

Abstract
Accumulating evidence suggests that mitochondria are important targets for the actions of estrogens and studies indicated that localization of ERβ in neuronal mitochondrial ERβ (mtERβ) might directly affect neuronal mitochondrial function in vitro. However, it is unknown what expression levels and how important of mtERβ in the human brain, particularly in the brain with Alzheimer’s disease (AD). In the present study, by using rapidly autopsied human brain tissue, we found that the frontal cortices of female AD patients exhibited significantly reduced mtERβ, along with reduced mitochondrial cytochrome C oxidase activity, and increased protein carbonylation compared to that in normal controls. The correlation between the mtERβ expression and mitochondrial cytochrome C oxidase activity in the female human brain is significant. To understand the possible mechanisms of mtERβ in AD-related mitochondrial dysfunction, using ERβKO mice as a model, we found that lacking of ERβ enhanced brain reactive oxygen species generation and reduced mitochondrial membrane potential under Aβ peptide insult compared to brain mitochondria from wild-type control mice. Our studies for the first time, demonstrated neuronal mtERβ expression in the human brain and the deficiency of mtERβ in the female AD brain is associated with the dysfunction of mitochondria. Our results from ERβKO mice demonstrated that ERβ depletion-induced mitochondrial dysfunction is mediated through increasing reactive oxygen generation and reduction of mitochondria membrane potential. These results indicate that ERβ depletion has the ability to impair mitochondrial function in mice and reduction of brain mtERβ may significantly contribute to the mitochondrial dysfunction involved in AD pathogenesis in women.
Keywords: brain mitochondria estrogen Receptor β (mtERβ), Alzheimer’s disease (AD), mitochondria dysfunction
Introduction
Evidence suggests that mitochondrial dysfunction and oxidative damage are involved in the pathogenesis of Alzheimer’s disease (AD) [1–2]. Defects in cytochrome C oxidase or complex IV (COX), the fourth complex in the mitochondrial electron transport chain, have been shown to be associated with AD[3]. Mitochondria are critical for meeting the high energy demands of the brain, but they also generate the majority of intracellular reactive oxygen species (ROS), which can cause oxidative damage to important cellular structures. Several studies have found that increases in ROS and oxidative stress are involved in age-related degenerative diseases like AD. More importantly, oxidative damage has been found to occur as one of the earliest events in the neuropathogenesis of AD, even before the onset of significant plaque pathology[3].
Estrogen has been shown to suppress mitochondrial oxidative stress, regulate energy metabolism, and regulate the expression of mitochondria-involved anti-apoptotic proteins like the bcl-2 family and bcl-xL, which suggests that brain mitochondria may be major targets of estrogen action in the central nervous system[4–7]. Estrogen is an established modulator of neuronal viability against a variety of insults in cultured cells, including oxidative stress and β-amyloid peptide (Aβ) [4–6]. Studies have found that many of these protective effects can be blocked by an estrogen receptor (ER) antagonist, ICI-182780 [5, 8], suggesting an ER-mediated mode of estrogen action.
Estrogen receptors (ERα and ERβ) are believed to be ligand-activated transcription factors belonging to the nuclear receptor superfamily [7, 9]; however, recent work has supported the idea that a second pool of ERα and ERβ, localized to the plasma membrane, also contributes to the actions of estrogen[10–12]. Unlike ERα, which clearly plays a major role as a transcription factor in the reproductive system and development for both males and females, the function of ERβ remains unclear. Very interestingly, recent reports have confirmed the localization of ERβ in the mitochondria of epithelial cells, ligament cells and murine neurons [13–17]. In rat primary cortical and hippocampal neurons, ERβ colocalizes almost exclusively with mitochondria and does not translocate into the nucleus upon E2 treatment [17], suggesting that estrogen can directly affect mitochondrial function through mitochondrial ERβ (mtERβ).
In this study, we examined the ERβ expression and localization patterns in brains from women with AD compared to those from age-matched, non-demented (ND) women. We then analyzed the correlation between mitochondrial ERβ expression and mitochondrial function. To explore the relationship between ERβ and brain mitochondrial function more directly, we compared mitochondrial function and tolerance to amyloid insult in brain mitochondria from female transgenic ERβ-knockout mice to those from age-matched, wild-type (WT) control mice.
Materials and methods
Chemicals
2,6-dichloroindophenol indophenol (DCPIP), tetramethoxypropane, cytochrome C, decylubiquinone, 5,5′,6,6′- tetrachloro -1,1′,3,3′- tetraethylbenzimidazol-carbocyanine iodine (JC-1), dichlorofluorescein-2,7-diacetate (DCF-DA) were purchased from Sigma Chemical Co. (St. Louis, MO). Aβ1-42 and Aβ25-35 were purchased from Anaspec Inc. (Fremont, CA). Other chemicals were all analytical grade reagents from local vendors.
Human Brains
Human brain tissues were obtained from autopsies of subjects enrolled in the Banner Sun Health Research Institute Brain Donation Program [18]. AD was clinically diagnosed using the criteria of the National Institute of Neurological Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association (NINCDS–ARDRDA) and was subsequently confirmed postmortem by neuropathologist examination. Postmortem intervals for brain samples averaged 2.8 h (range, 2–3.5 h). Both AD and ND control subjects were Caucasian females (mean age, 85.88±3.76 y and 85.50 ±5.81 y, respectively, n=8 for each group). Frozen frontal cortex samples were used for all studies. None of the participants in our study had a history of estrogen replacement therapy.
Experimental Mice
Mice were maintained in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals[19]. Heterozygous female ERβ transgenic mice (ERβ+/−) were crossed with homozygous ERβ knockout mice (ERβ−/−) to obtain litter-matched ERβ+/− and ERβ−/− mice. Age-matched, C57BL/6 (WT) mice were used as controls. Brain mitochondria from fifteen mice (females, 10 to 12 months of age, n=5 from each genotype) were prepared immediately following decapitation as previously described [20].
Isolation of mitochondria from human frontal cortex and mouse brain
Mitochondria from the human frontal cortex were isolated as described previously[20]. Briefly, gray matter of human frontal gyrus were dissected and homogenized in isolation medium (0.25 M sucrose, 0.5 mM EDTA-K, 10 mM Tris.Cl, pH 7.4), and then centrifuged 2000 g for 3 min. The pellet was processed using a hypertonic solution (4.6 M NaCl and 5 mM HEPES, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 26% glycerol (v/v), pH 7.9) to obtain the nuclear fraction; the supernatant was carefully decanted followed by centrifugation at 12,500 g for 8 min to obtain the crude mitochondrial pellet (the supernatant were collected as the cytosolic fraction), which was resuspended in 3% ficoll medium and purified by spinning in 6% ficoll medium (6% ficoll, 50 μM EDTA-K, 10 mM Tris, 0.24 M manitol, 60 mM sucrose). The resultant brown pellet was washed and resuspended to obtain the brain mitochondrial fraction, stored in −80°C before use. Brain mitochondria from the mouse samples were isolated using a similar protocol, except that the entire cerebrum was used. The measurements of oxygen consumption, ROS generation, and mitochondrial membrane potential were performed immediately following isolation, and the remaining mouse brain mitochondria were stored at −80 °C for use in the other bioassays. The protein level of each sample was determined using a Bio-Rad protein assay kit with BSA as standard.
