

Abstract
As brain testosterone plays both androgenic and estrogenic actions due to its conversion into estrogen via aromatase naturally, it is unclear that the age-related reduction of testosterone increased risk of Alzheimer's disease (AD) in men is mediated through androgen alone or both androgen and estrogen mechanisms. Our previous studies using a gene-based approach in mouse model to block the conversion of testosterone into estrogen (aromatase gene knock-out, ArKO), found a depletion of estrogen and increase in testosterone endogenously in males. Here, we use crossing the ArKO mice with APP23 transgenic mice, a mouse model of AD, to produce APP23/Ar+/− mice to study the estrogen-independent effect of testosterone on AD. We found a significant reduction in brain plaque formation, improved cognitive function and increase NEP activity in male APP23/Ar+/− mice compared with age-matched male APP23 controls. In addition, we found, for the first time, a reduction of β-secretase (BACE1) enzyme activity, mRNA level and protein expression in the male APP23/Ar+/− mice, suggesting that endogenous testosterone, independent from estrogen, may protect against AD in males via two major mechanisms, downregulation of BACE1 activities at transcriptional level to reduce β amyloid production and upregulation of NEP activities to enhance bate amyloid degradation.
Introduction
Recent studies have suggested that testosterone may play an important role in the prevention of Alzheimer's disease (AD) in men. One study found that men with AD have ∼50% less brain testosterone than do age-matched men without AD (Rosario et al., 2004). Decreased circulating testosterone in aged men is associated with impaired cognitive function and increased risk of dementia and AD (Hogervorst et al., 2001; Moffat et al., 2004; Rosario and Pike, 2008). Conversely, testosterone replacement therapy and high free testosterone levels in men are associated with improved verbal and spatial memory (Sternbach, 1998) and reduced risk of AD (Moffat et al., 2006). The association between circulating testosterone and AD has also been demonstrated in AD-like transgenic (APP23) mice (Rosario et al., 2006). However, the mechanism underlying this association remains unclear.
Because testosterone can be converted into estrogen by aromatase in vivo, it is difficult to determine whether the link between testosterone and AD neuropathology is due to the aromatization of testosterone into estrogen or is estrogen-independent—especially as studies have found that estrogen may be neuroprotective in women (Inestrosa et al., 1998; Farrag et al., 2002; Sherwin, 2003; Tsolaki et al., 2005) and may regulate amyloid β (Aβ), a hallmark of AD (Xu et al., 1998).
Aβ is generated from proteolytic cleavages of amyloid precursor protein (APP) by β- and γ-secretases (Hardy and Selkoe, 2002). β-site APP cleaving enzyme 1 (BACE1) plays a particularly important role in Aβ production, by cleaving APP at the N-terminal end of the Aβ sequence, producing a 14 kDa C-terminal fragment (CTF)β or C99 (Pinnix et al., 2001). BACE1 gene knock-out has been shown to prevent Aβ synthesis in mice (Cai et al., 2001; Luo et al., 2001; Roberds et al., 2001; Dominguez et al., 2005), and studies have found that BACE1 activity levels are significantly increased in sporadic AD cases (Fukumoto et al., 2002; Holsinger et al., 2002; Yang et al., 2003; Li et al., 2004), particularly in neurons around Aβ plaques (Zhao et al., 2007). Because of the role of BACE1 in Aβ production, the inhibition of BACE1 has been one of the most hopeful approaches for AD therapy in addition to amyloid β-peptide vaccination (Haass, 2004; Marjaux et al., 2004).
Impaired Aβ clearance may also contribute to the accumulation of Aβ in the AD brain. Studies have shown that insulin-degrading enzyme (IDE) and neprilysin (NEP) are involved in Aβ clearance. IDE degrades the intracellular domain of APP and is involved in clearing Aβ from the brain (Qiu et al., 1998; Mukherjee et al., 2000; Leissring et al., 2003). Genetic linkage studies found that late-onset AD loci are linked to sequences of the IDE gene (Bertram et al., 2000; Ertekin-Taner et al., 2000). Other studies have suggested that NEP, whose mRNA and protein levels are significantly decreased in AD brains compared with age-matched normal brains (Yasojima et al., 2001a,b), may be the principal Aβ-degrading enzyme (Iwata et al., 2001; Marr et al., 2003).
In this study, we used a genetic approach to deplete aromatase in male APP23 mice to investigate the estrogen-independent effect of testosterone on AD-related processes and to identify the mechanisms involved. We found that endogenous testosterone, independent of estrogen, protects against Aβ neuropathology via BACE1 reduction and NEP upregulation and improves cognitive function in the male double-transgenic mice.
Materials and Methods
Experimental mice.
All mice were maintained in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and all experimental protocols were approved by the local IRB. Generation of the B6, D2-TgN (Thy1-APP23Swe) line of transgenic mice (APP23) and the aromatase gene knock-out (ArKO) line of mice on a C57BL/6 background have been described previously (Sturchler-Pierrat et al., 1997; Azcoitia et al., 2001). Heterozygous APP23 transgenic mice (APP23+/−) were crossed with homozygous ArKO mice (Ar−/−), and the resulting APP23/Ar+/− (APP23+/−Ar+/−) offspring were intercrossed through brother-sister mating to obtain the genotypes for the present study. Age-matched littermates having the following genotypes were used as controls: APP23 (APP23+/−Ar+/+) and wild-type (WT) (APP23−/−Ar+/+). The line was maintained by crossing heterozygous transgenic mice with F1 breeders having a C57BL/6 background. Mice were housed two to four per cage in standard plastic cages with bedding and were maintained on a 12 h light/dark cycle with access to food (NIH-07 formula) and acidified and filtered water ad libitum.
Genotyping.
The mice were genotyped using PCR. Tail tissue was digested with Proteinase K overnight at 56°C, and genomic DNA was isolated using a DNeasy Tissue Kit (Qiagen) and amplified by PCR using the following primer pairs: pair 1: 5′-ACCACCGTGGAGCTCCTTCCCGTGAA-3′ and 5′-GCAACTGCAGTGTGTACTGTTTCTTC-3′, for APP; pair 2: 5′-CCTTGACGATCGTTCATAC-3′ and 5′-GAGAGTTCATGAGAGTCTGG-3′, for the aromatase mutated gene. PCR was performed at the following parameters: 94°C, 1 min; 65°C, 2 min; 72°C, 3 min; 35 cycles. The PCR products were separated by 1% agarose gel electrophoresis and detected by staining with ethidium bromide.
Tissue preparation and immunohistochemistry.
