

Abstract
Metabolic activities in normal cells rely primarily on mitochondrial oxidative phosphorylation (OXPHOS) to generate ATP for energy. Unlike in normal cells, glycolysis is enhanced and OXPHOS capacity is reduced in various cancer cells. It has long been believed that the glycolytic phenotype in cancer is due to a permanent impairment of mitochondrial OXPHOS, as proposed by Otto Warburg. This view is challenged by recent investigations which find that the function of mitochondrial OXPHOS in most cancers is intact. Aerobic glycolysis in many cancers is the combined result of various factors such as oncogenes, tumor suppressors, a hypoxic microenvironment, mtDNA mutations, genetic background and others. Understanding the features and complexity of the cancer energy metabolism will help to develop new approaches in early diagnosis and effectively target therapy of cancer.
Keywords: cancer, glycolysis, oxidative phosphorylation
Contents
Introduction
Relationships between glycolysis and OXPHOS are cooperative and competitive
Cancer cells have a diversity of energy production pathways
Alterations of oncogenes and tumor suppressors drive cancer cells to aerobic glycolysis
Conclusion
1. Introduction
Energy consumption from metabolic activities in normal cells relies primarily on mitochondrial oxidative phosphorylation (OXPHOS), which is efficient and generates more adenosine triphosphate (ATP) than glycolysis. However, one of the metabolic features of cancer cells is to avidly take up glucose for aerobic glycolysis. This inefficient pathway for energy production in cancer cells was first described by German scientist Otto Warburg in the 1920s, and is also known as the Warburg effect (1).
Warburg originally proposed that the aerobic glycolysis in cancer cells was due to a permanent impairment of mitochondrial OXPHOS. However, this view is challenged by recent investigations which found that defects of mitochondrial OXPHOS are not common in spontaneous tumors (2) and that the function of mitochondrial OXPHOS in most cancers is intact (1,3–7). Aerobic glycolysis in many cancers is a result driven by various factors, such as activation of oncogenes, loss of tumor suppressors, the hypoxic microenvironment, mitochondrial DNA (mtDNA) mutation and the tissue of origin.
Cancers are extremely heterogeneous diseases and each cancer has its individual metabolic features. Even in a single cancer, its constituent cells are also heterogeneous and metabolic phenotypes vary from one cell to another. Although aerobic glycolysis is often found in malignant tumors, OXPHOS still contributes to energy production in cancers, and may play a major role in energy production in some cancers (8). This article reviews the roles of glycolysis and OXPHOS in the energy metabolism of cancers.
2. Relationships between glycolysis and OXPHOS are cooperative and competitive
Before the introduction of free oxygen into the atmosphere, organisms on earth rely on glycolysis as an energy source. However, with the increase of atmospheric oxygen, cells begin to rely primarily on OXPHOS to produce energy since it generates more ATP per metabolite than the glycolytic pathway.
However, glycolysis, an ancient energy production pathway preserved in evolution, still affects energy metabolism in certain human organs and tissues, such as the brain, liver and muscle (9,10), and is also a major energy production pathway in anaerobic bacteria. In fact, glycolysis and OXPHOS are tightly coupled and serve as a molecular interconversion system. The process of glycolysis is carried out in the cytoplasm and only produces two ATPs. Pyruvate, the end product of glycolysis, is a fuel for OXPHOS. Under aerobic conditions pyruvate enters the mitochondria to be oxidized to acetyl CoA which combines with oxaloacetate to start the tricarboxylic acid (TCA) cycle and OXPHOS, which can produce 36 ATPs. Under anaerobic conditions, pyruvate is reduced to lactate by lactate dehydrogenase A (LDH-A) in the cytoplasm and then lactate is excreted into the extracellular space through monocarboxylate transporters (MCTs).
Since energy production is a response to energy demand in the cell, the ATP yield varies depending on cellular conditions and the microenvironment. Mammalian cells rely primarily on both glycolysis and OXPHOS to produce ATP at present. However, the contribution ratio of glycolysis versus OXPHOS for the total ATP yield varies in different cells, growth states and microenvironments. In normal conditions, the cell metabolism consumes energy, of which 70% is supplied by OXPHOS. In hypoxia, however, glycolysis becomes enhanced to compensate for the weakened function of OXPHOS. Therefore, glycolysis and OXPHOS cooperate to maintain the cellular energetic balance. Supposing the total ATP is a constant, if the function of OXPHOS is weakened, the function of glycolysis must be enhanced in order to maintain a balance of energy. In contrast, if the function of OXPHOS is normal, it will regulate glycolytic activity via different pathways to maintain a balance of energy (11). In contrast to normal cells, most cancer cells use glycolysis as a means of energy production whenever normoxia or hypoxia occurs, which is referred to as aerobic glycolysis (1).
