

Abstract
E-cadherin is a key cell-to-cell adhesion molecule associated with the invasion and metastasis of tumor cells; however, the molecular mechanisms are not entirely understood. In this study, we investigated whether downregulation of E-cadherin by E-cadherin-specific small intefering RNA (siRNA) was able to promote malignant phenotypes of prostate cancer cells through upregulating the metastasis-associated gene 1 (MTA1) in vitro. The expression levels of E-cadherin in human paired prostate adenocarcinoma cell lines, PC-3M-2B4 (2B4) and PC-3M-1E8 (1E8), were investigated using western blot analysis. The alteration of malignant phenotypes associated with decreasing E-cadherin expression were assessed in 2B4 cells using wound-healing assays, solid-phase adhesion assays, invasion assays and cytoskeletal staining. The expression of E-cadherin and MTA1 in normal, localized and metastatic prostate cancer cells was analyzed using immunohistochemistry. Downregulation of E-cadherin using an RNA interference approach led to the upregulation of MTA1 expression, decreased tumor cell adhesion ability as well as enhanced cell mobility, invasion and cellular polarity compared with the controls (P<0.05). E-cadherin regulated MTA1 in a time-dependent manner. The correlation between E-cadherin and MTA1 was inversed in the prostate cancer group (P<0.05; rs=−0.434). The data suggest that E-cadherin plays an important role in prostate cancer metastasis, which is likely to be due to the regulation of MTA1 expression. E-cadherin may combine with MTA1 and alter the malignant phenotype of prostate cancer cells. A combined testing strategy for detecting MTA1 and E-cadherin may be sufficient for selecting high-risk patients with metastasis. Therefore, E-cadherin and MTA1 may be potential powerful factors for the treatment of various types of cancer.
Keywords: E-cadherin, metastasis-associated gene 1, prostate carcinoma, metastasis
Introduction
At present, prostate cancer is recognized as one of the most important medical problems facing the male population. In the USA and Europe, prostate cancer is currently the second most common cause of cancer mortality in males (1,2). The latest statistics reveal that among males in the USA, an estimated 240,890 new cancer cases and 33,720 mortalities due to prostate cancer occurred in 2011 (1). Prostate cancer-related mortality is largely due to its high metastatic potential for bone and/or other organs (3,4). Clinically, the prevention and treatment of prostate cancer metastasis remains a significant challenge since the molecular mechanisms of prostate cancer invasion and metastasis are not well understood.
Metastasis is a complex and multi-step process in the progression of malignant cancer (5). Cell migration results in the spreading of cancer, which is the leading cause of cancer-related mortalities. In the process of cancer progression, certain cell adhesion molecules (CAMs) play a pivotal role in the development of recurrent, invasive and distant metastasis (6). A loss or reduction in the expression of CAMs, including cadherins, facilitates the detachment of single cancer cells from the tumor bulk (7,8). One of the key molecules critical for cell-to-cell adhesion is E-cadherin, a membrane glycoprotein located at cell adherent junctions (9,10). E-cadherin aids the assembly of epithelial cells and maintains the quiescence of cells within sheets by forming adherent junctions with adjacent epithelial cells (11). A number of studies have demonstrated that increased expression of E-cadherin is able to inhibit invasion and metastasis, while a reduced expression potentiates these phenotypes (11–14). In order for epithelial cells to develop into cancer cells, activation of the epithelial-mesenchymal transition (EMT) is required (15). EMT causes the epithelial cell layers to lose polarity and cell-cell contacts. It therefore triggers the remodeling of the cellular skeleton (16,17). Upregulation of E-cadherin is implicated by the activation of EMT (14,18), and E-cadherin is regarded as a main indicator of epithelial/mesenchymal phenotype switching (19).