Male mice of each genotype were anesthetized and their brains were quickly removed and bisected. One hemisphere was frozen for use in Western blot, ELISA, enzyme activity and RIA analyses, while the other hemisphere was prepared for immunohistochemical analyses. For immunohistochemistry, hemispheres were either fixed with 4% paraformaldehyde or were frozen at −80°C, then serially sectioned (15–30 μm in thickness) in the sagittal plane with a Leica CA 1900 cryostat. Eight to 10 sections (∼120 μm apart) were immunostained for Aβ (rabbit anti-Aβ-peptide; 1:250; Zymed), neuronal marker NeuN (mouse anti-NeuN; 1:500; Millipore Bioscience Research Reagents), or APP intracellular c-terminal fragment AICD (rabbit anti-APP-CT20; 1:1000; Calbiochem). After incubation in primary antisera, the sections were incubated with the appropriate species-specific biotinylated secondary antibodies for 30 min followed by ABC reagent (Vectastain Elite, Vector Laboratories) for 30 min at room temperature. Immunoreactivity was visualized using 3,3′-diaminobenzidine as the chromagen (DAB Substrate kit, Vector Laboratories). Aβ immunostaining was documented by digital imaging then processed with a Leica DMLS complementary software package (MagnaFire SP). Tissue sections containing CA1 and CA3 regions of the hippocampus were imaged at 20× magnification. ImagePro Plus software (Media Cybernetics) was used to count neurons and measure positive area. To standardize measurements, the field of interest was defined as the image taken at 20×.
Testosterone radioimmunoassay.
Testosterone levels were determined with a competitive radioimmunoassay (RIA) method (Diagnostic Systems Laboratories). Either serum or brain homogenate (200 μl) was mixed with 100 μl of anti-testosterone serum. Following an incubation of 4 h at 4°C, [I125] testosterone was added to this mixture, which was incubated for an additional 24 h at 4°C. Proteins were precipitated, removed after centrifugation, and the radioactivity of the remaining supernatant was counted with a gamma counter. The sensitivity of the testosterone RIA was 3.0 pg/ml.
Enzyme activities of IDE, NEP and BACE1.
IDE enzyme activity was measured as described previously (Song et al., 2003). In brief, brains were homogenized in 50 mm potassium phosphate buffer, pH 7.3, containing 200 μm PMSF and a proteinase inhibitor mix (Sigma-Aldrich). The samples were centrifuged, and the supernatant fraction was used for IDE activity measurement. The hydrolysis of fluorogenic substrate peptides (2 μm Abz-GGFLRKHGQED-Dnp as substrate in 20 mm potassium phosphate buffer, pH 7.3) was measured by detecting an increase in fluorescence (excitation at 318 nm and emission at 419 nm) that occurred upon peptide bond cleavage. The max velocity of IDE activity was calculated during the first 20 min and was indicated as fluorescence unit/min per microgram of protein. For the NEP activity assay, the mouse brains were homogenized in 100 mm MES buffer, pH 6.5, with proteinase inhibitors (Sigma-Aldrich). The homogenate was centrifuged at 20,000 × g for 45 min to separate the membrane fraction, and the supernatant was removed. The membrane pellet was resuspended in MES buffer and was directly used in the NEP activity assay as previously described (Li and Hersh, 1995). Activity assays for BACE were performed by using synthetic peptide substrates containing either the APPwt BACE site (MCA-Glu-Val-Lys-Met-Asp-Ala-Glu-Phe-(Lys-DNP)-OH) or APPsw, in which Lys-Met are substituted with NL. In the MV mutant, M is substituted with V. Substrates were used at 50 mm and reactions were performed in 50 mm [2-(N-morpholino)ethanesulfonic acid] with 50 mm acetic acid, pH 5.5. Enzymatic crude extracts and fluorescent-labeled peptides were incubated for various times at 37°C. The reaction mixtures were quenched and absorbed at 383 nm with a fluorescent plate reader.
Aβ ELISA.
To quantify the levels of soluble Aβ, mouse brains were homogenized in 0.1 m carbonate/50 mm NaCl buffer (pH 11.5) containing 10 μg/ml leupeptin and 20 μg/ml aprotinin. Lysates were centrifuged at 14,000 rpm and 4°C for 20 min, and the supernatants were used for measurement of soluble Aβ with an ELISA kit for human Aβ40 and Aβ42 (Invitrogen). The pellets were dissolved in 5 m guanidine (in 50 mm Tris-HCl, pH 8.0) for 4 h at room temperature and used for measurement of insoluble Aβ40 and Aβ42 with an ELISA kit, as above. The final values were normalized to the amount of loaded wet tissue and analyzed for significance using the Student's t test.
Western blot.
Aliquots of brain homogenates from APP23, APP23/Ar+/−, and wild-type mice were lysed with RIPA buffer, and 50–150 μg of total protein was subjected to SDS-PAGE (8–12% acrylamide). Separated proteins were then transferred onto polyvinylidene fluoride membranes. The blots were probed with the following antibodies: anti-BACE1 monoclonal antibody (R&D Systems), anti-APP C-terminal fragment C99 (APPC8,; 1:500; a kind gift from Dr. Dennis J. Selkoe, Harvard University School of Medicine, Boston, MA), anti-AICD (rabbit anti-APP-CT20; 1:1000; Calbiochem), anti-IDE polyclonal antibody (Oncogene Research Products), anti-NEP polyclonal antibody (Millipore Bioscience Research Reagents), and anti–β actin antibody (Sigma-Aldrich) as described previously (He et al., 2007).
RNA isolation and reverse transcription PCR.
To compare BACE1 expression levels, we used the following primers for reverse transcription (RT)-PCR: mouse BACE1 forward primer, 5′-AGACGCTACACATCCTGGTG-3′, and backward primer, 5′CCTGGGTGTAGGGCACATAC-3′. The amplified BACE1 fragment was 146 bp. Mouse s18 was used as a loading control: forward primer, 5′-CAGAAGGACGTGAAGGATGG-3′, and backward primer, 5′CAGTGGTCTTGGTGTGCTGA-3′. The amplified mouse s18 fragment was 159 bp. Total RNA was extracted from the brains of 18-month-old APP23 and APP23/Ar+/− mice (n = 5) using an RNeasy mini kit (Qiagen). RT-PCR was performed using a One-Step RT-PCR kit (Invitrogen) and the following PCR cycles: 50°C for 30 min, 94°C for 2 min, followed by 25 cycles at 94°C for 15 s, 49°C for 30 s, and 68°C for 1 min. Real-time RT-PCR was performed as described previously by Zhao et al. (2007). In addition, to study the effect of testosterone on BACE transcription activity in vitro, we transfected 293 cells with pB1P-A vector containing a BACE1 promoter (−1941 to +292) upstream as previous published (He et al., 2007). After cells treated with vehicle or testosterone at 2 or 4 nm for 24 h, we isolated RNA and performed RT-PCR or real-time RT-PCR as described above.
Hole-board behavioral task.
The same testing protocol as described by Brosnan-Watters and Wozniak (1997) was used with a few minor modifications (He et al., 2007). The testing area for the hole-board was done in an isolated laboratory room. Extra-maze spatial cues were provided in the form of the experimenter in front of the board, a computer on one side of the board, a large desk on the other side of the board, and a large window behind the board that provided an external light source. The rotating hole-board (San Diego Instruments) used in our experiment is a 46 × 46 cm white plastic square floor enclosed by 4 clear plastic sides. The floor has 4 holes 2.5 cm in diameter cut into it, spaced evenly near each corner of the board with 28 cm between each hole. Each hole is ∼2 cm deep with a metal screen covering the bottom.