3. Cancer cells have a diversity of energy production pathways
Cancer cells are different from most normal tissues in the energy metabolism and they take up glucose and glutamine at a high rate for aerobic glycolysis. In addition, cancers are extremely heterogeneous and each cancer is different in tissue origin and metabolic phenotype (3). Even in a single cancer, its constituent cells vary in metabolic phenotype (12). It is worth mentioning that metabolic phenotypes in cancer are plastic, and cancer tissues exhibit greater plasticity than normal tissues (13). Cancer cells may change their metabolic phenotypes to adapt to microenvironmental changes and the results of these changes give a selective advantage to cancer cells under the unfavorable environment (14,15).
Glycolysis versus OXPHOS
Due to their different origin and differentiation, not all cancers rely primarily on glycolysis, which contributes to total ATP at a rate of 1–64% in cancer cells (8). For example, Suganuma et al examined the energy metabolism of four leukemia cell lines using glycolysis inhibitor 2-deoxy-D-glucose (2-DG) and OXPHOS inhibitor oligomycin (16). They found that NB4 cells were more sensitive to 2-DG than the three other cell lines, hence they regarded NB4 as a ‘glycolytic’ leukemia cell line. Alternatively, THP-1 cells were resistant to 2-DG and sensitive to oligomycin, and were regarded as an ‘OXPHOS’ leukemia cell line (16). These results suggest that energy metabolic pathways are different in various cancers. We should first examine energy metabolic pathways in cancer cells when considering energy metabolism as a target for cancer therapy in order to obtain good therapeutic results.
Warburg considered that aerobic glycolysis in cancer cells was irreversibly impaired in its mitochondrial function. This view is challenged by recent investigations which found that the function of mitochondrial OXPHOS in many cancers is intact (1,3–7). Certain authors consider that the Warburg effect in cancer is due to enhanced glycolysis suppressing OXPHOS rather than defects in mitochondrial OXPHOS. If glycolysis is inhibited in cancer cells, the function of mitochondrial OXPHOS can be restored (4,17–19). For example, Fantin et al observed that when LDH-A was suppressed in cancer cells, OXPHOS function could be enhanced to compensate for reduced ATP by inhibited glycolysis. This observation suggests that most cancer cells reserve the capacity to produce ATP by OXPHOS. The glycolytic phenotype in cancer cells is due to OXPHOS being suppressed by active glycolysis rather than defects in mitochondrial function. Furthermore, they also found that proliferation and tumorigenicity of cancer cells were inhibited when LDH-A activity was suppressed, suggesting that enhanced OXPHOS is still not sufficient to meet the requirement of cancer growth and that LDH-A is a target of cancer therapy (4).
Since the glycolytic contribution to total ATP production does not generally exceed 50–60% (8), OXPHOS still substantially contributes to ATP production in tumor cells. Although four human malignant tumor cell lines (HL60, HeLa, 143B and U937) are cells which rely on OXPHOS to support the growth of cells (20), this phenotype is altered under hypoxia. For example, the OXPHOS contribution to total ATP production is normally 79 and 91% in cervical carcinoma HeLa cells and breast carcinoma MCF cells, respectively. This contribution, however, is reduced to 29 and 36% in hypoxia, respectively (21), suggesting that the glycolytic phenotype in cancer cells is primarily caused by hypoxia. In their retrospective review, Moreno-Sánchez et al point out that although glycolysis plays an important role in cancer energy metabolism, a considerable amount of cancers use OXPHOS as a pathway of energy production or a mixture of glycolysis and OXPHOS (17). In some cases, the function of OXPHOS in cancer cells is even higher than in adjacent stromal cells (22). Researchers from Singapore recently isolated intact mitochondria from human ovarian and peritoneal cancer tissues, which exhibited the specific activities of succinate, malate and glutamine dehydrogenases, and had the capacity of OXPHOS. The cells produced ATP, but in lower amounts than the human skeletal muscle (6).
Smolkova et al (19) proposed four waves of metabolic regulation during carcinogenesis. Wave 1: cancer stem cell (CSC) transformation, primarily due to oncogene-mediated signaling; Wave 2: hypoxia, inducing hypoxia-inducible factor (HIF), AMP-activated protein kinase (AMPK) and NF-κB signaling. In waves 1 and 2, the cell metabolism highly favors glycolysis and inhibits OXPHOS due to the oncogenic and hypoxic controls of gene reprogramming, i.e., the classic Warburg phenotype. Wave 3: aglycemia, nutrient shortage due to the high proliferation rate during malignancy. In this wave, the function of mitochondrial OXPHOS is partially restored due to gene reprogramming via the LKB1-AMPK-p53 pathway and/or the PI3K-Akt-mTOR pathway. Myc-mediated glutaminolysis is also involved in this process. Wave 4: mitochondria revival, retrograde signaling from revitalized mitochondria may constitute this wave of gene reprogramming. This working hypothesis indicates that the Warburg phenotype is not exclusive and that a decrease of mitochondrial function is not a general feature of cancer cells.