Immunohistochemistry
Samples of the human frontal cortex were fixed with 4% paraformaldehyde and were sectioned (30 μm in thickness) coronally with a Leica CA 1900 cryostat. Sections were incubated with block solution followed by incubation with anti-ERβ (ab3576, 1:200, abcam) and either anti-NeuN (MAB377, 1:400; Chemicon) or anti-VDAC (ab14734, 1:500, abcam) primary antibodies, and then fluorescent-labeled with 488 (green) or 594 (red) secondary antibodies against rabbit IgG or mouse IgG (1:1,000; Invitrogen). To quench auto fluorescence, sections were dyed with 0.3% Sudan black for 10 min before observation. A confocal microscope with a 10×and 40× PL FLUOTAR was used to capture images. The images were processed with Fluoview software (Olympus).
Western blot
Mitochondrial, cytosolic, and nuclear fractions (50 μg protein/sample) from brain samples were separated by 10% SDS-PAGE, transferred to PVDF membranes, and blocked with 5% non-fat milk. The PVDF membranes were incubated with the following primary antibodies overnight at 4 °C: rabbit anti-ERβ (ab3576, 1:1000, abcam), mouse anti-VDAC (1:2000, abcam), goat anti-COX 1, 2 (1:1000, sc-23982, sc-23983, Santa Cruz), mouse anti-COX 4 (1:1000, ab14744, abcam) and rabbit anti-PARP (1:1000, Santa Cruz). The membranes were then incubated with anti-mouse or anti-goat (1:8000, Santa Cruz) IgG labeled with horseradish peroxidase for 1 h and were visualized using an enhanced chemiluminescence Western blotting detection system (Millipore).
Measurement of complex II and IV activity and protein carbonyls
Assays of COX and succinate dehydrogenase (SDH, part of complex II) activity were performed as previously described [21]. Protein carbonyls were assayed using the Oxyselective kit (Cell Biolabs, Inc., San Diego, CA) according to the manufacturer’s instructions [22].
Determination of oxygen consumption
Mitochondrial oxygen consumption was determined using a Clark oxygen electrode (Hansatech, UK). A suspension of brain mitochondria containing 0.25 mg protein was added to 0.5 ml of assay buffer along with various concentrations of Aβ at 37 °C. The Aβ 1-42 were dissolved in DMSO to maintain soluble form and then diluted to 0.5 mM with PBS, incubated at 37°C for minimum 60 min to promote aggregation prior use [23]. For mitochondrial respiration assay, using 5 mM succinate as substrates, the rate of oxygen consumption was measured prior to and following the addition of 0.5 mM ADP. The rate of brain mitochondrial oxygen consumption was expressed in nmol O2/min/mg protein (reflecting the rate of oxygen consumption following the addition of ADP).
Measurement of ROS generation and mitochondrial membrane potential (MMP)
The ROS and MMP determinations were performed as previously described with some modification [24–27]. Briefly, for the ROS measurement, the mitochondria were suspended at 200 μg/ml in an assay buffer plus 5 mM succinate and 5 μM DCF-DA. The emission fluorescence at 530 nm with a 480 nm excitation was kinetically recorded for 30 min. In order to determine the amount of oxidants generated from per nmol oxygen consumed by mitochondria, the rate of fluorescence generation was normalized to the corresponding rate of mitochondrial oxygen consumption as measured by the oxygen electrode following the addition of 5 mM succinate. For the MMP assay, following the insult with Aβ25-35 or Aβ1-42, the mitochondria were suspended at 50 μg/ml in an assay buffer plus 5 mM succinate and 1.5 μg/ml JC-1. The MMP was measured as the fluorescence ratio of 590 nm to 530 nm.
Results
Reduced ERβ expression in female AD brains
To examine the ERβ expression in brains from female AD patients and gender-and age-matched ND individuals, we performed immunohistochemistry analysis on a total of 16 individuals (8 AD and 8 ND). Double-staining for ERβ and NeuN in the frontal cortices of AD and ND women revealed that less ERβ was expressed in neurons in the AD frontal cortex compared to ND controls at the same brain region. However, we did not find significant neuron loss in the cortex sections observed (figure 1A) while a significant reduction of neuronal ERβ expression was found in the AD samples than that in ND brains (figure 1B). Co-staining with anti-ERβ and anti-VDAC (mitochondrial marker) antibodies showed that the ERβ was colocalized with the mitochondria in the ND brain and that the mitochondrial ERβ expression was much weaker in the AD brain than that in the ND brain (figure 2).
Figure 1.
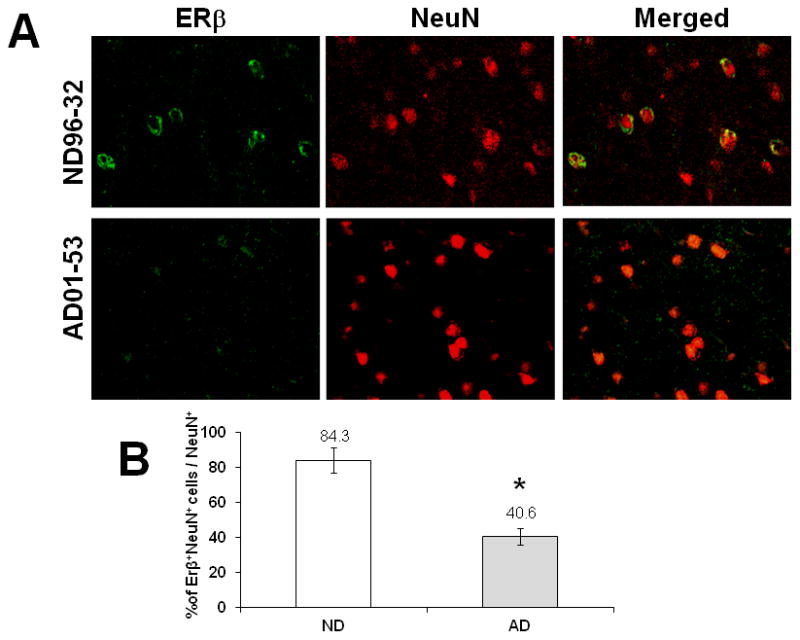
Reduction of ERβ expression in neurons of female AD patients. The brain sections of frontal cortex were prepared from postmortem AD and ND brains. Sections were incubated with anti-ERβ and anti-NeuN (neuronal marker) antibodies, followed by fluorescent-labeled secondary antibodies (A). The representative pictures were taken from one age-matched control brain (case No. 96-32, female) and one AD brain (case No. 01-53, female). The percentage of ERβ positive neurons to total neurons was compared between AD and ND (B). * indicates P<0.05 compare to ND.
Figure 2.
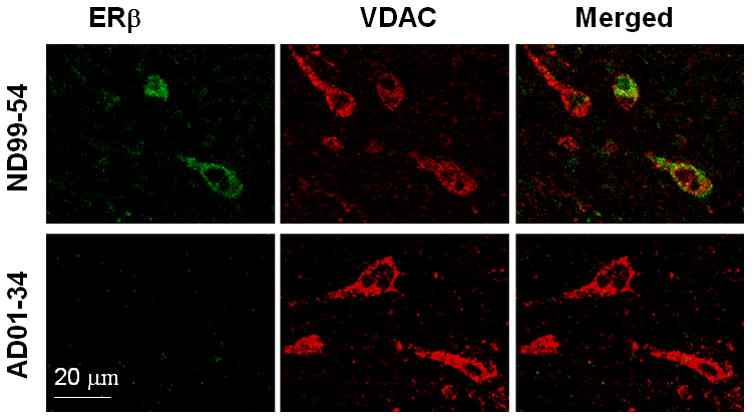
Reduction of mitochondrial ERβ in female AD patients. Sections of frontal cortex were incubated with anti-ERβ and anti-VDAC (mitochondria marker) antibodies, followed by fluorescent-labeled secondary antibodies. The representative pictures were taken from one age-matched ND brain (case No. 99-54, female) and one AD brain (case No. 01-34, female).