Testing was done over a 2 week period with 10 d of habituation and 2 d of data collection. Mice were handled for 1 min and placed in the hole-board for 3 min during each day of handling to acquaint them with the experimenter, hole-board, and procedure. During the first 5 d of habituation, the holes were covered and 8 pieces of sharp cheddar cheese (8 mm × 3 mm blocks) were scattered around the hole-board. In the last 5 d of habituation, the holes were uncovered and 4 pieces of cheese were used, one in each hole, to train the mice to poke their heads into the hole to retrieve the food. On the ninth day of habituation, each mouse was single-caged with only two pellets of rodent chow ∼2 cm in length. The amount of food eaten overnight on the ninth day was measured. On day 10, the food was removed from the cage and the mouse was fasted overnight before testing.
During data collection, a piece of cheese was placed in three of the holes beneath a screen, allowing the odor of the food to emanate from the hole but restricting access to it. A fourth piece of cheese was placed on top of the screen in the target hole, making it accessible to the mouse. Each mouse was given 10 trials in which to find the baited (target) hole. The first 2 trials are each 15 min long and the following 8 trials are each 5 min long. The first 2 trials are longer to allow the mouse time to get adjusted to the testing and explore the board. A trial consisted of placing the mouse in a start tube in the center of the hole-board, then removing the tube and allowing the mouse to explore. A trial ended when the mouse retrieved the cheese from the target hole or the trial time elapsed and the experimenter manually ended the trial. A computer was used to measure the response of each mouse via infrared beams at the bottom of each hole. A correct response was recorded when the mouse poked its head into the target hole, while an error was recorded when the mouse poked its head into a hole that was not the target. A good trial was defined as a trial in which the first head-poke was in the target hole.
The board was rotated and cleaned between each trial with water, and between each mouse with a mild Virex solution, precluding mice from using proximal visual, olfactory, or tactile cues to locate the target hole. After the first day of testing, the mouse was given ¼ the amount of food eaten between days 9 and 10. Retention testing or memory testing was done 24 h after the first day of data collection. The procedure performed was exactly the same as the first day with the only difference being that the mouse had a quarter the amount of its diet available the day before, instead of being fasted.
In brief, the memory performance was measured cumulatively across the 10 trials. Each mouse was awarded a score of “1” when its first poke was a correct poke (into the target hole), “0” when its first poke was incorrect but it made a correct poke within the given time, or “−1” when it failed to make a correct poke. Scores were averaged within each group, and each subsequent score was added to those before it, so that a perfect performance (first poke = correct poke for every trial) would yield a scale value score of 10 by the tenth trial.
Statistics.
Data were expressed as means ± SEM. Statistical analyses were performed either with ANOVA followed by least significant difference post hoc analysis (multiple comparisons) or unpaired t test (pairwise comparisons). Pearson's correlation coefficients were used for correlational analyses. The level of significance was set at p < 0.05.
Results
Genetic knock-out of aromatase in male APP23 mice increases endogenous testosterone levels and decreases estrogen levels in serum and in the brain
To examine the estrogen-independent effect of testosterone on AD pathology in males, we detected age-related changes in sex hormone levels in the brains and sera of male APP23/Ar+/−, APP23, and WT mice at 6, 12, and 18 months of age. The levels of testosterone in the serum and in the brain decreased gradually with age in all male mice regardless of genotype. However, the levels of testosterone in APP23/Ar+/− male mice of all ages were significantly higher than those in age-matched APP23 and WT male mice (Fig. 1A,B). Additionally, no estrogen could be detected in the APP23/Ar+/− mice (data not shown), suggesting that aromatase gene knock-out prevented the conversion of endogenous testosterone into estrogen.
Figure 1.

Testosterone levels in male APP23, APP23/Ar+/− and WT mice. A, B, Total testosterone was detected by RIA in serum (A) and brain (B) extractions from male APP23/Ar+/− (n = 10), APP23 (n = 10), and WT (n = 10) mice at 6, 12, and 18 months of age. *p < 0.05 and **p < 0.001 compared with WT and APP23 control mice.
APP23/Ar+/− male mice exhibit reduced amyloid accumulation and plaque formation in the brain compared with control mice
To study whether increased endogenous testosterone in APP23 male mice would alter APP processing and amyloid plaque formation in the brain, we used ELISA (Invitrogen) to detect Aβ40 and Aβ42 in the brains of APP23 and APP23/Ar+/− male mice at the ages of 3, 6, 9 and 18 months. As shown in Figure 2A, the ratio of Aβ42 to Aβ40 in APP23/Ar+/− mice was significantly reduced at 9 and 18 months of age compared with the ratio in age-matched APP23 mice, suggesting the possible role of testosterone in prohibiting Aβ42 accumulation. Immunohistochemical analysis of amyloid plaque formation and brain plaque density in 18-month-old APP23 (n = 11) and APP23/Ar+/− (n = 8) mice revealed a significant reduction in the numbers of plaques in the cortices of APP23/Ar+/− mice compared with APP23 mice (Fig. 2B,C).
Figure 2.

Analysis of amyloid levels, amyloid plaques and neuronal loss in the brains of 18-month-old APP23 and APP23/Ar+/− male mice. A, ELISA analysis of the reduced Aβ42/Aβ40 ratio in male APP23/Ar+/− mice compared with age-matched APP23. B, APP23 mice developed massive plaques in the cortex at the age of 18 months while small plaques were observed in the APP23/Ar+/− mice at the same age. C, The density of plaques from 19 mouse brain samples (APP23 n = 11 and APP23/Ar+/− n = 8) were analyzed. The density of plaques in APP23/Ar+/− mice were significantly less than in APP23 mice (**p < 0.001). D, Immunostaining of NeuN (1:1000) in the brain from an 18-month-old APP23 male mouse showed extensive neuronal loss in the CA3 region of the hippocampus (indicated in box) compared with the hippocampi from WT and APP23/Ar+/− male mice at the same age.
APP23/Ar+/− male mice exhibit less neuronal loss than do control mice
Another major hallmark of AD neuropathology is neuronal loss. To further examine the effect of endogenous testosterone on AD-like neuropathology, we performed immunohistochemical staining with a neuronal marker, NeuN, and compared neuron densities in the hippocampi of APP23 and APP23/Ar+/− male mice at 18 months of age. Male APP23/Ar+/− mice exhibited less neuronal loss than did male APP23 mice in the CA3 region of the hippocampus (Fig. 2D), suggesting that the increased endogenous testosterone in male APP23/Ar+/− mice not only prevented amyloid deposition in the brain, but also protected against age-related neuronal loss.