An emerging hypothesis of the so-called reverse Warburg effect also supports the function of OXPHOS in cancer cells (22). In the reverse Warburg effect, epithelial cancer cells induce aerobic glycolysis in carcinoma-associated fibroblasts (CAFs) which produce lactate, ketones and pyruvate that enter the TCA cycle in cancer cells for OXPHOS (22,23). Previously, it was consdiered that the behavior of cancer was determined by cancer cells, but more recently stromal cells have also been demonstrated to be involved in the growth of cancer cells. This is possible due to the co-evolution between cancer cells and stromal cells in tumorigenesis. For example, under the ‘education’ of cancer cells and inflammatory cytokines, stromal fibroblasts become CAFs, macrophages become tumor-associated macrophages (TAMs) and neutrophils become tumor-associated neutrophils (TANs), and so on. In fact, these tumor-associated stromal cells are already different from their original cells and have genetic and epigenetic changes which lead to alterations in expression and metabolic profiles. Therefore, tumor cells and stromal cells not only influence each other in cytokines and growth factors, but also in energy metabolites. The relationship between cancer cells and tumor-associated stromal cells is to promote each other, unlike the original inhibitory relationship between normal epithelial and stromal cells (24). These studies suggest that caution should be applied when using lactate in cancer patients, and also provide a theoretical basis for using stromal cells as a target of cancer therapy.
An increasing number of recent studies show that lactate released by glycolysis in hypoxic tumor cells and/or stromal cells is not discharged as a waste product, but can be taken up by oxygenated tumor cells as energy fuel. Lactate is converted to pyruvate by LDH-B and then enters the mitochondria for OXPHOS to generate ATP (3,14,22,25–28). Metabolic symbiosis is not cancer-specific. In fact, the cooperation between different cells in energy production is also observed in normal tissues. For example, neurons rely on OXPHOS to meet their energy demands, whereas astrocytes rely on glycolysis to meet their energy needs. Lactate released by astrocytes can be taken up by neurons, used as energy fuel and form the so-called astrocyte-neuron lactate shuttle (ANLS) (29).
Cancer cells prefer to use aerobic glycolysis for ATP production while still retaining the function of OXPHOS for the following reasons: i) Glycolysis is more suitable for cancer growth. Since proliferation of cancer tissues is faster than normal tissues, it not only needs energy, but also needs metabolic intermediates for the biosynthesis of macromolecules. Many intermediates from glycolysis and the truncated TCA cycle can be used to synthesize macromolecules, such as nucleic acids, lipids and proteins, which are required for cancer growth and proliferation (2,30). ii) Too efficient products of ATP may not a good thing for cancer cells. If cancer cells use high-efficiency glucose, ADP is converted to ATP. The high concentration of ATP will inhibit phosphofructokinase 1 (PFK1), the rate-limiting enzyme in glycolysis and pyruvate kinase 1 (PK1), and glycolysis will be inhibited. Inhibited glycolysis is unfavorable for cancer cell growth. Although glycolysis yields less ATP than OXPHOS, the speed of ATP generation in the former is quicker than in the latter, which is suited to the energy demands of rapid proliferation tissues such as cancer and embryonic tissues (11). Generally speaking, rapid proliferation tissues rely more on glycolysis for ATP production whereas differentiation tissues rely primarily on OXPHOS for energy production (13,31). If using glycolysis inhibitor 3-bromopyruvate (3-BP) treats tumors, it is more efficient for rapid growth of tumors than slow growth of tumors. iii) Hypoxia is often observed in cancer tissues, and glycolysis offers growth advantage of cancers under this hypoxic environment (4). Glycolysis produces lactate which is released into the extracellular space. An acidic microenvironment provides a growth advantage to cancer tissues over normal tissues and enhances the invasion and metastasis of cancer cells (32,33). In addition, lactic acidosis inhibits glycolysis and favors aerobic respiration as a means of energy generation (14). iv) Due to the decrease of mitochondrial OXPHOS, less reactive oxygen species (ROS) are generated, which are cytotoxic to cancer cells (34,35).
Although cancer cells may retain OXPHOS function, it does not mean that cancer cells have no defects in mitochondrial respiration. Enhanced glycolysis in certain cancers is due to an impairment of mitochondrial function (36,37), including decreased expression of mitochondrial oxidative enzymes and transporters, truncated TCA cycle, a lowering in the amount of mitochondria per cell and defective respiratory chain, an increased amount in the natural inhibitors of the mitochondrial ATP synthase and a higher sensitivity of mtDNA to oxidative stress (17,38).
Certain cancer cells may use glutamine as energy fuel
Although it is widely accepted that glucose is the dominant energy fuel for most cancers, it is not the only one. Glutaminolysis may be an alternative pathway for energy production in certain cancers since it is known that elevation in glutamine consumption is frequently observed in cancer (39,40). In 1979, Reitzer et al reported that glutamine, not sugar, was the major energy source for cultured HeLa cells (41). Since then, several reports have shown that glutamine may be used as the energy fuel for cancer cells (42–45). In contrast to glucose, glutamine as an energy fuel is only observed in a few cancer cell lines (27) and plays an important role in compensating for the shortage of glucose in some cases.
After entering cells via membrane transport ASCT2, glutamine is hydrolyzed to glutamate and ammonia by glutaminase (GLS). Glutamate combines with cysteine and glycine to form reduced glutathione (GSH) which is found in all human cells. GSH is involved in regulating the redox state and is a major antioxidant in cells.
