- and NFAT-DNA binding was 92 and 52 % lower, respectively, in the ZD than in the C group. In the ZD group, cytosolic NF-κB-DNA binding was 3-fold higher (p < 0.01) compared to the C and RF groups. Although a high AP-1-DNA binding was observed in the nuclear fraction from the ZD fetal brain compared to C, restrict feeding also caused the activation of AP-1 (Fig. 2). The nuclear/cytosolic ratio for OCT-DNA binding was similar among the groups.
Figure 1. A severe gestational zinc deficiency affects NF-κB, NFAT and AP-1 modulation in rat fetal brain total fractions.
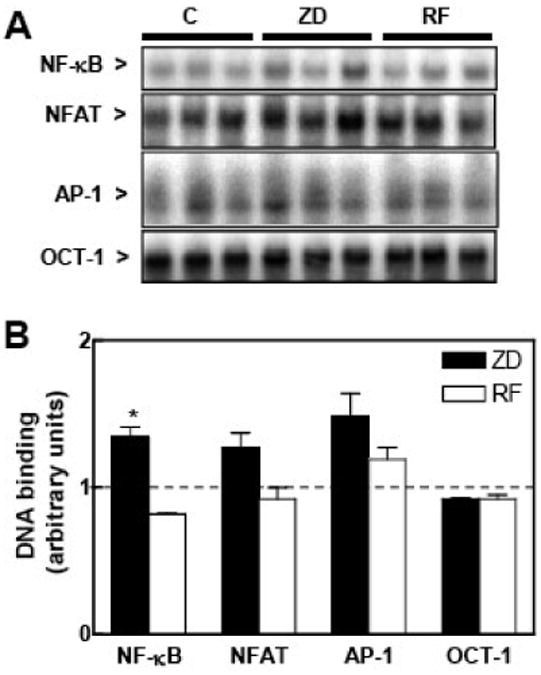
At GD 0, dams were fed control diets ad libitum (C) or at a restricted level (RF), or a zinc deficient (ZD) diet until GD 19. Total tissue fractions were prepared from fetal brains as described in the Materials and Methods section. A- EMSA for NF-κB, NFAT, AP-1 and OCT-1 in total fractions. B- After the EMSA assays, bands were quantitated, normalized to control levels (dotted line) and results for ZD (full bars) and RF (empty bars) groups are shown as means ± SEM of 4-6 animals per group. *Significantly different compared to the C and RF groups (p < 0.05, one way ANOVA test).
Figure 2. A severe gestational zinc deficiency affects the DNA binding of NF-κB, NFAT and AP-1 in nuclear and cytosolic fractions isolated from rat fetal brain.
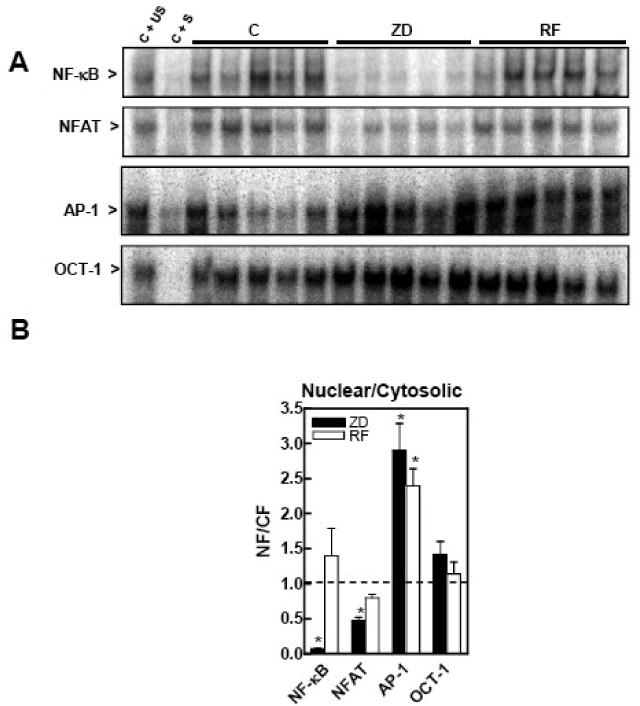
At GD 0, dams were fed control diets ad libitum (C) or at a restricted level (RF), or a zinc deficient (ZD) diet until GD 19. Nuclear and cytosolic fractions were prepared from fetal brains as described in the Materials and Methods section. A- EMSA for NF-κB, NFAT, AP-1 and OCT-1 in nuclear fractions. To determine the specificity of each transcription factor-DNA complex, the control nuclear fraction (C) was incubated in the presence of 100-fold molar excess of unlabeled oligonucleotide containing the consensus sequence for either the specific (C + S) or an unspecific (C + US) transcription factor before the binding assay. B- After the EMSA assays, bands were quantitated and the ratio nuclear/ cytosolic DNA binding was calculated. Results for ZD (full bars) and RF (empty bars) groups are normalized to control levels (dotted line) and are shown as means ± SEM of 5-6 animals per group. *Significantly different compared to the C group (p < 0.05, one way ANOVA test).