APP23/Ar+/− male mice exhibit decreased BACE1 protein expression and activity compared with control mice
To investigate whether the reduction of Aβ in APP23/Ar+/− male mice is partially mediated by the attenuation of BACE1, we used Western blot analysis to detect APP, BACE1, and C99 protein expression levels in the brains of APP23/Ar+/− and APP23 mice. Eighteen-month-old APP23/Ar+/− male mice exhibited a significant reduction in BACE1 and C99 protein levels compared with age-matched APP23 mice, while no difference in APP level was found (Fig. 3A,B). To examine the effect of testosterone on Aβ production at the cellular level, we also used immunohistochemical staining to detect APP intracellular c-terminal fragment ACID. The 18-month-old APP23/Ar+/− male mice exhibited a reduction in AICD compared with APP23 mice (Fig. 3A–C), suggesting that testosterone may attenuate Aβ production by downregulating BACE1 protein levels in the brain. In addition, we examined the BACE1 activity levels in APP23/Ar+/− mice and compared the activity levels with APP23 mice retaining aromatase. We found that BACE1 activity was significantly reduced in the male APP23/Ar+/ mice at age of 12 and 18 months as showed in Figure 3D.
Figure 3.
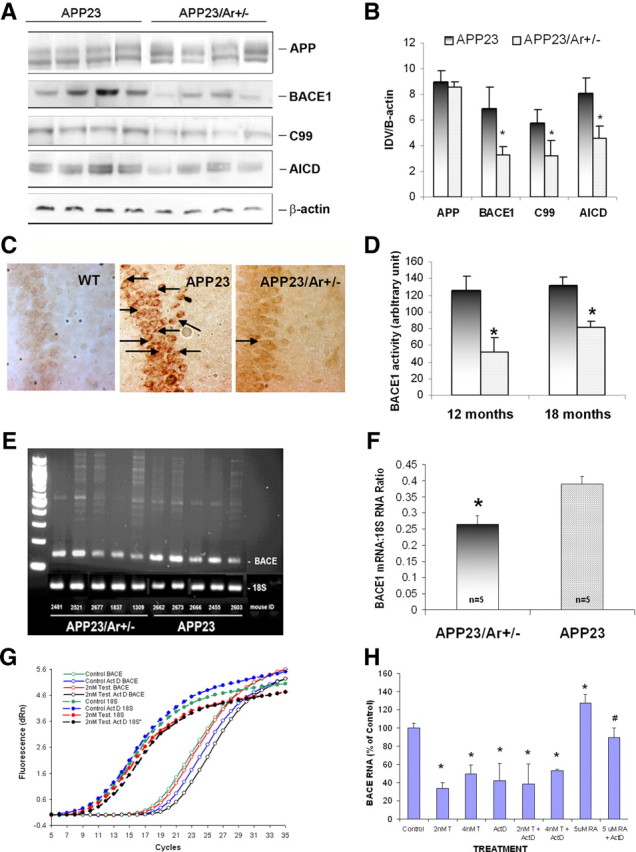
Testosterone downregulates BACE1 both in vivo and in vitro. At 18 months of age, the brains from male APP23 (n = 10) and APP23/Ar+/− (n = 8) mice were harvested and prepared for Western blot, immunostaining, enzyme activity analysis and mRNA measurement. A, The Western blot of APP, BACE1, c-terminal fragment, and C99 from 18-month-old mice is shown. B, The protein levels from the Western blot are presented as the ratio of the integrated optical density value (IDV) of the protein of interest to the IDV of β-actin. C, The levels of APP c-terminal fragment in the hippocampi of 18-month-old WT, APP23 and APP23/Ar+/− mice was detected by immunohistochemistry with anti-C-terminal antibody CT-20 (1:1000) and appears as intracellular dark brown staining as indicated by the arrows. D, BACE1 enzyme activity was measured from brain homogenize of 12- and 18-month-old APP23 and APP23/Ar+/− mice. E–H, The mRNA levels of BACE1 in 18-month old APP23 and APP23/Ar+/− male mice brains were detected by RT-PCR using 18S mRNA as a control (E), and the ratios of BACE1/18S mRNA were determined by densitometric analysis (F). The effect of testosterone (T) and actinomycin D (ActD) treatments on BACE1 and 18S mRNA levels in BACE transfected 293 cells was measured by real-time RT-PCR (G); the BACE1 mRNA levels were normalized to the 18S mRNA levels and were expressed relative to the vehicle-treated cells (H). *p < 0.05 compared with APP23 mice for B, D, and F; *p < 0.05 compared with vehicle-treated cells for H.
Reduction of BACE1 protein expression and activities in the brain of male APP23/Ar+/− mice is mediated through transcriptional mechanisms
To investigate whether reduced BACE1 protein levels in APP23/Ar+/− mice are partially caused by changes in BACE1 mRNA abundance, we performed RT-PCR and found that BACE1 mRNA levels were significantly decreased in 18-month-old APP23/Ar+/− mice compared with age-matched, APP23 male mice (n = 5 each genotype) (Fig. 3E,F). Our findings suggest that the reduction of BACE1 mRNA abundance may contribute to the decreased BACE1 activity exhibited by APP23/Ar+/− mice (Fig. 3D).
To further understand whether testosterone's regulation of BACE1 is mediated at the transcriptional level, we transfected the BACE1 cDNA into 293 cells, treated the cells with testosterone with or without actinomycin D, a transcription inhibitor, and detected BACE1 mRNA levels using real-time RT-PCR. As shown in Figure 3, G and H, the levels of BACE1 mRNA were reduced by testosterone treatment alone (2 or 4 nm), and that cotreatment with actinomycin D (1.7 μg/ml) and testosterone did not cause further reduction in mRNA levels while a significant reduction was found in cells treated with retinoic acid (our experimental positive control) combined with actinomycin D (data not show). Our data suggest that testosterone-induced reduction of BACE1 protein and activity is mediated through transcriptional mechanisms.
APP23/Ar+/− male mice exhibit increased protein expression of NEP and IDE as well as increased activity of NEP compared with control mice
To examine whether the reduction of amyloid plaque formation in APP23/Ar+/− male mice is partially mediated by the enhancement of Aβ clearance by NEP and IDE, we compared the protein levels and activities of NEP and IDE in 18-month-old APP23/Ar+/− and APP23 male mice. Our data showed a great increase in protein expression of NEP in 18-month-old APP23/Ar+/− male mice compared with age-matched APP23 and WT mice (Fig. 4A,B). The IDE protein levels in 18-month-old APP23/Ar+/− mice were also elevated compared with age-matched APP23 mice but were the same as the levels in age-matched WT mice (Fig. 4A,B). In addition, we examined the enzyme activities of NEP and IDE from the same mice. We found an enhancement of NEP activity in the male APP23/Ar+/− mice compared with age-matched APP23 mice from 6 to 18 months of age (Fig. 4D); however, the increase in mean IDE activity in APP23/Ar+/− mice compared with APP23 mice failed to reach statistical significance (Fig. 4C). Our data suggest that the testosterone-induced increase in NEP activity might enhance Aβ clearance in male APP23/Ar+/− mice.