Glutamate may also be converted into α-ketoglutarate (α-KG) and enters the TCA cycle to supply intermediates and energy for cell growth. It is particularly useful in the truncated TCA cycle which cannot efficiently use glucose to generate energy (2). This process provides fuel for the passive TCA cycle due to a lack of isocitrate (2,7,39,46–48). Therefore, it also demonstrates that cancer cells are able to use OXPHOS for ATP production.
Glutamine not only supplies energy in certain cancer cells, but also provides precursors (such as citrate) for the synthesis of lipids. For example, Mullen et al recently showed that tumor cells with mutations in complex I or III of the electron transport chain (ETC) used glutamine-dependent reductive carboxylation as the major pathway of citrate formation, suggesting that glutamine supports tumor growth in various pathways in defective mitochondria (49).
It is worth mentioning that elevation of glutamine consumption in cancer cells is closely related to Myc activation (see below). When mouse embryonic fibroblasts are introduced by the Myc gene, the transfected cells will increase glutaminolysis. If RNA interference (RNAi) downregulates Myc expression, the tumor cells will reduce their dependency on glutamine (39). Other research also supports the effects of Myc on glutaminolysis. For example, human fibroblasts in medium without glucose will die. This process is not related to Myc activity and occurs by a mechanism other than apoptosis. However, these cells in medium without glutamine induced Myc-dependent apoptosis (43), suggesting that interfering with glutaminolysis may obtain better therapeutic effects than interfering with glucose in certain cancers. In fact, the situation is more complex in vivo. Cells also obtain energy through other pathways, such as fatty acid, lactate and ketone oxidation, as well as unidentified sources (22,23,42,50).
4. Alterations of oncogenes and tumor suppressors drive cancer cells to aerobic glycolysis
Cancer cells still use aerobic glycolysis, despite it being an inefficient way to generate ATP. Increasing evidence shows that the alterations of oncogenes and tumor suppressors in tumorigenesis play a key role in aerobic glycolysis of cancer (Fig. 1) (51–53).
Figure 1.
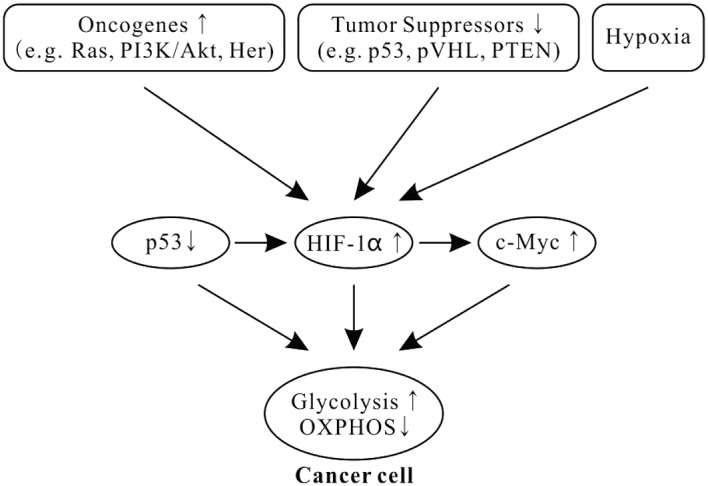
Alterations of oncogene and tumor suppressor and hypoxia drive cancer cells to aerobic glycolysis. The raised levels of HIF1 and c-Myc, and inactivation of p53 are very common in human cancers. HIF1, c-Myc and p53 form the ‘triad’ of transcription factors responsible for the glycolytic phenotype in cancer. HIF1α is induced by hypoxia or activated oncogenes (e.g. Ras, PI3K-Akt and Her) or inactivated tumor suppressors (e.g. p53, pVHL and PTEN) under normoxic conditions. In addition, HIF1 also enhances Myc expression. HIF, hypoxia-inducible factor.
Oncogene Ras mutations are often found in many types of human cancers and drive the metabolic phenotype of cancer cells toward aerobic glycolysis (54). Ras activates the mammalian target of rapamycin (mTOR) via the PI3K-Akt-mTOR signaling pathway and mTOR promotes glycolysis through inducing HIFs (34,55–58). HIFs, induced transcription factors to facilitate cellular adaptation to hypoxic environments, play critical roles in shifting from the OXPHOS to the glycolytic phenotype in cancer (Fig. 1) (34,55,59,60). HIFs are heterodimers consisting of an oxygen-labile α subunit and a stable β subunit. In mammals, HIFs have three isoforms: HIF1, HIF2 and HIF3. HIF1 is ubiquitously expressed in all cells, whereas HIF2 and HIF3 are selectively expressed in certain tissues (61). As a transcription factor, HIF1 regulates over one hundred different genes, including vascular endothelial growth factor (VEGF), hepatocyte growth factor receptor (c-Met), erythropoietin (epo), transforming growth factor-α (TGF-α), platelet-derived growth factor-β (PDGF-β) and glucose transporter GLUT1, and influences many cellular activities, including angiogenesis, glycolysis and cell survival.