To further examine the protein expression of ERβ in each brain cell fraction, we performed western blot analysis on the samples from the female AD and ND frontal cortex (figure 3A). The PARP and β-actin were employed as nucleus and general marker of the fractions, respectively. For the mitochondrial fraction, we used VDAC and MFN1 as markers for mitochondrial out membrane and plasma membrane in this study [28]. The mitochondrial fraction obtained by density gradient in the present study was purified successfully with little nuclear remnant. Consistent with our immunohistochemical data, the expression of ERβ was significantly reduced in the mitochondrial fraction. Moreover, a reduction of ERβ protein expression was also found in the whole brain lysates, nuclear, and cytosolic fractions of the AD female brains compared to the ND female brains (figure 3).
Figure 3.
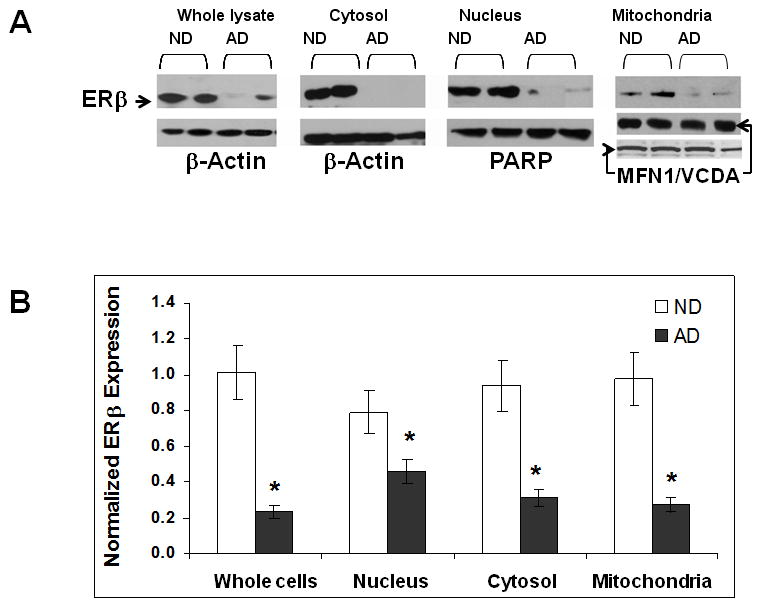
Less ERβ detected in various cell fractions of female AD brains. The mitochondrial, nuclear, and cytosolic cell fractions from the frontal cortex were prepared, and the levels of ERβ were measured using western blot. (A) Representative images of ERβ expression in whole brain lysate, and cytosolic, nucleus and mitochondria fractions with beta-actin, PARP, VDAC (middle band) and MFN1 (lower band) serving as internal controls. (B) Quantitative result of ERβ expression in each fraction as well as in the whole brain lysate. *p<0.05 vs. ND.
The ERβ reduction correlated with mitochondrial dysfunction and oxidative stress in female AD brains
As shown in figure 4, both SDH and COX activities were significantly compromised in mitochondria isolated from the frontal cortices of the AD group (45% and 32% reduction in SDH and COX activity compared to the ND group, respectively). To understand the relationship between mtERβ and mitochondrial function in the AD, we also performed a correlation analysis between mitochondrial complex activities and expression of ERβ (mtERβ and total brain homogenate ERβ) as shown in figure 5. Our data as showed in figure 5B, both COX and SDH activities are significant associated with the expression of mtERβ in the female brain samples combined both AD and ND (p=0.006 and 0.036 for COX and SDH, respectively). However, the COX activity is not only significantly associated with mtERβ (p=0.014), but also with the total ERβ (p=0.007) in the AD brain, suggesting a disease specific effect of ERβ in AD (figure 5B). In contrast, the SDH activity showed a weak association with mtERβ (p=0.066) but strong correlation with total ERβ (p=0.041) in the AD brain, indicating a general role of ERβ, rather than mtERβ, in the AD mitochondrial pathology in the female brain.
Figure 4.
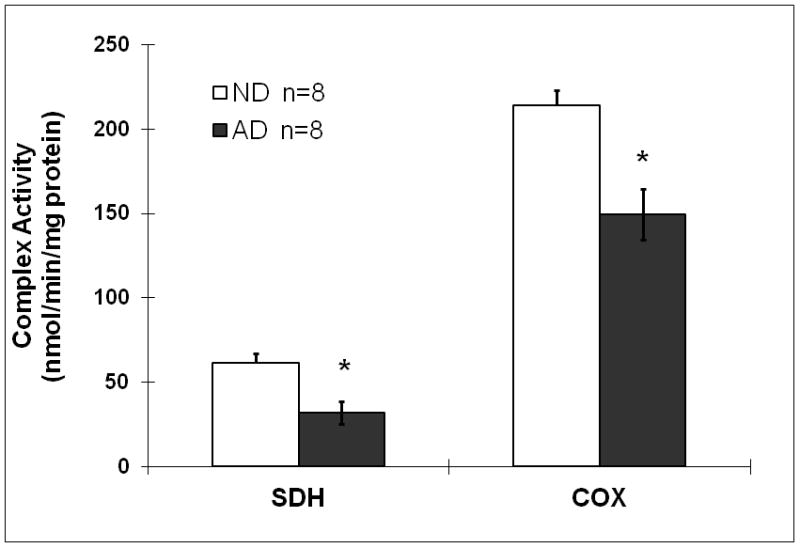
Reduced brain SDH and COX activities in female AD patients. The brain mitochondria were prepared from frozen tissues of postmortem brain (AD n=8, ND n=8), followed by spectrometric determination of SDH and COX activities. Data were standardized with protein values. *p<0.05 vs. ND.
Figure 5.
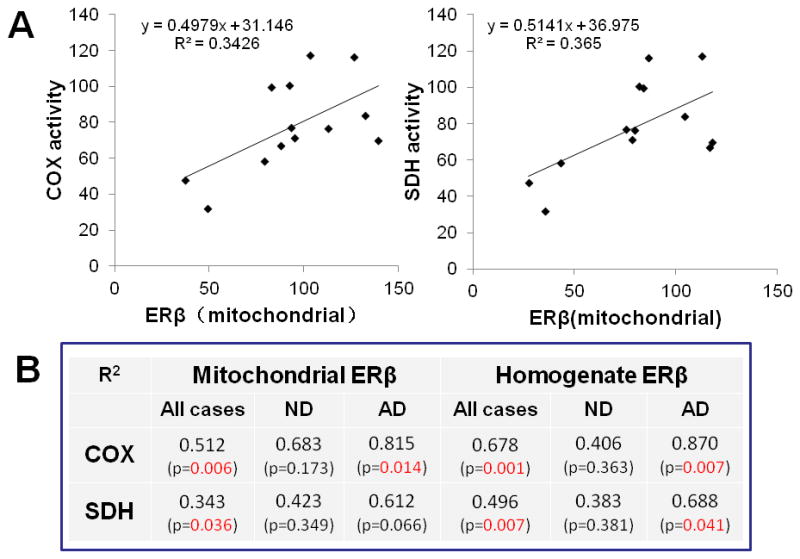
Positive correlation between ERβ expression and activities of COX and SDH in human brain mitochondria. The correlation analysis was performed between mtERβ expression and COX or SDH activity in both AD and ND (A). The correlation between the complex activities and expression of ERβ (mtERβ and total ERβ) were analyzed in AD and ND independently (B). The density of ERβ blot represented mtERβ or total ERβ expression and were calibrated with VDAC and β-actin blot, respectively.
To further understand which subunits of COX were involved in the alteration of COX activity in AD brains, we detected the three major subunits of COX by Western blots. Out results showed that COX 1, and COX 2, which are encoded by mitochondrial DNA, were significantly reduced in the AD group compared to the ND group, while COX 4, which is encoded by nuclear DNA, remained at a constant level (figure 6).