Marginal zinc nutrition during gestation affects the modulation of transcription factors AP-1, NF-κB and NFAT in fetal brain
The overall activation of NF-κB and AP-1, measured as the DNA binding in fetal brain total fractions by EMSA, was significantly higher (25 and 70%, respectively) in the MZD compared to the C group (Fig. 3A). NFAT- and OCT-1-DNA binding in total fractions was similar between the MZD and C groups. Upstream events in the NF-κB and AP-1 signaling cascade were next evaluated, measuring the activation (as the phosphorylated forms) of IκB and of the MAPKs p38, JNK1/2 and ERK1/2 by Western blot. Although the levels of phosphorylated IκB were similar in MZD and C fetal brain (Fig. 3B), the content of total IκB was significantly lower (25%, p < 0.05) in fetal brain from the MZD compared to the C group (1.00 ± 0.1 and 0.75 ± 0.05, respectively). The levels of phosphorylation were higher for the MAPKs p38 and JNK1/2 (69 and 70%, respectively) and lower (40%) for ERK1/2 in fetal brain from the MZD than in the C rats (Fig.3B). IκB and MAPK activation was only characterized in the C and MZD groups because the patterns of NF-κB and AP-1 regulation measured by EMSA are similar in ZD and MZD fetal brain. Furthermore, marginal zinc nutrition is the condition that more closely represents what occurs in human populations.
Figure 3. A marginal zinc nutrition during gestation affects NF-κB, NFAT, AP-1 transcription factors, IκB and MAPKs in fetal brain total fractions.
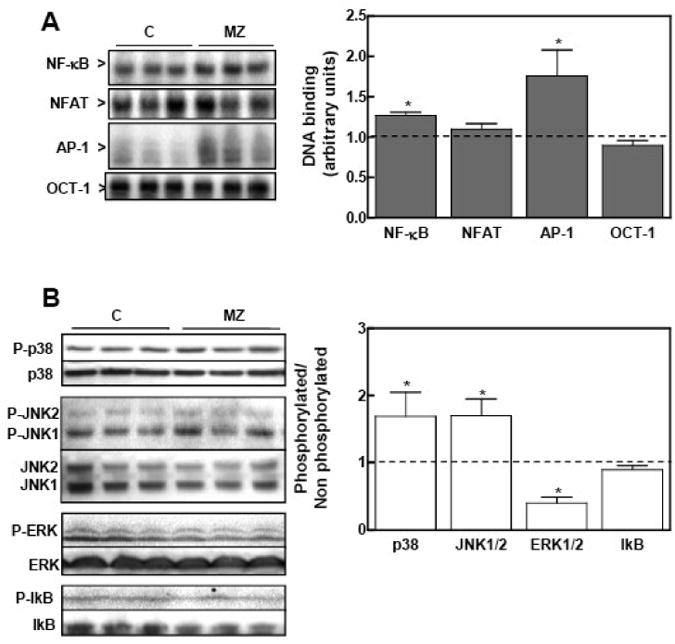
At GD 0, dams were fed ad libitum a control (C) or a marginal zinc (MZD) diet until GD 19. Total fetal brain tissue fractions were prepared as described in the Materials and Methods section. A- Transcription factor-DNA binding. Left panel: EMSA for NF-κB, NFAT, AP-1 and OCT-1 in total tissue fractions. Right panel: after the EMSA assays, bands were quantitated, normalized to control levels (dotted line) and results for the MZD group (gray bars) are shown as means ± SEM of 4-6 animals per group. *Significantly different compared to the C group (p < 0.05, one way ANOVA test). B- Immunoblots for IκB and MAPKs. Left panel: Western blots for phosphorylated JNK1/2 (p-JNK1/2), phosphorylated-p38 (p-p38), phosphorylated ERK1/2 (p-ERK1/2), phosphorylated IκB (p-IκBα) and their non- phosphorylated forms. Right panel: After quantitation, results were expressed as the ratio phosphorylated/non-phosphorylated protein. Results for the MZD group were normalized to control values (dotted line) and are shown as means ± SEM of 6 animals per group. *Significantly different compared to the C group (p < 0.05, one way ANOVA test).