Figure 4.

Protein expression and enzyme activity of IDE and NEP in APP23 and APP23/Ar+/− mice at various ages. A, Brain samples from APP23, APP23/Ar+/−, and WT mice at age of 18 months old were examined for IDE and NEP protein expression by Western blot. B, The relative amounts of IDE and NEP protein were determined by standard scanning densitometry. C, D, The enzyme activity levels of IDE (C) and NEP (D) in brains from APP23, APP23/Ar+/−, and WT mice at ages of 6, 12 and 18 months were detected. The brains were homogenized and incubated with fluorogenic substrate peptides. The increase in fluorescence occurred upon peptide bond cleavage and was detected during the first 20 min as fluorescence unit/min microgram protein. *p < 0.05 compared with APP23 mice; #p < 0.05 compared with WT mice.
APP23/Ar+/− male mice display enhanced cognitive function compared with control mice
We evaluated the effect of aging and testosterone levels on the cognitive function of male APP23/Ar+/−, APP23, and WT mice using a hole-board memory task as described in previous publications (Dodart et al., 2002; He et al., 2007). Cognitive performance was evaluated by ability of the mice to quickly and successfully to recognize the target hole. In brief, each mouse was awarded a score of “1” when its first poke was a correct poke (into the target hole), “0” when its first poke was incorrect but it made a correct poke within the given time, or “−1” when it failed to make a correct poke. Although the scores of WT and APP23 mice did not change significantly with age (data not shown), the APP23/Ar+/− male mice showed a great improvement in memory performance at the ages of 12 and 18 months compared with 6 months of age (Fig. 5A). At age of 18 months, WT animals showed initial good performance (trials 1–3) which then leveled off, showing that the animals, while not failing to find the target, were not improving in their ability to locate the correct hole on the first try. The APP23 mice did steadily worse in their ability to locate the target, while the APP23/Ar+/− showed improved learning compared with both WT and APP23 mice (Fig. 5B).
Figure 5.

Hole-board performance of APP23/Ar+/−, APP23, and WT male mice at various ages. Male APP23/Ar+/−, APP23, and WT mice were tested in the hole-board learning and memory paradigm (see Materials and Methods) at 6, 12, and 18 months of age. A, The scale value scores of male APP23/Ar+/− mice were higher at 12 and 18 months of age than at 6 months of age; and p < 0.05 compared with 6 months old. B–D, At the age of 18 months, APP23/Ar+/− mice (n = 6) demonstrated significantly better scale value scores (B), percentages of good trials (C), and target latencies (D) than did WT (n = 10) and APP23 (n = 7) mice. Data were expressed as mean of each score per trial (±SE) for the 10 trials of testing. The APP23/Ar+/− male mice not only found the target holes with fewer mistakes, but also found the target holes more quickly. *p < 0.05 compared with age-matched APP23 mice; #p < 0.05 compared with age-matched WT mice; &p < 0.05 compared to 6-month-old APP/Ar+/− mice.
These results are reiterated in Figure 5C, which shows the percentage of good trials (defined as trials in which the first poke was into the target hole) for APP23/Ar+/−, APP23, and WT male mice at 6, 12, and 18 months of age. The percentage of good trials for 6-month-old mice did not vary by genotype. As expected, 18-month-old APP23 mice tended to have a lower percentage of good trials compared with age-matched WT mice and compared with the percentage of good trials for 6-month-old APP23 mice. The APP23/Ar+/− mice, on the other hand, showed an increase in percentage of good trials at 12 and 18 months of age compared with age-matched WT and APP23 mice and compared with the percentage of good trials for their 6-month-old APP23/Ar+/− counterparts (Fig. 5C).
We also measured the target latencies (i.e., the amount of time it took for the mice to find the target hole) averaged over all 10 trials. The target latency for a trial in which the mouse never found the target hole was recorded at the time-limit of the trial. The 12- and 18-month-old APP23/Ar+/− male mice had significantly shorter target latencies than did age-matched WT and APP23 mice (Fig. 5D), showing that aged APP23/Ar+/− were able to recognize the target hole not only more reliably (Fig. 5B,C) but also more quickly (Fig. 5D) than age-matched WT and APP23 mice.
Discussion
Recent studies have suggested that testosterone reduction might be a risk factor for AD in aged males (Hogervorst et al., 2001; Moffat et al., 2004; Rosario et al., 2004; Rosario and Pike, 2008). Consistent with this hypothesis, research data has shown that testosterone treatment can reduce AD neuropathology both in clinical observation (Moffat et al., 2006) and in cell culture research (Gouras et al., 2000). Because testosterone can be converted into estrogen by aromatase, some researchers have suggested that the effects of testosterone on preventing AD may be a secondary effect of estrogen (estrogen-dependent) and/or may be estrogen-independent (Li and Shen, 2005). To differentiate between the estrogen-dependent and -independent mechanisms of testosterone's association with AD neuropathology, previous studies have used nonaromatized androgen dihydrotestosterone (DHT) as estrogen-independent agent and have found that DHT treatment caused a reduction in amyloid levels in the brains of male mice that was similar to the effect of testosterone treatment (Ramsden et al., 2003), suggesting that estrogen-independent mechanisms may be involved. However, recent studies showed that the DHT metabolite 5α-androgestan-3β,17β-diol can stimulate estrogen receptor subtype β (ERβ) (Lund et al., 2006). Therefore, there is a need for a more specific model to investigate the possible estrogen-independent pathways of testosterone's action on amyloid deposition.
Here, we used genetic knock-out of aromatase in APP transgenic mice as a model to study the estrogen-independent effect of testosterone on AD-like neuropathology and on cognition. This model was advantageous because it allowed us to study the effects of increased testosterone levels in males while avoiding a corresponding increase in estrogen generated from the aromatization of testosterone. As expected, the APP23/Ar+/− male mice showed elevated levels of endogenous testosterone and reduced levels of endogenous estrogen compared with age-matched APP23 male mice due to the depletion of aromatase. Therefore, the improvements in AD-like neuropathology and cognitive function that were exhibited in the male APP23/Ar+/− mice compared with the APP23 mice are most likely due to the estrogen-independent action of testosterone rather than due to the aromatization of testosterone into estrogen.
We predicted that if testosterone plays a role in preventing amyloid deposition independently from estrogen, we should see a decrease in amyloid levels in the APP23/Ar+/− male mice. Indeed, we found reductions not only in amyloid plaque formation but also in the Aβ42/Aβ40 ratio and in neuronal loss in the male APP23/Ar+/− mice compared with age-matched APP23 control mice, suggesting that increased endogenous testosterone limited amyloid deposition via an estrogen-independent mechanism.