Figure 6.
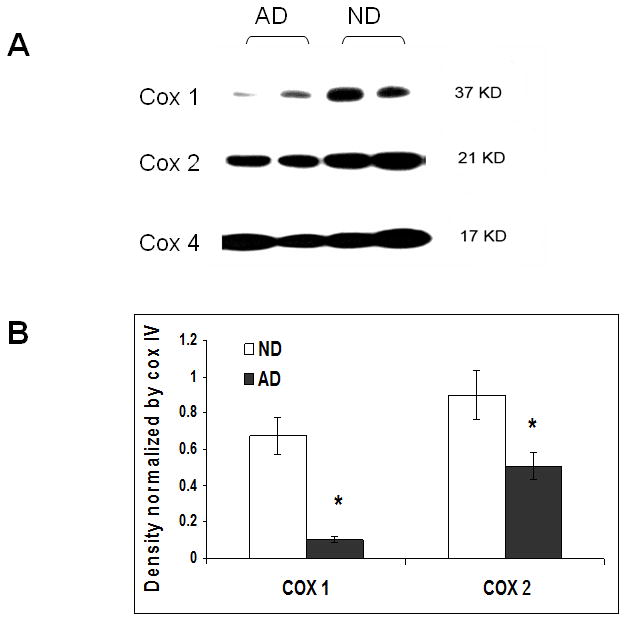
Decreased expression of mitochondrial DNA encoding subunits of COX relative to nuclear DNA encoding subunit. Mitochondria were isolated from the frontal cortices of AD and ND female brains, and the expression of COX 1, 2 and 4 were measured via western blot. (A) Representative image of COX 1, 2 and 4 blot in brain mitochondria. (B) Density of COX 1 and COX 2 normalized by the density of COX 4. * p<0.05 vs. ND.
To find out whether oxidative stress is enhanced in AD brain due to mitochondrial dysfunction, we measured the level of protein carbonylation, which represents the level of intracellular protein oxidation by ROS. Not surprisingly, elevated levels of protein carbonyls were found in AD brain compared to ND controls, including in the cytosolic, nuclear, and mitochondrial cell fractions (figure 7), suggesting that the female AD patients had more oxidative stress due to mitochondria impairment than that in female ND individuals. To examine whether the increased protein oxidative damage in AD is related to the SDH, the only enzyme participates in both citric acid cycle and the electron transport chain in the mitochondria, we also analyzed the correlation between the level of protein carbonyl and SDH activity as shown in figure 7C. Our data showed no significant association between the SDH activity and level of protein carbonylation, suggesting an SDH-independent protein oxidative damage in the female AD patients.
Figure 7.
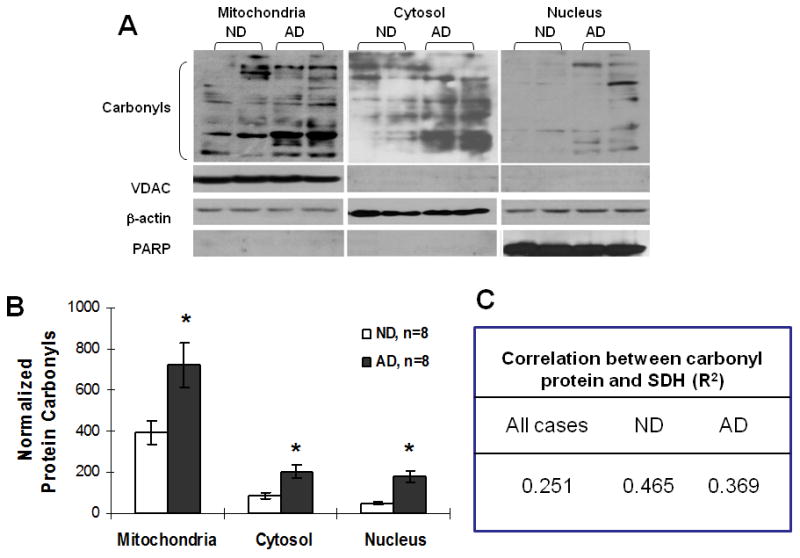
Increased oxidative stress (protein carbonyls) in AD brain. The cell fractions were prepared from frozen tissues of postmortem AD and control brains, followed by an assay for protein carbonyls. (A) Representative images of protein carbonyls blot in mitochondrial, cytosolic, and nuclear cell fractions. (B) Quantitative result of protein carbonyls in each fraction, values were normalized with respect to the internal controls of VDAC, beta-actin and PARP blot. (C) Correlation between carbonyl protein and SDH activity. *p<0.05 vs. ND.
Lacking ERβ increased ROS generation and decreased mitochondrial membrane potential (MMP) in brain mitochondria of ERβ-deficient mice
Our findings from human brain samples, the positive correlation between ERβ expression and mitochondrial function encouraged us to explore ERβ’s role in regulating mitochondrial activity. Using ERβ gene knockout mice, we detected brain mitochondrial oxygen consumption, mitochondrial ROS generation and MMP in WT, ERβ +/− and ERβ −/− mice (female, 10–12 months, n=5 for each genotypes). Interestingly, the basal level of brain mitochondrial oxygen consumption showed no difference among the three genotypes of mice, but treatment with Aβ25-35 (100 μM) caused a great reduction of O2 consumption in the ER+/− mice compare to that in WT animals (p=0.012, figure 8). All the tested dosages of Aβ25-35 and Aβ1-42 significantly depressed brain mitochondrial oxygen consumption compared to non-treated controls for each of the genotypes of mice (P<0.05 in all instances). Between WT and ERβ −/− mice, no significant difference was found between the responses to Aβ insult.
Figure 8.
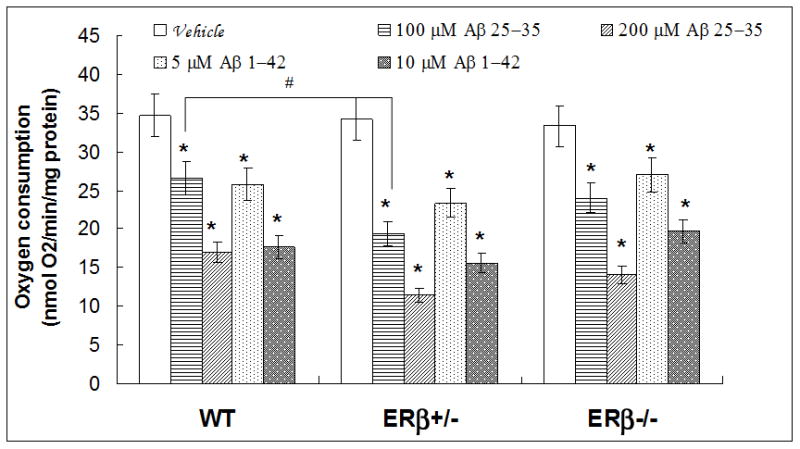
Oxygen consumption in brain mitochondria of WT, ERβ+/− and ERβ−/− mice. Brain mitochondria were isolated from WT, ERβ+/− and ERβ−/− mice (n=5 for each genotype). Oxygen consumption (state III) was measured with an oxygen electrode following Aβ25-35 or Aβ1-42 insult. * p<0.05, vs. vehicle; # p<0.05, vs. WT with Aβ25-35 (100uM) insults.