In energy metabolism, HIF1 induces GLUT1 and GLUT3 expression and upregulates 9 of the 10 enzymes that function in glycolysis (52). It also inhibits the conversion of pyruvate to acetyl-CoA via the activation of pyruvate dehydrogenase kinase 1 (PDK1), leading to a decrease of mitochondrial OXPHOS.
Recent studies show that mTOR upregulation of pyruvate kinase M2 (PKM2) is critical for aerobic glycolysis and tumor growth (62). M type pyruvate kinase (PK) has two isoforms: PKM1 and PKM2. PKM1 is expressed in most adult tissue, whereas PKM2 is only expressed in embryonic and proliferating tissues. When cells are transformed, PKM1 expression is inhibited, and PKM2 expression is restored. The metabolic switch from OXPHOS to aerobic glycolysis in tumor tissues is considered to be a shift from PKM1 to PKM2 expression (63). mTOR upregulates PKM2 via HIF1 and Myc (62), being consistent with Myc upregulation of glycolysis. Myc can also upregulate the PKM2 expression via polypyrimidine tract-binding protein (PTB), heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) and hnRNPA2 (64). Notably, new data also show that PKM2 may be a strong partner for HIF1 transcription activity in hypoxia, and since PKM2 gene transcription is also activated by HIF1, this suggests that PKM2 participates in a positive feedback loop of HIF1 transcription in cancer cells (65). However, another recent study showed no evidence of a shift from PKM1 to PKM2 expression during tumorigenesis. Researchers found that PKM2 was the prominent isoform in all analyzed cancer samples and cell lines but PKM2 was also found in matched control tissues (66). Therefore, whether a shift from PKM1 to PKM2 expression occurs during tumorigenesis remains to be clarified.
In addition to mediating HIFs, mTOR directly upregulates the basic glycolytic processes, from glucose uptake to lactate formation (56,58,67). When glucose uptake goes beyond that required by cancer cells, it results in pyruvate being reduced to lactate which is excreted into the extracellular space.
The oncogene Myc, frequently overexpressed in human cancers, is a master transcription factor which regulates over 15% of human genes, including cell cycle, metabolism (glucose, glutamine, protein and lipid), ribosome biogenesis, RNA (miRNA, tRNA and rRNA), mitochondrial biogenesis, apoptosis and transformation. Myc stimulates the Warburg effect in cancers in two aspects (46,48). Firstly, Myc upregulates the expression of glucose transporter GLUT and LDH-A (1); and secondly, Myc promotes glutamine metabolism, including glutamine uptake and glutaminolysis to provide energy for use by cells (39). The upregulation of the glutamine metabolism by c-Myc is related to microRNA-23a/b (miR-23a/b), which targets GLS. c-Myc stimulates the glutamine metabolism and cell proliferation by repressing miR-23a/b (68).
Compared with Ras, which regulates glycolysis via the PI3K-Akt pathway, increase of the glutamine metabolism by Myc does not depend on the PI3K-Akt pathway. Using the PI3K-Akt pathway inhibitors suppressed the glucose metabolism, but not the glutamine metabolism, suggesting that the oncogenes Ras and Myc cooperatively maintain transforming cells via the two key nutrients glucose and glutamine, complementing each other (39). In order to engage in replicative division, a cell must duplicate its genome, proteins and lipids and assemble the components into daughter cells; in short, it becomes a factory for macromolecular biosynthesis. These activities require that cells take up extracellular nutrients such as glucose and glutamine and allocate them into metabolic pathways that convert them into biosynthetic precursors. Many intermediates from glycolysis, truncated TCA and glutaminolysis are used in cell biosynthesis during the cancer cell metabolic switch to glycolysis by Ras and Myc (40,57).
In addition, HIF1 also binds to a DNA motif in the promoter of Myc and enhances the transcription of Myc (Fig. 1). HIF1 cooperates with c-Myc to promote aerobic glycolysis by induction of hexokinase 2 (HK2), converting glucose to glucose 6-phosphate, and PDK1, a negative regulator of pyruvate dehydrogenase (PDH) (69).
Tumor suppressor p53, one of the most common gene mutations in human cancers, is a transcription factor and widely regulates diverse biological functions, including the cellular energy metabolism. It plays a pivotal role in balancing between glycolysis and OXPHOS (70,71). p53 combined with HIF1 and c-Myc has been described as the ‘triad’ of transcription factors responsible for the glycolytic phenotype in cancer (Fig. 1) (1,69,72).
In normal conditions, p53 downregulates the expression of GLUT1, GLUT4 and HK2, and upregulates the expression of TP53-induced glycolysis and apoptosis regulator (TIGAR) and synthesis of cytochrome c oxidase 2 (SCO2) (Fig. 2) and apoptosis-inducing factor (AIF) (73). Thus, the basic effects of p53 on the cellular energy metabolism are to inhibit glycolysis and promote OXPHOS (70,73). TIGAR, an enzyme that decreases the levels of fructose-2, 6-bisphosphate (Fru-2, 6-P2) by dephosphorylating, inhibits glycolytic activity (74). Fru-2, 6-P2 is an important allosteric effector (+) of PFK1, one of key regulatory enzymes of glycolysis (Fig. 2). SCO2 facilitates the assembly of cytochrome c oxidase complex in the mitochondrial ETC complex IV. AIF is essential for ETC complex I function (75). p53 deficiency leads to reduced SCO2 and AIF activity, resulting in mitochondrial OXPHOS impairment (76).