The transcription factor-DNA binding was next investigated in nuclear and cytosolic fractions (Fig. 4). The DNA binding of NF-κB- and NFAT was lower (61 and 41%, respectively), that of AP-1 was higher (45%), and that of OCT-1 remained unchanged in fetal brain nuclear fractions isolated from the MZD rats compared to the C group. NF-κB-DNA binding in cytosolic fractions was markedly higher (2.2-fold) in MZD than in C fetal brain. The nuclear/cytosolic ratio of DNA binding from fetal brain was significantly lower for NF-κB and NFAT, higher for AP-1, and remained unchanged for OCT-1, in the MZD compared to C group (Fig. 4B). In support of a zinc deficiency-induced alteration in NFAT and NF-κB nuclear transport, the nuclear content of NFATc4 and p50 was lower in nuclear fractions from MZD fetal brain than in controls (Fig. 4C).
Figure 4. A marginal zinc nutrition during gestation affects the DNA binding of NF-κB, NFAT and AP-1 in fetal brain nuclear and cytosolic fractions.
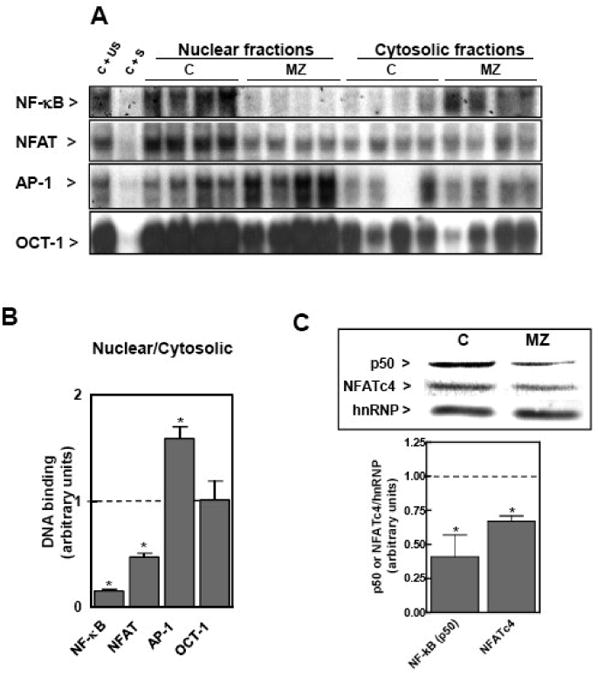
At GD 0, dams were fed ad libitum a control (C) o or a marginal zinc (MZD) diet until GD 19. Nuclear and cytosolic fractions were prepared from fetal brains as described in the Materials and Methods section. A- EMSA for NF-κB, NFAT, AP-1 and OCT-1 in nuclear fractions. To determine the specificity of each transcription factor-DNA complex, the control nuclear fraction (C) was incubated in the presence of 100-fold molar excess of unlabeled oligonucleotide containing the consensus sequence for either the specific (C + S) or an unspecific (C + US) transcription factor before the binding assay. B- After the EMSA assays, bands were quantitated and the ratio nuclear/cytosolic DNA binding was calculated. Results for the MZD group were normalized to control values (dotted line) and are shown as means ± SEM of 6 animals per group. *Significantly different compared to the C group (p < 0.05, one way ANOVA test). C- Western blots for NFATc4, p50 and hnRNP in nuclear fractions. After quantitation, results were expressed by the hnRNP content as loading control. Results for the MZD group were normalized to control values (dotted line) and are shown as means ± SEM of 3 animals per group. *Significantly different compared to the C group (p < 0.05, one way ANOVA test).
Discussion
Gestational zinc deficiency can occur under several different conditions including low dietary zinc intake (2), poor availability of zinc from food sources (4), exposure to several toxicants (33), and in proinflammatory processes (34, 35). In several of these conditions either teratogenicity or altered behavior has been reported in the offspring.