To examine the effect of ERβ on Aβ-induced ROS generation, we measured ROS level in WT, ERβ+/− and ERβ−/− mice. Our results showed that the effects of Aβ1-42 and Aβ25-35 on mitochondrial ROS levels varied by genotype while no difference was found in the baseline ROS levels among all three genotypes of mice. Treatment of brain mitochondria with Aβ25-35 (200 μM) resulted in higher ROS production in WT mice compare to vehicle treatment (P=0.0002). However, mitochondria from ERβ−/− mice was the most sensitive to amyloid insult; treatment with either Aβ1-42 (10 μM) or Aβ25-35 (200 μM) induced substantial increases in ROS (vs. ERβ−/− without insult, 72% increase p=0.003 and 195% increase p=0.01, respectively), and furthermore the increase of ROS in response to 200 μM Aβ25-35 was double that in WT mice (P=0.05, figure 9). No other difference was found between WT and ERβ knockout mice with the same level of Aβ insult.
Figure 9.
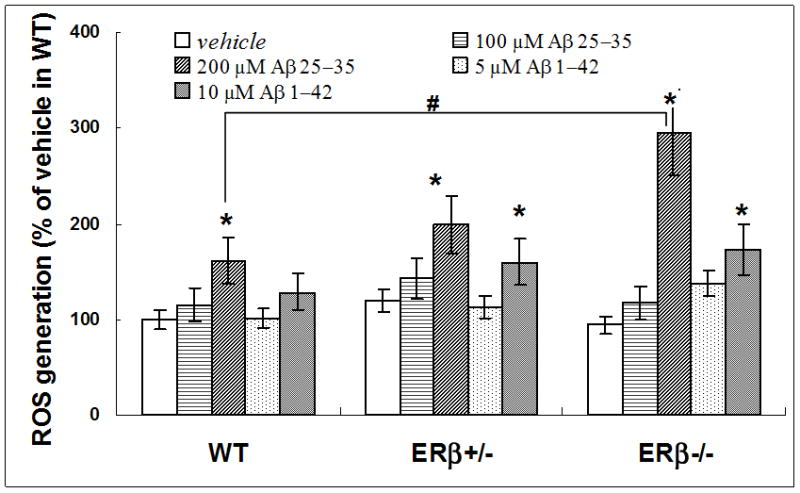
Higher ROS generation in brain mitochondria from ERβ-knockout mice challenged with Aβ. Brain mitochondria were isolated from WT, ERβ+/− and ERβ−/− mice (n=5 for each genotype). Mitochondrial ROS generation was measured with DCFDA staining following Aβ25-35 or Aβ1-42 insult. * p<0.05 vs. vehicle; # p<0.05, vs. WT Aβ25-35 (200μM) insults.
Compared with age matched WT mice, MMP declined slightly in brain mitochondria of ERβ+/− mice (15% loss, p=0.04) and was greatly reduced in ERβ−/−mice (36% loss, p=0.0045) as shown in figure 10, suggesting a direct impact of ERβ deficiency on brain mitochondrial function. To examine whether the effect of ERβ on mitochondrial function, such as MMP, is associated with AD associated amyloid pathology, the isolated the brain mitochondrial were challenged in vitro with Aβ1-42 or Aβ25-35. MMP of brain mitochondria declined in a dose-dependent manner in mice of all three genotypes (figure 10). MMP showed a progressive decrease in ERβ+/− and ERβ−/− mice in comparison with WT mice with the same Aβ insult, but the effect was not significant, suggesting a non-specific effect of ERβ on amyloid-induced mitochondria impairment, at least in MMP.
Figure 10.
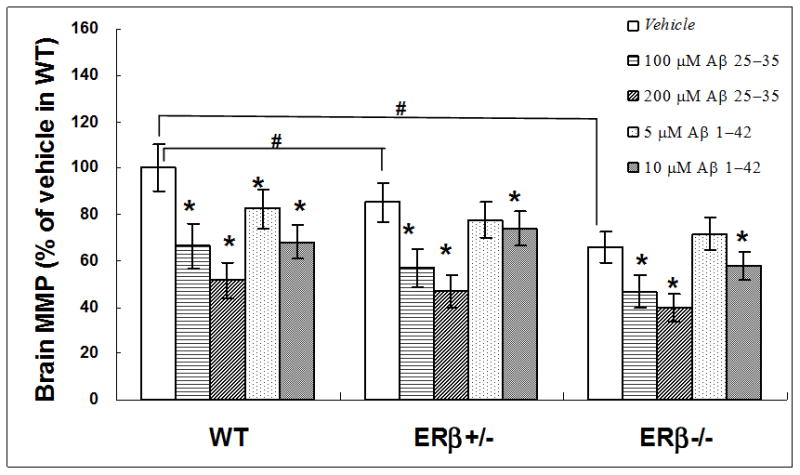
Damaged mitochondrial membrane potential (MMP) in brain mitochondria from ERβKO mice challenged with β amyloid. Brain mitochondria were isolated from WT, ERβ+/− and ERβ−/− mice (n=5 for each genotype). MMP were measured with JC-1 staining following Aβ25-35 or Aβ1-42 insult. * p<0.05, vs. vehicle; # p<0.05 vs. WT vehicle.
Discussion
While mitochondrial impairment in AD has been well established, recent studies suggest that mitochondrial dysfunction in the brain appears much earlier than many other neuropathological changes in AD [3]. As mitochondria play critical roles in maintaining normal living cells, extensive studies have shown that many molecules could detrimentally regulate the function of mitochondria, leading to some abnormality or damage of the mitochondria, including molecules of interest such as amyloid precursor protein[1], β amyloid[29], sex hormones, including estrogen and estrogen receptors [30,31]
Evidence indicates that women have a higher risk of developing AD than do men[32]. In a previous study, we found greatly reduced estrogen levels in rapidly–acquired postmortem brains from women with AD compared with those from age- and gender-matched normal control subjects [33]. Our results suggested that deficits in brain estrogen significantly impact AD pathogenesis in females. Using a gene-based approach, we also demonstrated that the depletion of endogenous estrogen through aromatase gene knockout in AD-like transgenic mice causes early onset of neuropathology and increased Aβ deposition compared to control animals [33]. It is unclear whether the early onset of AD-like pathology induced by estrogen deficiency is mediated through an estrogen receptor-dependent mitochondrial impairment pathway.
Here, we first examined the expression of ERβ in the brains of aged female AD patients and ND controls. Using immunohistochemical staining, we found that neurons from the ND frontal cortex exhibited extensive ERβ expression (figure 1). However, compared to ND controls, neurons from the female AD frontal cortex showed very little ERβ expression, suggesting a reduction of ERβ in female AD brain. To examine whether the reduction of ERβ in AD is due to the loss of neurons in aged AD patients, we also examined total number of neurons as well as neurons express ERβ in the frontal cortex of ND and AD. Our data showed that the ratio of ERβ positive neurons was 40.6 percent and 84.3 percent in AD and ND, respectively, suggesting the reduction of neuronal ERβ in AD is independent from the loss of neurons during aging.
While the mechanisms of estrogen action on mitochondria remain unclear, accumulating studies have shown that estrogen may exert neuroprotective effects via both estrogen receptor-dependent and -independent forms [32]. It is generally accepted that the majority of estrogen actions are mediated through ERα and ERβ. While both ERα and ERβ are located in the nucleus, where they act as important transcription factors, recent evidence has shown that the majority of ERβ is also located extra-nuclearly and is responsible for various non-nuclear actions of estrogen [14,17,32, 34,35]. Although previous studies have demonstrated mitochondrial localization of ERβ in neurons from various mammals[17], little is known regarding expression and function of ERβ in the mitochondria of human neurons.