Figure 2.

p53 modulates cell energy metabolism. p53 represses the expression of glucose transporter genes (e.g., GLUT1 and GLUT4) and increases the levels of TIGAR, an inhibitor of glycolysis, in the cytoplasm. In addition, p53 increases the levels of SCO2 in the inner membrane of the mitochondria, consequently promoting mitochondrial respiration. TIGAR, TP53-induced glycolysis and apoptosis regulator; SCO2, synthesis of cytochrome c oxidase 2.
Moreover, p53 is also a potent negative regulator of HIF1α (Fig. 1). Activation of p53 blocks the accumulation of HIF1α in normoxia and hypoxia (77), and inhibits HIF1 by inducing microRNA-107 (78).
The p53-deficient cells produced significantly higher levels of lactate, indicating a shift from OXPHOS to glycolysis in energy production. For example, glycolysis contributing to the total amount of ATP is 40% in human colon cancer cell line HCT116 with wild-type (+/+) p53, rising to 66% in homozygous (−/−) p53 (71). Inactivation of p53 leads to favor aerobic glycolysis is in many aspects, including increase of glucose uptake and HIF-1α, and decrease of TIGAR, SCO2 and AIF expression. Moreover, mutant p53 also increases the activity of two other glycolytic enzymes, HK2 and phosphoglycerate mutase (PGM), catalyzing 3-phosphoglycerate (3PG) to 2-phosphoglycerate (2PG) (71,73,79).
5. Conclusion
Cancers have different metabolic phenotypes of energy for several reasons. First, cancers are heterogeneous diseases and their genetic heterogeneity determines metabolic heterogeneity (15). Even from a single cancer, its constituent cells are also heterogeneous and reflect differences in metabolic phenotype from one cell to another. The different subclones within a cancer benefit each other in the metabolism and form a metabolic symbiont (26). This phenomenon is not cancer-specific, and cancer cells also use other physiological mechanisms to support their rapid growth. Second, cancer cells continuously reprogram to adapt to environmental pressures and alteration of growth conditions. The result is that the ratio between glycolysis and OXPHOS to yield total ATP, the ratio between glucose and glutamine to yield total ATP, or the ratio between glucose/glutamine and fatty acid to yield total ATP, are continuously changing. The result of these changes is that the unfavorable environment provides a selective advantage to cancer cells.
Acknowledgments
This study was in part supported by a grant from the Ministry of Education in China (Grant No. 20110092110043). Dr Peng Gao is acknowledged for the artwork of the figures.
References
- 1.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 2.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Griguer CE, Oliva CR, Gillespie GY. Glucose metabolism heterogeneity in human and mouse malignant glioma cell lines. J Neurooncol. 2005;74:123–133. doi: 10.1007/s11060-004-6404-6. [DOI] [PubMed] [Google Scholar]
- 4.Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 5.Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134:703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 6.Lim HY, Ho QS, Low J, Choolani M, Wong KP. Respiratory competent mitochondria in human ovarian and peritoneal cancer. Mitochondrion. 2011;11:437–443. doi: 10.1016/j.mito.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 7.Scott DA, Richardson AD, Filipp FV, Knutzen CA, Chiang GG, Ronai ZA, Osterman AL, Smith JW. Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J Biol Chem. 2011;286:42626–42634. doi: 10.1074/jbc.M111.282046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zu XL, Guppy M. Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun. 2004;313:459–465. doi: 10.1016/j.bbrc.2003.11.136. [DOI] [PubMed] [Google Scholar]
- 9.Brooks GA. Cell-cell and intracellular lactate shuttles. J Physiol. 2009;587:5591–5600. doi: 10.1113/jphysiol.2009.178350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaishnavi SN, Vlassenko AG, Rundle MM, Snyder AZ, Mintun MA, Raichle ME. Regional aerobic glycolysis in the human brain. Proc Natl Acad Sci USA. 2010;107:17757–17762. doi: 10.1073/pnas.1010459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science. 2001;292:504–507. doi: 10.1126/science.1058079. [DOI] [PubMed] [Google Scholar]
- 12.Miccheli A, Tomassini A, Puccetti C, Valerio M, Peluso G, Tuccillo F, Calvani M, Manetti C, Conti F. Metabolic profiling by 13C-NMR spectroscopy: [1,2–13C2]glucose reveals a heterogeneousmetabolism in human leukemia T cells. Biochimie. 2006;88:437–448. doi: 10.1016/j.biochi.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 13.Berridge MV, Herst PM, Tan AS. Metabolic flexibility and cell hierarchy in metastatic cancer. Mitochondrion. 2010;10:584–588. doi: 10.1016/j.mito.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 14.Chen JL, Lucas JE, Schroeder T, Mori S, Wu J, Nevins J, Dewhirst M, West M, Chi JT. The genomic analysis of lactic acidosis and acidosis response in human cancers. PLoS Genet. 2008;4:e1000293. doi: 10.1371/journal.pgen.1000293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta. 2010;1805:105–117. doi: 10.1016/j.bbcan.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suganuma K, Miwa H, Imai N, Shikami M, Gotou M, Goto M, Mizuno S, Takahashi M, Yamamoto H, Hiramatsu A, et al. Energy metabolism of leukemia cells: glycolysis versus oxidative phosphorylation. Leuk Lymphoma. 2010;51:2112–2119. doi: 10.3109/10428194.2010.512966. [DOI] [PubMed] [Google Scholar]