Although the deleterious effects of zinc deficiency on behavior and cognition are accepted and reported to occur in human populations, there is limited knowledge on the mechanisms underlying those effects. The modulation of AP-1, NF-κB and NFAT has been found to be altered in neuronal cells in culture (15-18). Given the participation of these transcription factors in the regulation of various cellular events including cell proliferation, differentiation, apoptosis and synaptic plasticity; they are critical for brain development and function (20-26). To further understand the relevance of appropriate zinc nutrition for normal neurodevelopment, this work investigated the potential impact of marginal or deficient zinc nutrition during gestation on the modulation of transcription factors AP-1, NF-κB and NFAT in GD 19 fetal brains. We demonstrate that suboptimal zinc nutrition, even at marginal levels, during gestation has a major impact on those fetal brain signals. Therefore, a deregulation of transcription factors AP-1, NF-κB and NFAT, could underlie the brain teratogenesis triggered by severe gestational zinc deficiency, and the behavioral, locomotor and cognitive abnormalities associated with marginal zinc nutrition (36).
In the present study, AP-1-DNA binding was higher in total and nuclear fractions isolated from fetal brain from rats fed a zinc deficient diet or the control diet at a restricted intake compared to those fed a control diet ad libitum. These results indicate that AP-1 activation is triggered both, by gestational zinc deficiency and by undernutrition. However, rats fed marginal zinc diets showed no differences in food intake compared to controls, but AP-1 was activated in the fetal brain. This indicates that AP-1 activation in fetal brain can specifically occur as a consequence of inadequate maternal zinc nutrition. Thus, the activation of AP-1 upon gestational food restriction could be in part secondary to impaired zinc nutrition and to other factors associated with general maternal undernutrition. As previously observed in IMR-32 neuroblastoma cells (15), in Zn deficient fetal brain AP-1 activation occurs downstream the activation of MAPKs p38 and JNK. The signal mediating p38 and JNK phosphorylation in IMR-32 cells was found to be an increase in the steady state levels of cell oxidants (15, 19). Studies in JNK knock out mice showed that these MAPK are critical for neurodevelopment having pro- and antiapoptotic actions (37, 38). Although the role of p38 on neurodevelopment is still poorly defined, p38 exerts a negative control of neural stem cell proliferation (39) and could modulate the activity of transcription factor PAX-6 which controls eye and brain development (40). Significantly, embryos of knock out mice for MEKK 4, a kinase upstream p38 and JNK with strong expression in the developing neuroepithelium, present spina bifida and exencephaly (41). An increased expression of JNK-1 may cause neuronal cell death following excitotoxin-induced injury (42). In addition, phosphorylation of JNK and its consequent activation of c-jun leads to neuronal death (43, 44). Also in agreement with previous findings in zinc deficient IMR-32 cells, lower levels of ERK phosphorylation were observed in fetal brain from rats fed marginal zinc diets. In zinc deficient neuroblastoma cells, ERK inhibition is independent of oxidative stress and could have a critical role in the associated decrease in neuronal proliferation and apoptotic death (15) (Adamo et al, unpublished results). Therefore the deregulation of fetal brain MAPKs, and the activation of AP-1 caused by zinc deficiency could, through modifications in the normal neurodevelopmental pattern of cell proliferation and apoptosis.
Transcription factors NF-κB and NFAT are widely distributed in the nervous system. Although their target genes are still not fully defined, NF-κB and NFAT regulate central processes to neurodevelopment (22, 23, 26, 45). NF-κB is highly expressed during neurodevelopment and in the mature mouse brain in areas of active neurogenesis which suggests its involvement in cell proliferation (46). NF-κB protects neurons from different pro-apoptotic stimuli (reviewed in (23)) and NF-κB inactivation by proteasome inhibitors triggers apoptosis in various areas of the central nervous system (47) and in IMR-32 cells (18). The antiapoptotic action of NF-κB can be related to its role in the regulation of several pro-survival genes. Furthermore, a large body of evidence indicates that NF-κB modulates synaptic plasticity, and memory (reviewed in (48). With regard to NFAT, its binding to DNA in combination with other transcription factors indicates its function as an integrator of different signaling cascades. Calcineurin-NFAT are known to regulate axonal growth, plasticity, and neuronal survival. Mice bearing mutations in NFATc2, c3 and c4 present remarkable alterations in neurotrophins- or netrins-induced axonal out growth (21). Furthermore, NFAT protects cerebellum granule cells from apoptosis (49), and regulates the expression of brain-derived neurotrophic factor, a modulator of neuronal survival and proliferation (50).