To examine whether mtERβ is expressed in neuronal mitochondria from the human brain as and whether the level of in mtERβ expression correlates with AD pathology, we investigated the expression and subcellular location of ERβ in the brains from ND and AD individuals using immunohistochemistry and western blotting. Co-staining the brain samples with an anti-ERβ monoclonal antibody and the mitochondrial marker VDAC or MFN1 revealed the novel finding that ERβ colocalizes with mitochondria in the brains of normal aged females and that mtERβ expression is significantly reduced in the female AD brain, even though the density of mitochondrial staining is similar to that of the ND brain (figure 2). This reduction in mtERβ in AD brain was further confirmed by western blotting, which demonstrated a significant reduction of ERβ in the mitochondria fraction as well as in all other cell fractions of the AD brain samples, even in the whole brain lysates, compared to ND brain samples. These results suggest that the reduction of ERβ in the female AD brain is not limited to neuronal mitochondria, but is present throughout the cells from frontal cortex.
To further characterize the differences between female AD and ND mitochondria, we measured complex II and IV activities. Our study found that activity of brain SDH (complex II) and COX (complex IV) was reduced in the female AD patients compared to the ND individuals. (figure 4). Our result is consistent with other reports of AD-related impairment of SDH and COX activity [36,37], and with findings relating to the effects of estrogen on COX [35]. To understand whether the reduction of mtERβ in AD is associated with mitochondrial impairment in female AD patients, we examined the correlation between the complex activity and mtERβ expression in the brain (figure 5). Our data showed for the first time to our knowledge, a significant association between the reduction of mtERβ expression and decrease in COX activity in AD brain samples, not in ND human subjects (figure 5B). However, the COX activity is not only significantly associated with mtERβ, but also with the total ERβ in the AD brain, not in ND, suggesting a disease specific effect of ERβ in the female AD. In contrast, the SDH activity showed a weak association with mtERβ but strong correlation with total ERβ in the AD brain. Our data suggest that the relationship between mtERβ and mitochondrial activities in the female AD brain is largely reflected AD pathology status on both ERβ and COX activity.
In mammalian mitochondrial respiratory chains, COX is made up of 13 subunits. Three of the subunits (COX 1, COX 2, and COX 3) are encoded by mitochondrial DNA, while the other 10 subunits are encoded by nuclear DNA, such as COX 4. To further investigate and understand the molecular changes in COX activity in AD, we examined the levels of mitochondrial DNA encoded subunits (COX 1 and COX 2) with the expression of a nuclear DNA encoded subunit (COX 4) from both AD and ND samples. Interestingly, we found significant decreases only in the subunits encoded by mitochondrial DNA (COX 1 and COX 2), but not in the subunit encoded from nuclear DNA (COX 4). Our results suggest that COX subunits encoded by mitochondrial DNA are selectively compromised in the female AD frontal cortex (figure 6). Thus, the impairment of COX activity in AD patients may be related to an impairment of protein synthesis in dysfunctional mitochondria, and mtERβ might play roles in regulation of COX activity. Our data corresponds to previous observations that AD pathology is accompanied by a decrease in expression and activity of enzymes involved in mitochondrial bioenergetics[38,39], but also demonstrates a possible linkage between mtERβ and the electron transport chain complex activity in the female AD brain. In addition to the lowered mitochondrial bioenergetic capacity, decreased COX activity, overproduction of reactive oxygen species and higher oxidative stress are also characteristics of AD brains.
To further investigate the oxidative damage due to mitochondrial dysfunction in the female AD brain, we measured the expression of mitochondrial protein carbonyls, an indication of protein oxidation in brain mitochondria. As shown in figure 7, the protein carbonyl levels in the mitochondrial fractions from AD samples were significantly higher than those from the ND samples. Our data provided additional evidence that female AD patients exhibit increased oxidative stress, especially mitochondrial oxidative damage, in their brains compared to ND individuals. In addition, in this present study, we showed no significant association between the elevated protein carbonylation level and reduced SDH activity in AD, suggesting a SDH-independent protein oxidative damage in the female AD patients.
Although female AD patients showed a significant reduction of ERβ along with enhanced mitochondrial impairment in the brain, it is unknown whether it is the ERβ deficiency in females that promotes this mitochondrial dysfunction exhibited in AD brains. Here, using a genetic approach, we examined the effects of ERβ on various indicators of mitochondrial function in female WT, ERβ+/− or ERβ−/− mice. Isolated mitochondria from mouse brain were treated with or without Aβ, which is associated with neuropathology in AD and were found to induce mitochondrial impairment [36].
To determine the cellular contribution from ERβ to mitochondrial function, we detected the O2 consumption from freshly isolated mitochondria from mice brains and showed that the O2 consumption rates in response to vehicle were similar in both WT and ERβ+/− or ERβ−/− mice, indicating the baseline of O2 consumption was not affected by a direct impairment of ERβ. When exposed to Aβ, a greater attenuation in state 3 O2 consumption rates was observed in ERβ+/−, not ERβ−/−, compared to that in WT mice (figure 8). Because our data showed that only ERβ+/−, not ERβ−/−mice, enhanced sensitivity of response to Aβ in mitochondria, and based on the that receptors may form ERα (αα) or ERβ (ββ) homodimers or ERαβ (αβ) heterodimers in many cells [40], the action of ERβ in protecting against Aβ-induced reduction of O2 consumption might need of participation of ERα, possible in the form of the αβ heterodimer. In contract to O2 consumption, as shown in figure 9, the mitochondria from ERβ−/− mice showed a great increase in ROS generation after being challenged with high dosages of Aβ1-42 or Aβ25-35 compared to vehicle treatment. These data suggest that the effect of ERβ on mitochondria structure and function might be mediated through the formation of homodimers or heterodimers of ER. No ROS changes were found between WT and ERβ-manipulated mice treated with vehicle. These findings suggest that depletion of ERβ does not disturb mitochondrial respiration in physiological normal condition directly, but it may enhance the vulnerability of brain mitochondria to Aβ-induced mitochondrial oxidative stress, as indicated by decreased oxygen consumption and increased ROS generation in vitro. These results are in agreement with recent studies showing that Aβ and Bax (a member of Bcl-2 family) can be transported from the cytosol into the mitochondria and damage mitochondrial structure and function, reducing oxygen consumption and increasing ROS levels. Estrogen was shown to prevent the translocation of apoptosis protein such as Bax from the cytosol to the mitochondria, and prevent Aβ-induced mitochondrial impairments [41–43]. Although it is unknown whether the reduced oxygen consumption rates and enhanced ROS generation found in ERβ+/− or ERβ−/− mice were mediated through the inhibition of translocation into mitochondria, our study for the first time suggested that ERβ might be involved in the estrogen-induced neuroprotective actions in AD via regulating mitochondria function in vitro.
While reduction of MMP is an index of mitochondria degeneration due to mitochondrial membrane collapse [37], we examined the MMP in the mice with or without ERβ expression. Our data demonstrated that MMP showed a decreasing trend along with the ERβ depletion (figure 10). This suggests that ERβ might be an essential component of the neuronal mitochondria and contribute to the maintenance of normal membrane potential. Consistent with the O2 and ROS dysfunction, MMP exhibited significant loss following exposure to Aβ in mice of all three genotypes (figure 10). These results suggest that ERβ might be more involved in retaining normal MMP rather than protecting against Aβ-induced mitochondria MMP loss.
Although further investigation is required, our novel detection of ERβ expression in neuronal mitochondria in the female human brain, along with the direct correlation of mtERβ expression with COX activity and the significant decrease of mtERβ and total ERβ in the AD brain, suggests a possible connection between ERβ and mitochondrial dysfunction in AD. This hypothesis is supported by our experiments involving transgenic ERβ knockout mice, which show that ERβ deficiency leads to mitochondrial dysfunction in the brain by decreasing MMP and increasing mitochondrial vulnerability to Aβ-induced ROS generation. Together, the results of this study indicate that ERβ deficiency may play an important role in AD pathogenesis in females by contributing to mitochondrial dysfunction.