One of the major enzymes responsible for Aβ production is BACE1. To examine whether testosterone plays a role in the regulation of BACE1, we detected levels of BACE1 protein from the brains of male APP23/Ar+/− and APP23 mice. Our data showed that BACE1 protein levels are reduced in APP23/Ar+/− compared with APP23 mice at the age of 18 months (Fig. 3A,B), suggesting that the inhibitory action of testosterone on Aβ production involves the reduction of BACE1 protein expression. To confirm the effect of testosterone on BACE1 activity, in addition to examine the protein levels of C99 and APP c-terminal fragments (both of which result from the cleavage of APP by BACE1), we also measured BACE1 enzyme activity. We found a reduction of BACE1 enzyme activity as well as lower levels of C99 and intracellular c-terminal fragments in APP23/Ar+/− male mice compared with APP23 mice (Fig. 3A–D), suggesting that BACE1 activity was reduced in the APP23/Ar+/− mice. This is the first time, to our knowledge, that testosterone has been shown to regulate Aβ levels by reducing BACE1 activity in the brain.
To further understand molecular mechanisms by which testosterone regulates BACE1 expression in APP23/Ar+/− mice, we performed RT-PCR and found that the level of BACE1 mRNA is significantly reduced in APP23/Ar+/− male mice at 18 months of age compared with that in age-matched APP23 mice (Fig. 3E,F), suggesting the testosterone-induced reduction of BACE1 activity and protein expression may be mediated at the transcriptional level. To confirm the action of testosterone on BACE1 mRNA, we also transfected BACE1 cDNA into 293 cells and found that testosterone treatment alone caused a decrease in BACE1 mRNA levels (Fig. 3G,H). To study the stability of mRNA, we also treated the cells with testosterone and actinomycin D and found no further reduction of BACE1 mRNA compared with cells receiving testosterone alone while actinomycin D did block retinoic acid-induced BACE1 transcription. Because we found that testosterone reduces the activity, protein expression, and mRNA abundance of BACE1, we conclude that the effect of testosterone on BACE1 regulation is more likely mediated at the transcriptional level through estrogen-independent mechanism.
To examine whether increased endogenous testosterone promotes Aβ clearance, we examined protein levels and enzyme activities of IDE and NEP in the brains of APP23/Ar+/−, APP23, and WT mice. Our results showed that NEP protein levels and activities were increased in APP23/Ar+/− mice compared with APP23 control mice (Fig. 4B,D), which is in line with a recent study which showed that androgen (DHT) treatment upregulates NEP protein expression in APP transfected cells (Yao et al., 2008). The effect of testosterone on NEP protein expression might be mediated by promoting NEP transcription via stimulation of the androgen response element located on the NEP promoter (Shen et al., 2000; Zheng et al., 2006). Although IDE protein expression was increased in male APP23/Ar+/− mice compared with age-matched APP23 mice (Fig. 4B), the differences in IDE enzyme activity failed to reach significance (Fig. 4C). Further investigation of the connection between androgen and IDE-mediated Aβ degradation is needed.
The effect of testosterone on learning and memory has been controversial. Many studies have found that androgens improve spatial learning and memory (Flood and Roberts, 1988; Alexander et al., 1994; Janowsky et al., 1994; Ishunina et al., 2002; Raber et al., 2002). However, other studies have reported that androgens have the opposite effect (Goudsmit et al., 1990; Gouchie and Kimura, 1991; Galea et al., 1995; Hampson, 1995; Naghdi et al., 2001). One possible explanation for this discrepancy is that some studies only looked at serum levels of testosterone rather than the levels of testosterone in the brain. Because our APP23/Ar+/− male mice exhibited increased testosterone levels in the brain as well as in the serum (Fig. 1A,B), the effect of systemic testosterone levels versus brain-localized testosterone levels is not an issue.
To investigate the estrogen-independent effect of endogenous testosterone on cognitive function in aged male mice, we examined cognitive function in male APP23/Ar+/−, APP23, and WT mice at various ages by hole-board testing. The hole-board test is a measurement of spatial learning and memory, both of which have been attributed to hippocampal function (Kesner et al., 1993; Dodart et al., 2002). At the age of 6 months, there were no differences in performance among the three genotypes (Fig. 5C,D), suggesting that the increase of endogenous testosterone at young ages does not alter cognitive function in males. At the age of 18 months, however, male APP23/Ar+/− mice showed a great improvement in spatial memory compared with APP23 and WT mice (Fig. 5B–D). The 12- and 18-month old APP23/Ar+/− mice not only found the target hole with greater accuracy (Fig. 5B,C), but they also found the target hole in less time (Fig. 5D). In contrast, APP23 mice, at 18 months old, showed a great reduction of memory performance and made more mistakes than did WT or APP23/Ar+/− mice (Fig. 5B). Together, these data suggest that increased endogenous testosterone, independent of estrogen, not only prevents Aβ-induced impairment of spatial memory and cognitive function, but also may prevent deficits due to normal aging.
In summary, the genetic knock-out of aromatase in male APP23 mice leads to increased endogenous testosterone levels in the serum and brain without a corresponding increase in estrogen (Fig. 1A,B) and prevents the development of AD-like neuropathology as measured by the Aβ42/Aβ40 ratio, the level of amyloid plaque formation, and the extent of neuronal loss in the hippocampus (Fig. 2A–D). Furthermore, the estrogen-independent mechanisms by which testosterone reduces plaque formation in the brain involves the reduction of Aβ production through attenuation of BACE1 enzyme activity via downregulation of BACE1 protein expression at transcription level (Fig. 3A–H) as well as the promotion of Aβ clearance through the enhancement of NEP protein expression and enzyme activity (Fig. 4B,D). The APP23/Ar+/− mice also showed improved spatial memory and cognitive function compared with APP23 and WT mice (Fig. 5B–D), suggesting that endogenous testosterone in males is essential in preventing age- and AD-related cognitive declines.
Footnotes
This work was funded in part by the Alzheimer's Association, the Arizona Disease Control Research Commission, American Health Assistance Foundation, and the National Institutes of Health. We thank Dr. Zheming Lei from the University of Louisville, School of Medicine for his help with the testosterone assay.