We have observed in IMR-32 cells and in rat cortical neurons, that zinc deficiency has a dual effect on the NF-κB signaling cascade. Zinc deficiency causes the activation of NF-κB initial steps, including IκBα phosphorylation and degradation. Both events are triggered by the increase in neuronal oxidants associated with zinc deficiency (19). The nuclear transport of the active NF-κB is impaired secondary to alterations in tubulin polymerization (16). As a consequence, lower expression of NFκB-regulated genes occurs in zinc deficient IMR-32 cells (16). A similar altered modulation, high activation in total cell extracts and decreased nuclear transport and dependent gene expression, was observed for transcription factor NFAT in zinc deficient IMR-32 cells (17). Similarly, in GD 19 fetal brain from rats fed zinc deficient or marginal zinc diets, NF-κB-DNA binding was high in total fractions and low in nuclear fractions, compared to controls. The fact that the results of NFAT-DNA binding in fetal brain total fractions differed from our findings in IMR-32 cells may be explained by the brain cellular heterogeneity. However, the findings in nuclear fractions are consistent with an altered nuclear transport of both active transcription factors. They are also in agreement with the hypothesis that the altered microtubule polymerization associated with zinc deficiency can affect transcription factors such as NF-κB and NFAT that after activation in the cytosol are transported into the nucleus. In this regard, we showed that a functional cytoskeleton is required for the nuclear transport of NFAT and NF-κB in neuronal cells (16, 17). Moreover, the in vitro tubulin polymerization kinetics is impaired in brain supernatants from GD 19 fetuses from dams fed zinc deficient diets throughout gestation (32). The finding of low protein levels of NFATc4 and p50 (a NF-κB component) in nuclear fractions from MZD fetal brain supports the concept that the nuclear transport is being affected. Of important physiological implications, Chowanadisai et al. (13) demonstrated that a suboptimal Zn nutrition during development alters the expression of several NF-κB-dependent genes, including the NMDA receptor subunits NR1, NR2A, NR2B.
There is still limited knowledge on the genes regulated by NF-κB, NFAT and AP-1 in the nervous system. Their role in modulating cellular proliferation, apoptosis and differentiation has been widely described in different tissues. A highly coordinated process involving the regulation of cellular proliferation and survival, and cellular crosstalk is required during the development of the nervous system (51). Thus, the present results demonstrating that gestational suboptimal zinc nutrition has a major impact on fetal brain NF-κB, NFAT and AP-1 modulation, provides new mechanistic insights on the adverse effects of zinc deficiency on neurodevelopment. Of major significance for human populations, the observed alterations occur even at marginal dietary zinc levels Further studies are needed to link these findings to alterations in developmental patterns of neuronal proliferation, differentiation, synaptic plasticity and apoptosis, as well as on their impact on behavior and cognition. Alterations in those tightly coordinated processes by developmental zinc deficiency can cause changes in brain structure/function (e.g. brain circuitry and connectivity) that can affect children's behavior and cognition and/or increase the risk for adult neurological disorders.
Acknowledgments
This work was supported by grants from the University of California and NIH (grant # HD 01743), USA.
Abbreviations
- EMSA
electrophoretic mobility shift assay
- MAPKs
mitogen-activated protein kinases
- GD
gestation day
- hnRNP