Table 1.
related information of human cases in the study
| Case ID | expired age (Year) | Clinical diagnosis | Disease Duration (Year) | Pathology summary | Plaque density |
|---|---|---|---|---|---|
| 01-34 | 89 | AD | 26 | AD; Cerebral white matter rarefaction (CWMR); Alzheimer type II astrocytosis | frequent |
| 01-53 | 87 | AD | 6 | AD; Cerebral white matter rarefaction | frequent |
| 01-17 | 78 | AD; stroke | 11 | AD; Large old infarction of left lateral temporal and occipital lobes, caudate nucleus, putamen and internal capsule, with Wallerian degeneration of ipsilateral corticospinal tract; Acute infarction, left lateral temporal and pariet | frequent |
| 01-07 | 85 | AD | 9 | AD; Small acute infarct, and microscopic subacute infarct, left postcentral gyrus; Subacute infarct, left superior frontal gyrus; Two microscopic foci of cerebellar cortical sclerosis/old infarct; Capillary telangiectasia, right cing | frequent |
| 01-02 | 89 | AD | 11 | AD; Cerebral white matter rarefaction, frontal lobe; Old lacunar infarcts, left head of caudate nucleus/putamen/internal capsule and left thalamus; Etat crible, caudate nucleus and thalamus; Probable Candidal microabscesses, cerebrum | frequent |
| 01-09 | 83 | AD | 26 | AD; Cerebral white matter rarefaction; Subacute lacunar infarct, basal pons; Etat crible, putamen and thalamus; Single microscopic focus of cerebellar cortical sclerosis; Hyperostosis frontalis interna | frequent |
| 00-25 | 95 | AD | 14 16 |
AD; Large old infarct, left temporal and occipital lobe; Cerebral white matter rarefaction, frontal, temporal and occipital lobes; Several old lacunar infarcts, left and right putamen and left thalamus; Focal cerebellar cortical scle | frequent |
| 01-10 | 78 | AD | AD; Cerebral white matter rarefaction; Old lacunar infarct, left caudate nucleus | frequent | |
| 01-14 | 78 | Not AD | Control; Argyrophilic grains, mesial temporal lobe; Cerebral white matter rarefaction, mild | sparse | |
| 99-54 | 86 | Not AD | Control (brain showing only normal aging changes) | zero | |
| 97-50 | 88 | Not AD, MCI | Control (non-motoric); Mild cognitive impairment; Incidental Lewy bodies in substantia nigra; Alzheimer type II glia, consistent with metabolic encephalopathy | zero | |
| 96-32 | 85 | Not AD | Control; cerebral amyloid angiopathy, mild (CAA) | zero | |
| 99-27 | 88 | Not AD; MCI | Control (MCI); psychologic testing consistent with borderline dementia/ MCI; extensive periventricular white matter rarefaction, frontal & parietal lobes; cerebral amyloid angiopathy; old lacunar infarct, left globus pallidus; mild to moderate histologic | sparse | |
| 00-49 | 86 | Not AD | Control; Recent small infarctions in left frontal, left temporal, left cerebellar cortex and left basal pons; Old cortical microinfarction, left precentral gyrus; Etat crible & mineralization of globus pallidus; Alzheimer type II astrocytosis; Argyrophili | zero | |
| 99-58 | 91 | Not AD; possible MCI | Control (Mild cognitive impairment); cerebral amyloid angiopathy; cerebral white matter rarefaction; restless legs syndrome (history) | sparse | |
| 96-13 | 85 | Not AD, essential tremor, spasmodic dysphonia | Control; Spasmodic dysphonia (history); Essential tremor | zero |
MCI: mild cognition impairment.
Acknowledgments
This work was supported by grants from the Alzheimer’s Association IIRG-07-59510 and IIRG-09-61521, American Health Assistance Foundation Grant G2006-118, and National Institute of Health (NIH R01AG032441-01, R01AG025888-01). We are grateful to the Banner Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona for the provision of human brain tissue. The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research
References
- 1.Lin MT, Beal MF. Alzheimer’s APP mangles mitochondria. Nat Med. 2006;12:1241–1243. doi: 10.1038/nm1106-1241. [DOI] [PubMed] [Google Scholar]
- 2.Moreira PI, Cardoso SM, Santos MS, Oliveira CR. The key role of mitochondria in Alzheimer’s disease. J Alzheimers Dis. 2006;9:101–110. doi: 10.3233/jad-2006-9202. [DOI] [PubMed] [Google Scholar]
- 3.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 4.Pike CJ. Estrogen modulates neuronal Bcl-xL expression and beta-amyloid-induced apoptosis: relevance to Alzheimer’s disease. J Neurochem. 1999;72:1552–1563. doi: 10.1046/j.1471-4159.1999.721552.x. [DOI] [PubMed] [Google Scholar]
- 5.Yao M, Nguyen TV, Pike CJ. Estrogen regulates Bcl-w and Bim expression: role in protection against beta-amyloid peptide-induced neuronal death. J Neurosci. 2007;27:1422–1433. doi: 10.1523/JNEUROSCI.2382-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nilsen J, Brinton RD. Mitochondria as therapeutic targets of estrogen action in the central nervous system. Curr Drug Targets CNS Neurol Disord. 2004;3:297–313. doi: 10.2174/1568007043337193. [DOI] [PubMed] [Google Scholar]
- 7.Jensen EV, Suzuki T, Kawashima T, Stumpf WE, Jungblut PW, DeSombre ER. A two-step mechanism for the interaction of estradiol with rat uterus. Proc Natl Acad Sci U S A. 1968;59:632–638. doi: 10.1073/pnas.59.2.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Razmara A, Duckles SP, Krause DN, Procaccio V. Estrogen suppresses brain mitochondrial oxidative stress in female and male rats. Brain Res. 2007;1176:71–81. doi: 10.1016/j.brainres.2007.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greene GL, Gilna P, Waterfield M, Baker A, Hort Y, Shine J. Sequence and expression of human estrogen receptor complementary DNA. Science. 1986;231:1150–1154. doi: 10.1126/science.3753802. [DOI] [PubMed] [Google Scholar]
- 10.Levin ER. Cell localization, physiology, and nongenomic actions of estrogen receptors. J Appl Physiol. 2001;91:1860–1867. doi: 10.1152/jappl.2001.91.4.1860. [DOI] [PubMed] [Google Scholar]
- 11.Chambliss KL, Yuhanna IS, Anderson RG, Mendelsohn ME, Shaul PW. ERbeta has nongenomic action in caveolae. Mol Endocrinol. 2002;16:938–946. doi: 10.1210/mend.16.5.0827. [DOI] [PubMed] [Google Scholar]
- 12.Li R, Shen Y. Estrogen and brain: synthesis, function and diseases. Front Biosci. 2005;10:257–267. doi: 10.2741/1525. [DOI] [PubMed] [Google Scholar]
- 13.Chen JQ, Russo PA, Cooke C, Russo IH, Russo J. ERbeta shifts from mitochondria to nucleus during estrogen-induced neoplastic transformation of human breast epithelial cells and is involved in estrogen-induced synthesis of mitochondrial respiratory chain proteins. Biochim Biophys Acta. 2007;1773:1732–1746. doi: 10.1016/j.bbamcr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 14.Herrick SP, Waters EM, Drake CT, McEwen BS, Milner TA. Extranuclear estrogen receptor beta immunoreactivity is on doublecortin-containing cells in the adult and neonatal rat dentate gyrus. Brain Res. 2006;1121:46–58. doi: 10.1016/j.brainres.2006.08.084. [DOI] [PubMed] [Google Scholar]