- 17.Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007;274:1393–1418. doi: 10.1111/j.1742-4658.2007.05686.x. [DOI] [PubMed] [Google Scholar]
- 18.Jose C, Bellance N, Rossignol R. Choosing between glycolysis and oxidative phosphorylation: a tumor’s dilemma? Biochim Biophys Acta. 2011;1807:552–561. doi: 10.1016/j.bbabio.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 19.Smolková K, Plecitá-Hlavatá L, Bellance N, Benard G, Rossignol R, Ježek P. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int J Biochem Cell Biol. 2011;43:950–968. doi: 10.1016/j.biocel.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Herst PM, Berridge MV. Cell surface oxygen consumption: a major contributor to cellular oxygen consumption in glycolytic cancer cell lines. Biochim Biophys Acta. 2007;1767:170–177. doi: 10.1016/j.bbabio.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 21.Rodríguez-Enríquez S, Carreño-Fuentes L, Gallardo-Pérez JC, Saavedra E, Quezada H, Vega A, Marín-Hernández A, Olín-Sandoval V, Torres-Márquez ME, Moreno-Sánchez R. Oxidative phosphorylation is impaired by prolonged hypoxia in breast and possibly in cervix carcinoma. Int J Biochem Cell Biol. 2010;42:1744–1751. doi: 10.1016/j.biocel.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 22.Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, Frank PG, Flomenberg N, Howell A, Martinez-Outschoorn UE, et al. Ketones and lactate ‘fuel’ tumor growth and metastasis: evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010;9:3506–3514. doi: 10.4161/cc.9.17.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 24.Kiaris H, Chatzistamou I, Kalofoutis Ch, Koutselini H, Piperi Ch, Kalofoutis A. Tumour-stroma interactions in carcinogenesis: basic aspects and perspectives. Mol Cell Biochem. 2004;261:117–122. doi: 10.1023/b:mcbi.0000028746.54447.6c. [DOI] [PubMed] [Google Scholar]
- 25.Koukourakis MI, Giatromanolaki A, Harris AL, Sivridis E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: a metabolic survival role for tumor-associated stroma. Cancer Res. 2006;66:632–637. doi: 10.1158/0008-5472.CAN-05-3260. [DOI] [PubMed] [Google Scholar]
- 26.Feron O. Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol. 2009;92:329–333. doi: 10.1016/j.radonc.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 27.Sandulache VC, Ow TJ, Pickering CR, Frederick MJ, Zhou G, Fokt I, Davis-Malesevich M, Priebe W, Myers JN. Glucose, not glutamine, is the dominant energy source required for proliferation and survival of head and neck squamous carcinoma cells. Cancer. 2011;117:2926–2938. doi: 10.1002/cncr.25868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porporato PE, Dhup S, Dadhich RK, Copetti T, Sonveaux P. Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Front Pharmacol. 2011;2:49. doi: 10.3389/fphar.2011.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 30.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 31.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer. 2008;8:56–61. doi: 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- 33.Vaupel P. Metabolic microenvironment of tumor cells: a key factor in malignant progression. Exp Oncol. 2010;32:125–127. [PubMed] [Google Scholar]
- 34.Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8:705–713. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 35.Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, Unterman T, Hay N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008;14:458–470. doi: 10.1016/j.ccr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chandra D, Singh KK. Genetic insights into OXPHOS defect and its role in cancer. Biochim Biophys Acta. 2011;1807:620–625. doi: 10.1016/j.bbabio.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Owens KM, Kulawiec M, Desouki MM, Vanniarajan A, Singh KK. Impaired OXPHOS complex III in breast cancer. PLoS One. 2011;6:e23846. doi: 10.1371/journal.pone.0023846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.López-Ríos F, Sánchez-Aragó M, García-García E, Ortega AD, Berrendero JR, Pozo-Rodríguez F, López-Encuentra A, Ballestín C, Cuezva JM. Loss of the mitochondrial bioenergetic capacity underlies the glucose avidity of carcinomas. Cancer Res. 2007;67:9013–9017. doi: 10.1158/0008-5472.CAN-07-1678. [DOI] [PubMed] [Google Scholar]
- 39.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA. 2008;105:18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem. 1979;254:2669–2676. [PubMed] [Google Scholar]