References
- Alexander GM, Packard MG, Hines M. Testosterone has rewarding affective properties in male rats: implications for the biological basis of sexual motivation. Behav Neurosci. 1994;108:424–428. doi: 10.1037//0735-7044.108.2.424. [DOI] [PubMed] [Google Scholar]
- Azcoitia I, Sierra A, Veiga S, Honda S, Harada N, Garcia-Segura LM. Brain aromatase is neuroprotective. J Neurobiol. 2001;47:318–329. doi: 10.1002/neu.1038. [DOI] [PubMed] [Google Scholar]
- Bertram L, Blacker D, Mullin K, Keeney D, Jones J, Basu S, Yhu S, McInnis MG, Go RC, Vekrellis K, Selkoe DJ, Saunders AJ, Tanzi RE. Evidence for genetic linkage of Alzheimer's disease to chromosome 10q. Science. 2000;290:2302–2303. doi: 10.1126/science.290.5500.2302. [DOI] [PubMed] [Google Scholar]
- Brosnan-Watters G, Wozniak DF. A rotating holeboard procedure for testing drug effects on spatial learning and memory in mice. Brain Res Brain Res Protoc. 1997;1:331–338. doi: 10.1016/s1385-299x(97)00007-x. [DOI] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Dominguez D, Tournoy J, Hartmann D, Huth T, Cryns K, Deforce S, Serneels L, Camacho IE, Marjaux E, Craessaerts K, Roebroek AJ, Schwake M, D'Hooge R, Bach P, Kalinke U, Moechars D, Alzheimer C, Reiss K, Saftig P, De Strooper B. Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J Biol Chem. 2005;280:30797–30806. doi: 10.1074/jbc.M505249200. [DOI] [PubMed] [Google Scholar]
- Ertekin-Taner N, Graff-Radford N, Younkin LH, Eckman C, Baker M, Adamson J, Ronald J, Blangero J, Hutton M, Younkin SG. Linkage of plasma Abeta42 to a quantitative locus on chromosome 10 in late-onset Alzheimer's disease pedigrees. Science. 2000;290:2303–2304. doi: 10.1126/science.290.5500.2303. [DOI] [PubMed] [Google Scholar]
- Farrag AK, Khedr EM, Abdel-Aleem H, Rageh TA. Effect of surgical menopause on cognitive functions. Dement Geriatr Cogn Disord. 2002;13:193–198. doi: 10.1159/000048652. [DOI] [PubMed] [Google Scholar]
- Flood JF, Roberts E. Dehydroepiandrosterone sulfate improves memory in aging mice. Brain Res. 1988;448:178–181. doi: 10.1016/0006-8993(88)91116-x. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- Galea LA, Kavaliers M, Ossenkopp KP, Hampson E. Gonadal hormone levels and spatial learning performance in the Morris water maze in male and female meadow voles, Microtus pennsylvanicus. Horm Behav. 1995;29:106–125. doi: 10.1006/hbeh.1995.1008. [DOI] [PubMed] [Google Scholar]
- Gouchie C, Kimura D. The relationship between testosterone levels and cognitive ability patterns. Psychoneuroendocrinology. 1991;16:323–334. doi: 10.1016/0306-4530(91)90018-o. [DOI] [PubMed] [Google Scholar]
- Goudsmit E, Van de Poll NE, Swaab DF. Testosterone fails to reverse spatial memory decline in aged rats and impairs retention in young and middle-aged animals. Behav Neural Biol. 1990;53:6–20. doi: 10.1016/0163-1047(90)90729-p. [DOI] [PubMed] [Google Scholar]
- Gouras GK, Xu H, Gross RS, Greenfield JP, Hai B, Wang R, Greengard P. Testosterone reduces neuronal secretion of Alzheimer's beta-amyloid peptides. Proc Natl Acad Sci U S A. 2000;97:1202–1205. doi: 10.1073/pnas.97.3.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C. Take five—BACE and the gamma-secretase quartet conduct Alzheimer's amyloid beta-peptide generation. EMBO J. 2004;23:483–488. doi: 10.1038/sj.emboj.7600061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson E. Spatial cognition in humans: possible modulation by androgens and estrogens. J Psychiatry Neurosci. 1995;20:397–404. [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer's mice. J Cell Biol. 2007;178:829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogervorst E, Williams J, Budge M, Barnetson L, Combrinck M, Smith AD. Serum total testosterone is lower in men with Alzheimer's disease. Neuro Endocrinol Lett. 2001;22:163–168. [PubMed] [Google Scholar]
- Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G. Increased expression of the amyloid precursor beta-secretase in Alzheimer's disease. Ann Neurol. 2002;51:783–786. doi: 10.1002/ana.10208. [DOI] [PubMed] [Google Scholar]
- Inestrosa NC, Marzolo MP, Bonnefont AB. Cellular and molecular basis of estrogen's neuroprotection. Potential relevance for Alzheimer's disease. Mol Neurobiol. 1998;17:73–86. doi: 10.1007/BF02802025. [DOI] [PubMed] [Google Scholar]
- Ishunina TA, Fisser B, Swaab DF. Sex differences in androgen receptor immunoreactivity in basal forebrain nuclei of elderly and Alzheimer patients. Exp Neurol. 2002;176:122–132. doi: 10.1006/exnr.2002.7907. [DOI] [PubMed] [Google Scholar]
- Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, Gerard C, Hama E, Lee HJ, Saido TC. Metabolic regulation of brain Abeta by neprilysin. Science. 2001;292:1550–1552. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- Janowsky JS, Oviatt SK, Orwoll ES. Testosterone influences spatial cognition in older men. Behav Neurosci. 1994;108:325–332. doi: 10.1037//0735-7044.108.2.325. [DOI] [PubMed] [Google Scholar]
- Kesner RP, Bolland BL, Dakis M. Memory for spatial locations, motor responses, and objects: triple dissociation among the hippocampus, caudate nucleus, and extrastriate visual cortex. Exp Brain Res. 1993;93:462–470. doi: 10.1007/BF00229361. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, Frosch MP, Selkoe DJ. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–1093. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- Li C, Hersh LB. Neprilysin: assay methods, purification, and characterization. Methods Enzymol. 1995;248:253–263. doi: 10.1016/0076-6879(95)48018-8. [DOI] [PubMed] [Google Scholar]
- Li R, Shen Y. Estrogen and brain: synthesis, function and diseases. Front Biosci. 2005;10:257–267. doi: 10.2741/1525. [DOI] [PubMed] [Google Scholar]
- Li R, Lindholm K, Yang LB, Yue X, Citron M, Yan R, Beach T, Sue L, Sabbagh M, Cai H, Wong P, Price D, Shen Y. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer's disease patients. Proc Natl Acad Sci U S A. 2004;101:3632–3637. doi: 10.1073/pnas.0205689101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund TD, Hinds LR, Handa RJ. The androgen 5alpha-dihydrotestosterone and its metabolite 5alpha-androstan-3beta, 17beta-diol inhibit the hypothalamo-pituitary-adrenal response to stress by acting through estrogen receptor beta-expressing neurons in the hypothalamus. J Neurosci. 2006;26:1448–1456. doi: 10.1523/JNEUROSCI.3777-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R. Mice deficient in BACE1, the Alzheimer's beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci. 2001;4:231–232. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- Marjaux E, Hartmann D, De Strooper B. Presenilins in memory, Alzheimer's disease, and therapy. Neuron. 2004;42:189–192. doi: 10.1016/s0896-6273(04)00218-1. [DOI] [PubMed] [Google Scholar]