- 15.Jonsson D, Nilsson J, Odenlund M, Bratthall G, Broman J, Ekblad E, Lydrup ML, Nilsson BO. Demonstration of mitochondrial oestrogen receptor beta and oestrogen-induced attenuation of cytochrome c oxidase subunit I expression in human periodontal ligament cells. Arch Oral Biol. 2007;52:669–676. doi: 10.1016/j.archoralbio.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 16.Yager JD, Chen JQ. Mitochondrial estrogen receptors--new insights into specific functions. Trends Endocrinol Metab. 2007;18:89–91. doi: 10.1016/j.tem.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 17.Yang SH, Liu R, Perez EJ, Wen Y, Stevens SM, Jr, Valencia T, Brun-Zinkernagel AM, Prokai L, Will Y, Dykens J, Koulen P, Simpkins JW. Mitochondrial localization of estrogen receptor beta. Proc Natl Acad Sci U S A. 2004;101:4130–4135. doi: 10.1073/pnas.0306948101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beach TG, Sue LI, Walker DG, Roher AE, Lue L, Vedders L, Connor DJ, Sabbagh MN, Rogers J. The Sun Health Research Institute Brain Donation Program: description and experience, 1987–2007. Cell Tissue Bank. 2008;9:229–245. doi: 10.1007/s10561-008-9067-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci USA. 1993;90:11162–11166. doi: 10.1073/pnas.90.23.11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lai JC, Clark JB. Preparation of synaptic and nonsynaptic mitochondria from mammalian brain. Methods Enzymol. 1979;55:51–60. doi: 10.1016/0076-6879(79)55008-3. [DOI] [PubMed] [Google Scholar]
- 21.Long J, Liu C, Sun L, Gao H, Liu J. Neuronal mitochondrial toxicity of malondialdehyde: inhibitory effects on respiratory function and enzyme activities in rat brain mitochondria. Neurochem Res. 2009;34:786–794. doi: 10.1007/s11064-008-9882-7. [DOI] [PubMed] [Google Scholar]
- 22.Levine RL, Garland D, Oliver CN, Amici A, Climent I, Lenz AG, Ahn BW, Shaltiel S, Stadtman ER. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990;186:464–478. doi: 10.1016/0076-6879(90)86141-h. [DOI] [PubMed] [Google Scholar]
- 23.Li R, Yang L, Lindholm K, Konishi Y, Yue X, Hampel H, Zhang D, Shen Y. Tumor necrosis factor death receptor signaling cascade is required for amyloid-beta protein-induced neuron death. J Neurosci. 2004;24:1760–1771. doi: 10.1523/JNEUROSCI.4580-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hagen TM, Liu J, Lykkesfeldt J, Wehr CM, Ingersoll RT, Vinarsky V, Bartholomew JC, Ames BN. Feeding acetyl-L-carnitine and lipoic acid to old rats significantly improves metabolic function while decreasing oxidative stress. Proc Natl Acad Sci U S A. 2002;99:1870–1875. doi: 10.1073/pnas.261708898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cossarizza A, Ceccarelli D, Masini A. Functional heterogeneity of an isolated mitochondrial population revealed by cytofluorometric analysis at the single organelle level. Exp Cell Res. 1996;222:84–94. doi: 10.1006/excr.1996.0011. [DOI] [PubMed] [Google Scholar]
- 26.Moreira PI, Santos MS, Moreno AM, Seica R, Oliveira CR. Increased vulnerability of brain mitochondria in diabetic (Goto-Kakizaki) rats with aging and amyloid-beta exposure. Diabetes. 2003;52:1449–1456. doi: 10.2337/diabetes.52.6.1449. [DOI] [PubMed] [Google Scholar]
- 27.Zamzami N, Metivier D, Kroemer G. Quantitation of mitochondrial transmembrane potential in cells and in isolated mitochondria. Methods Enzymol. 2000;322:208–213. doi: 10.1016/s0076-6879(00)22021-1. [DOI] [PubMed] [Google Scholar]
- 28.Etoh S, Matsui H, Tokuda M, Itano T, Nakamura M, Hatase O. Purification and immunohistochemical study of actin in mitochondrial matrix. Biochem Int. 1990;20:599–606. [PubMed] [Google Scholar]
- 29.Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M. ABAD directly links A {beta} to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304:448. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 30.Vina J, Lloret A, Valles SL, Borras C, Badia MC, Pallardo FV, Sastre J, Alonso MD. Effect of gender on mitochondrial toxicity of Alzheimer’s Abeta peptide. Antioxid Redox Signal. 2007;9:1677–1690. doi: 10.1089/ars.2007.1773. [DOI] [PubMed] [Google Scholar]
- 31.Veiga S, Melcangi RC, Doncarlos LL, Garcia-Segura LM, Azcoitia I. Sex hormones and brain aging. Exp Gerontol. 2004;39:1623–1631. doi: 10.1016/j.exger.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 32.Green PS, Simpkins JW. Neuroprotective effects of estrogens: potential mechanisms of action. Int J Dev Neurosci. 2000;18:347–358. doi: 10.1016/s0736-5748(00)00017-4. [DOI] [PubMed] [Google Scholar]
- 33.Yue X, Lu M, Lancaster T, Cao P, Honda S, Staufenbiel M, Harada N, Zhong Z, Shen Y, Li R. Brain estrogen deficiency accelerates Abeta plaque formation in an Alzheimer’s disease animal model. Proc Natl Acad Sci U S A. 2005;102:19198–19203. doi: 10.1073/pnas.0505203102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen JQ, Yager JD. Estrogen’s effects on mitochondrial gene expression: mechanisms and potential contributions to estrogen carcinogenesis. Ann N Y Acad Sci. 2004;1028:258–272. doi: 10.1196/annals.1322.030. [DOI] [PubMed] [Google Scholar]
- 35.Chen JQ, Yager JD, Russo J. Regulation of mitochondrial respiratory chain structure and function by estrogens/estrogen receptors and potential physiological/pathophysiological implications. Biochim Biophys Acta. 2005;1746:1–17. doi: 10.1016/j.bbamcr.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 36.Canevari L, Clark JB, Bates TE. β-Amyloid fragment 25–35 selectively decreases complex IV activity in isolated mitochondria. FEBS letters. 1999;457:131–134. doi: 10.1016/s0014-5793(99)01028-5. [DOI] [PubMed] [Google Scholar]
- 37.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 38.Ansari MA, Scheff SW. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol. 2010;69:155–167. doi: 10.1097/NEN.0b013e3181cb5af4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blass JP, Sheu RK, Gibson GE. Inherent abnormalities in energy metabolism in Alzheimer disease. Interaction with cerebrovascular compromise. Ann N Y Acad Sci. 2000;903:204–221. doi: 10.1111/j.1749-6632.2000.tb06370.x. [DOI] [PubMed] [Google Scholar]
- 40.Li X, Huang J, Yi P, Bambara RA, Hilf R, Muyan M. Single-chain estrogen receptors (ERs) reveal that the ERalpha/beta heterodimer emulates functions of the ERalpha dimer in genomic estrogen signaling pathways. Mol Cell Biol. 2004;24:7681–7694. doi: 10.1128/MCB.24.17.7681-7694.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nilsen J, Chen S, Irwin RW, Iwamoto S, Brinton RD. Estrogen protects neuronal cells from amyloid beta-induced apoptosis via regulation of mitochondrial proteins and function. BMC Neurosci. 2006;7:74. doi: 10.1186/1471-2202-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, Leinonen V, Ito A, Winblad B, Glaser E, Ankarcrona M. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A. 2008;105:13145–13150. doi: 10.1073/pnas.0806192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reddy PH. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp Neurol. 2009;218:286–292. doi: 10.1016/j.expneurol.2009.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]