heterogeneous nuclear ribonucleoprotein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Scheplyagina LA. Impact of the mother's zinc deficiency on the woman's and newborn's health status. J Trace Elem Med Biol. 2005;19:29–35. doi: 10.1016/j.jtemb.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 2.Briefel RR, Bialostosky K, Kennedy-Stephenson J, McDowell MA, Ervin RB, Wright JD. Zinc intake of the U.S. population: findings from the third National Health and Nutrition Examination Survey, 1988-1994. J Nutr. 2000 May;130:1367S–73S. doi: 10.1093/jn/130.5.1367S. [DOI] [PubMed] [Google Scholar]
- 3.Wada L, King JC. Trace element nutrition during pregnancy. Clin Obstet Gynecol. 1994 Sep;37:574–86. doi: 10.1097/00003081-199409000-00010. [DOI] [PubMed] [Google Scholar]
- 4.Walsh CT, Sandstead HH, Prasad AS, Newberne PM, Fraker PJ. Zinc: health effects and research priorities for the 1990s. Environ Health Perspect. 1994 Jun;102(2):5–46. doi: 10.1289/ehp.941025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caulfield LE, Zavaleta N, Shankar AH, Merialdi M. Potential contribution of maternal zinc supplementation during pregnancy to maternal and child survival. Am J Clin Nutr. 1998 Aug;68:499S–508S. doi: 10.1093/ajcn/68.2.499S. [DOI] [PubMed] [Google Scholar]
- 6.Bentley ME, Caulfield LE, Ram M, Santizo MC, Hurtado E, Rivera JA, Ruel MT, Brown KH. Zinc supplementation affects the activity patterns of rural Guatemalan infants. J Nutr. 1997 Jul;127:1333–8. doi: 10.1093/jn/127.7.1333. [DOI] [PubMed] [Google Scholar]
- 7.Gardner JM, Powell CA, Baker-Henningham H, Walker SP, Cole TJ, Grantham-McGregor SM. Zinc supplementation and psychosocial stimulation: effects on the development of undernourished Jamaican children. Am J Clin Nutr. 2005 Aug;82:399–405. doi: 10.1093/ajcn.82.2.399. [DOI] [PubMed] [Google Scholar]
- 8.Penland JG, Sandstead HH, Alcock NW, Dayal HH, Chen XC, Li JS, Zhao F, Yang JJ. A preliminary report: effects of zinc and micronutrient repletion on growth and neuropsychological function of urban Chinese children. J Am Coll Nutr. 1997 Jun;16:268–72. doi: 10.1080/07315724.1997.10718684. [DOI] [PubMed] [Google Scholar]
- 9.Arnold LE, DiSilvestro RA. Zinc in attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2005 Aug;15:619–27. doi: 10.1089/cap.2005.15.619. [DOI] [PubMed] [Google Scholar]
- 10.Brown KH, Peerson JM, Rivera J, Allen LH. Effect of supplemental zinc on the growth and serum zinc concentrations of prepubertal children: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2002 Jun;75:1062–71. doi: 10.1093/ajcn/75.6.1062. [DOI] [PubMed] [Google Scholar]
- 11.Keen CL, Taubeneck MW, Daston GP, Gershwin ME, Ansari A, Rogers J. Primary and secondary Zn deficiency as factors contributing to abnormal central nervous system development. Dev Brain Dysfunct. 1995;8:79–89. [Google Scholar]
- 12.Rogers JM, Keen CL, Hurley LS. Zinc deficiency in pregnant Long-Evans hooded rats: teratogenicity and tissue trace elements. Teratology. 1985 Feb;31:89–100. doi: 10.1002/tera.1420310111. [DOI] [PubMed] [Google Scholar]
- 13.Chowanadisai W, Kelleher SL, Lonnerdal B. Maternal zinc deficiency reduces NMDA receptor expression in neonatal rat brain, which persists into early adulthood. J Neurochem. 2005 Jul;94:510–9. doi: 10.1111/j.1471-4159.2005.03246.x. [DOI] [PubMed] [Google Scholar]
- 14.Golub MS, Takeuchi PT, Keen CL, Hendrickx AG, Gershwin ME. Activity and attention in zinc-deprived adolescent monkeys. Am J Clin Nutr. 1996 Dec;64:908–15. doi: 10.1093/ajcn/64.6.908. [DOI] [PubMed] [Google Scholar]
- 15.Zago MP, Mackenzie GG, Adamo AM, Keen CL, Oteiza PI. Differential Modulation of MAP Kinases by Zinc Deficiency in IMR-32 Cells: Role of H(2)O(2) Antioxid Redox Signal. 2005 Nov-Dec;7:1773–82. doi: 10.1089/ars.2005.7.1773. [DOI] [PubMed] [Google Scholar]
- 16.Mackenzie GG, Keen CL, Oteiza PI. Microtubules are required for NF-kappaB nuclear translocation in neuroblastoma IMR-32 cells: modulation by zinc. J Neurochem. 2006 Oct;99:402–15. doi: 10.1111/j.1471-4159.2006.04005.x. [DOI] [PubMed] [Google Scholar]