- Marr RA, Rockenstein E, Mukherjee A, Kindy MS, Hersh LB, Gage FH, Verma IM, Masliah E. Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J Neurosci. 2003;23:1992–1996. doi: 10.1523/JNEUROSCI.23-06-01992.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat SD, Zonderman AB, Metter EJ, Kawas C, Blackman MR, Harman SM, Resnick SM. Free testosterone and risk for Alzheimer disease in older men. Neurology. 2004;62:188–193. doi: 10.1212/wnl.62.2.188. [DOI] [PubMed] [Google Scholar]
- Moffat SD, Elkins W, Resnick SM. Age differences in the neural systems supporting human allocentric spatial navigation. Neurobiol Aging. 2006;27:965–972. doi: 10.1016/j.neurobiolaging.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Mukherjee A, Song E, Kihiko-Ehmann M, Goodman JP, Jr, Pyrek JS, Estus S, Hersh LB. Insulysin hydrolyzes amyloid beta peptides to products that are neither neurotoxic nor deposit on amyloid plaques. J Neurosci. 2000;20:8745–8749. doi: 10.1523/JNEUROSCI.20-23-08745.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naghdi N, Nafisy N, Majlessi N. The effects of intrahippocampal testosterone and flutamide on spatial localization in the Morris water maze. Brain Res. 2001;897:44–51. doi: 10.1016/s0006-8993(00)03261-3. [DOI] [PubMed] [Google Scholar]
- Pinnix I, Musunuru U, Tun H, Sridharan A, Golde T, Eckman C, Ziani-Cherif C, Onstead L, Sambamurti K. A novel gamma-secretase assay based on detection of the putative C-terminal fragment-gamma of amyloid beta protein precursor. J Biol Chem. 2001;276:481–487. doi: 10.1074/jbc.M005968200. [DOI] [PubMed] [Google Scholar]
- Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A, Hersh LB, Selkoe DJ. Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem. 1998;273:32730–32738. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- Raber J, Bongers G, LeFevour A, Buttini M, Mucke L. Androgens protect against apolipoprotein E4-induced cognitive deficits. J Neurosci. 2002;22:5204–5209. doi: 10.1523/JNEUROSCI.22-12-05204.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden M, Nyborg AC, Murphy MP, Chang L, Stanczyk FZ, Golde TE, Pike CJ. Androgens modulate beta-amyloid levels in male rat brain. J Neurochem. 2003;87:1052–1055. doi: 10.1046/j.1471-4159.2003.02114.x. [DOI] [PubMed] [Google Scholar]
- Roberds SL, Anderson J, Basi G, Bienkowski MJ, Branstetter DG, Chen KS, Freedman SB, Frigon NL, Games D, Hu K, Johnson-Wood K, Kappenman KE, Kawabe TT, Kola I, Kuehn R, Lee M, Liu W, Motter R, Nichols NF, Power M, et al. BACE knock-out mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer's disease therapeutics. Hum Mol Genet. 2001;10:1317–1324. doi: 10.1093/hmg/10.12.1317. [DOI] [PubMed] [Google Scholar]
- Rosario ER, Pike CJ. Androgen regulation of beta-amyloid protein and the risk of Alzheimer's disease. Brain Res Rev. 2008;57:444–453. doi: 10.1016/j.brainresrev.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario ER, Chang L, Stanczyk FZ, Pike CJ. Age-related testosterone depletion and the development of Alzheimer disease. JAMA. 2004;292:1431–1432. doi: 10.1001/jama.292.12.1431-b. [DOI] [PubMed] [Google Scholar]
- Rosario ER, Carroll JC, Oddo S, LaFerla FM, Pike CJ. Androgens regulate the development of neuropathology in a triple transgenic mouse model of Alzheimer's disease. J Neurosci. 2006;26:13384–13389. doi: 10.1523/JNEUROSCI.2514-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen R, Sumitomo M, Dai J, Hardy DO, Navarro D, Usmani B, Papandreou CN, Hersh LB, Shipp MA, Freedman LP, Nanus DM. Identification and characterization of two androgen response regions in the human neutral endopeptidase gene. Mol Cell Endocrinol. 2000;170:131–142. doi: 10.1016/s0303-7207(00)00326-9. [DOI] [PubMed] [Google Scholar]
- Sherwin BB. Estrogen and cognitive functioning in women. Endocr Rev. 2003;24:133–151. doi: 10.1210/er.2001-0016. [DOI] [PubMed] [Google Scholar]
- Song ES, Juliano MA, Juliano L, Hersh LB. Substrate activation of insulin-degrading enzyme (insulysin). A potential target for drug development. J Biol Chem. 2003;278:49789–49794. doi: 10.1074/jbc.M308983200. [DOI] [PubMed] [Google Scholar]
- Sternbach H. Age-associated testosterone decline in men: clinical issues for psychiatry. Am J Psychiatry. 1998;155:1310–1318. doi: 10.1176/ajp.155.10.1310. [DOI] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsolaki M, Grammaticos P, Karanasou C, Balaris V, Kapoukranidou D, Kalpidis I, Petsanis K, Dedousi E. Serum estradiol, progesterone, testosterone, FSH and LH levels in postmenopausal women with Alzheimer's dementia. Hell J Nucl Med. 2005;8:39–42. [PubMed] [Google Scholar]
- Xu H, Gouras GK, Greenfield JP, Vincent B, Naslund J, Mazzarelli L, Fried G, Jovanovic JN, Seeger M, Relkin NR, Liao F, Checler F, Buxbaum JD, Chait BT, Thinakaran G, Sisodia SS, Wang R, Greengard P, Gandy S. Estrogen reduces neuronal generation of Alzheimer beta-amyloid peptides. Nat Med. 1998;4:447–451. doi: 10.1038/nm0498-447. [DOI] [PubMed] [Google Scholar]
- Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, Li R, Shen Y. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- Yao M, Nguyen TV, Rosario ER, Ramsden M, Pike CJ. Androgens regulate neprilysin expression: role in reducing beta-amyloid levels. J Neurochem. 2008;105:2477–2488. doi: 10.1111/j.1471-4159.2008.05341.x. [DOI] [PubMed] [Google Scholar]
- Yasojima K, McGeer EG, McGeer PL. Relationship between beta amyloid peptide generating molecules and neprilysin in Alzheimer disease and normal brain. Brain Res. 2001a;919:115–121. doi: 10.1016/s0006-8993(01)03008-6. [DOI] [PubMed] [Google Scholar]
- Yasojima K, Akiyama H, McGeer EG, McGeer PL. Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of beta-amyloid peptide. Neurosci Lett. 2001b;297:97–100. doi: 10.1016/s0304-3940(00)01675-x. [DOI] [PubMed] [Google Scholar]
- Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O'Connor T, Logan S, Maus E, Citron M, Berry R, Binder L, Vassar R. beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J Neurosci. 2007;27:3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng R, Shen R, Goodman OB, Jr, Nanus DM. Multiple androgen response elements cooperate in androgen regulated activity of the type 1 neutral endopeptidase promoter. Mol Cell Endocrinol. 2006;259:10–21. doi: 10.1016/j.mce.2006.07.004. [DOI] [PubMed] [Google Scholar]