- 42.Guppy M, Leedman P, Zu X, Russell V. Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem J. 2002;364:309–315. doi: 10.1042/bj3640309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol. 2007;178:93–105. doi: 10.1083/jcb.200703099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, DeBerardinis RJ. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009;69:7986–7993. doi: 10.1158/0008-5472.CAN-09-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ko YH, Lin Z, Flomenberg N, Pestell RG, Howell A, Sotgia F, Lisanti MP, Martinez-Outschoorn UE. Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells: Implications for preventing chemotherapy resistance. Cancer Biol Ther. 2011;12:1085–1097. doi: 10.4161/cbt.12.12.18671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009;15:6479–6483. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuneva M. Finding an ‘Achilles’ heel’ of cancer: the role of glucose and glutamine metabolism in the survival of transformed cells. Cell Cycle. 2008;7:2083–2089. doi: 10.4161/cc.7.14.6256. [DOI] [PubMed] [Google Scholar]
- 48.Zheng J. Features of energy metabolism and clinical application in cancer growth. Chin J Cell Biol. 2011;33:1158–1165. [Google Scholar]
- 49.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G, Elstrom RL, Thompson CB. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene. 2005;24:4165–4173. doi: 10.1038/sj.onc.1208622. [DOI] [PubMed] [Google Scholar]
- 51.Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- 52.Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010;330:1340–1344. doi: 10.1126/science.1193494. [DOI] [PubMed] [Google Scholar]
- 53.Bayley JP, Devilee P. The Warburg effect in 2012. Curr Opin Oncol. 2012;24:62–67. doi: 10.1097/CCO.0b013e32834deb9e. [DOI] [PubMed] [Google Scholar]
- 54.Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y, Ogasawara M, Trachootham D, Feng L, Pelicano H, et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012;22:399–412. doi: 10.1038/cr.2011.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yecies JL, Manning BD. mTOR links oncogenic signaling to tumor cell metabolism. J Mol Med. 2011;89:221–228. doi: 10.1007/s00109-011-0726-6. [DOI] [PubMed] [Google Scholar]
- 59.Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, Simon MC, Thompson CB. The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007;21:1037–1049. doi: 10.1101/gad.1529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weljie AM, Jirik FR. Hypoxia-induced metabolic shifts in cancer cells: moving beyond the Warburg effect. Int J Biochem Cell Biol. 2011;43:981–989. doi: 10.1016/j.biocel.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 61.Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8:967–975. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, Wang Y, Jing Y, Yang H, Chen R, et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci USA. 2011;108:4129–4134. doi: 10.1073/pnas.1014769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 64.David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364–368. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bluemlein K, Grüning NM, Feichtinger RG, Lehrach H, Kofler B, Ralser M. No evidence for a shift in pyruvate kinase PKM1 to PKM2 expression during tumorigenesis. Oncotarget. 2011;2:393–400. doi: 10.18632/oncotarget.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 68.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, Dang CV. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dang CV, Kim JW, Gao P, Yustein J. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008;8:51–56. doi: 10.1038/nrc2274. [DOI] [PubMed] [Google Scholar]
- 70.Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 71.Ma W, Sung HJ, Park JY, Matoba S, Hwang PM. A pivotal role for p53: balancing aerobic respiration and glycolysis. J Bioenerg Biomembr. 2007;39:243–246. doi: 10.1007/s10863-007-9083-0. [DOI] [PubMed] [Google Scholar]
- 72.Yeung SJ, Pan J, Lee MH. Roles of p53, MYC and HIF-1 in regulating glycolysis - the seventh hallmark of cancer. Cell Mol Life Sci. 2008;65:3981–3999. doi: 10.1007/s00018-008-8224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang PY, Zhuang J, Hwang PM. p53: exercise capacity and metabolism. Curr Opin Oncol. 2012;24:76–82. doi: 10.1097/CCO.0b013e32834de1d8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 75.Vahsen N, Candé C, Brière JJ, Bénit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, et al. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou S, Kachhap S, Singh KK. Mitochondrial impairment in p53-deficient human cancer cells. Mutagenesis. 2003;18:287–292. doi: 10.1093/mutage/18.3.287. [DOI] [PubMed] [Google Scholar]
- 77.Yang J, Ahmed A, Poon E, Perusinghe N, de Haven Brandon A, Box G, Valenti M, Eccles S, Rouschop K, Wouters B, Ashcroft M. Small-molecule activation of p53 blocks hypoxia-inducible factor 1alpha and vascular endothelial growth factor expression in vivo and leads to tumor cell apoptosis in normoxia and hypoxia. Mol Cell Biol. 2009;29:2243–2253. doi: 10.1128/MCB.00959-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yamakuchi M, Lotterman CD, Bao C, Hruban RH, Karim B, Mendell JT, Huso D, Lowenstein CJ. p53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc Natl Acad Sci USA. 2010;107:6334–6339. doi: 10.1073/pnas.0911082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, Martinez D, Carnero A, Beach D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65:177–185. [PubMed] [Google Scholar]