- 17.Mackenzie GG, Oteiza PI. Zinc and the cytoskeleton in the neuronal modulation of transcription factor NFAT. J Cell Physiol. 2007 Jan;210:246–56. doi: 10.1002/jcp.20861. [DOI] [PubMed] [Google Scholar]
- 18.Mackenzie GG, Zago MP, Keen CL, Oteiza PI. Low intracellular zinc impairs the translocation of activated NF-kappa B to the nuclei in human neuroblastoma IMR-32 cells. J Biol Chem. 2002 Sep 13;277:34610–7. doi: 10.1074/jbc.M203616200. [DOI] [PubMed] [Google Scholar]
- 19.Mackenzie GG, Zago MP, Erlejman AG, Aimo L, Keen CL, Oteiza PI. alpha-Lipoic acid and N-acetyl cysteine prevent zinc deficiency-induced activation of NF-kappaB and AP-1 transcription factors in human neuroblastoma IMR-32 cells. Free Radic Res. 2006 Jan;40:75–84. doi: 10.1080/10715760500312305. [DOI] [PubMed] [Google Scholar]
- 20.Graef IA, Chen F, Crabtree GR. NFAT signaling in vertebrate development. Curr Opin Genet Dev. 2001 Oct;11:505–12. doi: 10.1016/s0959-437x(00)00225-2. [DOI] [PubMed] [Google Scholar]
- 21.Graef IA, Wang F, Charron F, Chen L, Neilson J, Tessier-Lavigne M, Crabtree GR. Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell. 2003 May 30;113:657–70. doi: 10.1016/s0092-8674(03)00390-8. [DOI] [PubMed] [Google Scholar]
- 22.Albensi BC, Mattson MP. Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse. 2000 Feb;35:151–9. doi: 10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 23.Mattson MP, Culmsee C, Yu Z, Camandola S. Roles of nuclear factor kappaB in neuronal survival and plasticity. J Neurochem. 2000 Feb;74:443–56. doi: 10.1046/j.1471-4159.2000.740443.x. [DOI] [PubMed] [Google Scholar]
- 24.Gutierrez H, Hale VA, Dolcet X, Davies A. NF-kappaB signalling regulates the growth of neural processes in the developing PNS and CNS. Development. 2005 Apr;132:1713–26. doi: 10.1242/dev.01702. [DOI] [PubMed] [Google Scholar]
- 25.Raivich G, Behrens A. Role of the AP-1 transcription factor c-Jun in developing, adult and injured brain. Prog Neurobiol. 2006 Apr;78:347–63. doi: 10.1016/j.pneurobio.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 26.Mattson MP, Meffert MK. Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006 May;13:852–60. doi: 10.1038/sj.cdd.4401837. [DOI] [PubMed] [Google Scholar]
- 27.Keen CL, Peters JM, Hurley LS. The effect of valproic acid on 65Zn distribution in the pregnant rat. J Nutr. 1989 Apr;119:607–11. doi: 10.1093/jn/119.4.607. [DOI] [PubMed] [Google Scholar]
- 28.Clegg MS, Keen CL, Lonnerdal B, Hurley LS. Influence of ashing techniques in the analysis of trace elements in animal tissue. I. Wet ashing. Biol Trace Element Res. 1981;3:107–15. doi: 10.1007/BF02990451. [DOI] [PubMed] [Google Scholar]
- 29.Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983 Mar 11;11:1475–89. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Osborn L, Kunkel S, Nabel GJ. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc Natl Acad Sci U S A. 1989 Apr;86:2336–40. doi: 10.1073/pnas.86.7.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 32.Oteiza PI, Cuellar S, Lonnerdal B, Hurley LS, Keen CL. Influence of maternal dietary zinc intake on in vitro tubulin polymerization in fetal rat brain. Teratology. 1990 Jan;41:97–104. doi: 10.1002/tera.1420410110. [DOI] [PubMed] [Google Scholar]
- 33.Taubeneck MW, Daston GP, Rogers JM, Keen CL. Altered maternal zinc metabolism following exposure to diverse developmental toxicants. Reprod Toxicol. 1994 Jan-Feb;8:25–40. doi: 10.1016/0890-6238(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 34.Taubeneck MW, Daston GP, Rogers JM, Gershwin ME, Ansari A, Keen CL. Tumor necrosis factor-alpha alters maternal and embryonic zinc metabolism and is developmentally toxic in mice. J Nutr. 1995 Apr;125:908–19. doi: 10.1093/jn/125.4.908. [DOI] [PubMed] [Google Scholar]
- 35.Coyle P, Tran N, Fung JN, Summers BL, Rofe AM. Maternal dietary zinc supplementation prevents aberrant behaviour in an object recognition task in mice offspring exposed to LPS in early pregnancy. Behav Brain Res. 2009 Jan 30;197:210–8. doi: 10.1016/j.bbr.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 36.Golub MS, Keen CL, Gershwin ME, Hendrickx AG. Developmental zinc deficiency and behavior. J Nutr. 1995 Aug;125:2263S–71S. doi: 10.1093/jn/125.suppl_8.2263S. [DOI] [PubMed] [Google Scholar]
- 37.Karin M. Mitogen-activated protein kinase cascades as regulators of stress responses. Ann N Y Acad Sci. 1998 Jun 30;851:139–46. doi: 10.1111/j.1749-6632.1998.tb08987.x. [DOI] [PubMed] [Google Scholar]
- 38.Karin M, Gallagher E. From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life. 2005 Apr-May;57:283–95. doi: 10.1080/15216540500097111. [DOI] [PubMed] [Google Scholar]
- 39.Sato K, Hamanoue M, Takamatsu K. Inhibitors of p38 mitogen-activated protein kinase enhance proliferation of mouse neural stem cells. J Neurosci Res. 2008 Aug 1;86:2179–89. doi: 10.1002/jnr.21668. [DOI] [PubMed] [Google Scholar]
- 40.Yan Q, Liu WB, Qin J, Liu J, Chen HG, Huang X, Chen L, Sun S, Deng M, et al. Protein phosphatase-1 modulates the function of Pax-6, a transcription factor controlling brain and eye development. J Biol Chem. 2007 May 11;282:13954–65. doi: 10.1074/jbc.M611476200. [DOI] [PubMed] [Google Scholar]
- 41.Chi H, Sarkisian MR, Rakic P, Flavell RA. Loss of mitogen-activated protein kinase kinase kinase 4 (MEKK4) results in enhanced apoptosis and defective neural tube development. Proc Natl Acad Sci U S A. 2005 Mar 8;102:3846–51. doi: 10.1073/pnas.0500026102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schauwecker PE. Seizure-induced neuronal death is associated with induction of c-Jun N-terminal kinase and is dependent on genetic background. Brain Res. 2000 Nov 24;884:116–28. doi: 10.1016/s0006-8993(00)02888-2. [DOI] [PubMed] [Google Scholar]
- 43.Eilers A, Whitfield J, Babij C, Rubin LL, Ham J. Role of the Jun kinase pathway in the regulation of c-Jun expression and apoptosis in sympathetic neurons. J Neurosci. 1998 Mar 1;18:1713–24. doi: 10.1523/JNEUROSCI.18-05-01713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ham J, Eilers A, Whitfield J, Neame SJ, Shah B. c-Jun and the transcriptional control of neuronal apoptosis. Biochem Pharmacol. 2000 Oct 15;60:1015–21. doi: 10.1016/s0006-2952(00)00372-5. [DOI] [PubMed] [Google Scholar]
- 45.Mattson MP. NF-kappaB in the survival and plasticity of neurons. Neurochem Res. 2005 Jun-Jul;30:883–93. doi: 10.1007/s11064-005-6961-x. [DOI] [PubMed] [Google Scholar]
- 46.Denis-Donini S, Caprini A, Frassoni C, Grilli M. Members of the NF-kappaB family expressed in zones of active neurogenesis in the postnatal and adult mouse brain. Brain Res Dev Brain Res. 2005 Jan 1;154:81–9. doi: 10.1016/j.devbrainres.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 47.Taglialatela G, Kaufmann JA, Trevino A, Perez-Polo JR. Central nervous sys DNA fragmentation induced by the inhibition of nuclear factor kappa B. Neurorep. 1998 Feb 16;9:489–93. doi: 10.1097/00001756-199802160-00024. [DOI] [PubMed] [Google Scholar]
- 48.Romano A, Freudenthal R, Merlo E, Routtenberg A. Evolutionarily-conser role of the NF-kappaB transcription factor in neural plasticity and memory. Eu Neurosci. 2006 Sep;24:1507–16. doi: 10.1111/j.1460-9568.2006.05022.x. [DOI] [PubMed] [Google Scholar]
- 49.Benedito AB, Lehtinen M, Massol R, Lopes UG, Kirchhausen T, Rao A, Bonni A. The transcription factor NFAT3 mediates neuronal survival. J Biol Chem. 2005 Jan 28;280:2818–25. doi: 10.1074/jbc.M408741200. [DOI] [PubMed] [Google Scholar]
- 50.Groth RD, Mermelstein PG. Brain-derived neurotrophic factor activation of NF (nuclear factor of activated T-cells)-dependent transcription: a role for the transcrip factor NFATc4 in neurotrophin-mediated gene expression. J Neurosci. 2003 Sep 3;23:8125–34. doi: 10.1523/JNEUROSCI.23-22-08125.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hidalgo A, ffrench-Constant C. The control of cell number during central nervous system development in flies and mice. Mech Dev. 2003 Nov;120:1311–25. doi: 10.1016/j.mod.2003.06.004. [DOI] [PubMed] [Google Scholar]