The metastasis-associated gene 1 (MTA1) was originally identified by the differential screening of a cDNA screening library using highly metastatic mammary adenocarcinoma cell lines (20). MTA1 appears to interact with, or may even be a member of, the histone deacetylase (HDAC) complex, and acts as a co-activator of this complex (21). Studies have demonstrated that MTA1 overexpression is associated with the adhesion, invasion and metastasis of certain cancer cells (22–24), and with a higher tumor grade, the development, microvascular invasion and poor prognosis in a number of malignant cancer types (25). Through repression of the estrogen receptor α (ERα), hypoxia-inducible factor-1α (HIF-1α) and p53 protein, MTA1 converts cancer cells into a more aggressive phenotype (26). Moreover, MTA1 has been identified to determine EMT phenotypes mainly through downregulating the expression of E-cadherin, which leads to EMT (27,28). E-cadherin can be upregulated using MTA1 small interfering RNA (siRNA) in melanoma cells, which was also confirmed in our previous study in cervical cancer cells (29,30). MTA1 and E-cadherin are involved in the EMT process (28), since the loss of E-cadherin expression has been demonstrated to increase cancer metastasis progresses (13,14,31), and tumor cells with increased expression of MTA1 indicate more invasive phenotypes (32). Negative feedback regulation is crucial for cells to determine their fate and maintain function during gene regulation. Our other study (unpublished data) provided information concerning the regulation of E-cadherin expression by MTA1 when controlling malignant phenotypes in prostate cancer. Whether E-cadherin has an effect on MTA1 expression has not yet been elucidated.
In the present study, we examined whether the expression of MTA1 is an important contributing factor to the metastasis induced by E-cadherin loss in vitro. In addition, we investigated the correlation between E-cadherin and MTA1 expression and location in prostate cancer and metastatic prostate cancer tissue samples. We identified that loss of E-cadherin expression changes the malignant phenotype of prostate cancer cells through an MTA1-dependent pathway.
Materials and methods
Reagents and antibodies
All reagents were of analytical grade and commercially purchased. Primary antibodies against MTA1 were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). E-cadherin was obtained from Epitomics Inc. (Burlingame, CA, USA). α-tublin, β-actin and alkaline phosphatase-conjugated anti-rabbit/mouse/goat IgGs were purchased from Sigma (Deisenhofen, Germany). FITC/Cy3-conjugated IgG was obtained from Proteintech Group Inc. (Chicago, IL, USA). 4,6-Diamino-2-pheylindoledi (DAPI), fibronectin (FN) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma. The SP histostain-plus kit was obtained from ZhongShan Biotech Co. (Beijing, China). All other chemicals were of analytical grade and obtained from standard suppliers.
Cell lines
The human paired poorly-metastatic prostate adenocarcinoma cell lines PC-3M-2B4 (2B4) and highly-metastatic prostate adenocarcinoma cell lines PC-3M-1E8 (1E8) were kindly provided by Dr Jie Zheng (Beijing University, Beijing, China) (33). All cells were cultured in RPMI-1640 medium (Invitrogen Life Technologies, Carlsbad, CA, USA) with 10% (v/v) fetal bovine serum (FBS; Sijiqing Co., Hangzhou, China), 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C and 5% CO2 using a humidified incubator (Heraeus, Hanau, Germany).
Western blot analysis
For western blot analysis, cells were lysed in RIPA buffer containing 50 mM Tris/HCl (pH 7.2), 150 mM NaCl, 1% NP-40, 0.1% SDS and 0.5% (w/v) sodium deoxycholate. Equivalent amounts of cell extracts (150 μg) were separated on 10% SDS-polyacrylamide gel (SDS-PAGE) and transferred onto a polyvinylidene difluoride membrane (PVDF). Filters were blocked in 25 mM Tris (pH 8.0) containing 125 mM NaCl, 0.1% Tween 20 and 5% skimmed milk for 1 h, and then incubated with the diluted primary antibody (MTA1, 1:200; E-cadherin, 1:2000; β-actin, 1:1000) at 4°C overnight. After incubation, the secondary antibodies were added (1:1000 dilution). The immunoreactive bands were visualized with alkaline phosphatase and 5-bromo-4-chloro-3-indolyl-phosphate/nitro blue tetrazolium (BCIP/NBT).
Cell transfection
Cell lines were cultured in 6-well tissue culture plates and flasks at 37°C and 5% CO2 using a humidified incubator (Heraeus). Cells were transfected with 200 pmol/ml of siRNA duplex using 5 μl Lipofectamine 2000 when cells were 20% confluent following the manufacturer’s instructions of Lipofectamine™ 2000 (Invitrogen Life Technologies) siRNA treatment. The controls included untransfected cells and transfected cells with a scrambled negative control siRNA (Ruibo Biotechnology Co., Guangzhou, China). For plasmid transfection, 4×105 cells/6-well plate were prepared using 4 μg of plasmid and 10 μl Lipofectamine 2000 per well. The full length PCMV-SPORT6 E-cadherin plasmid was purchased from Yrbio (Hunan, China). It was reconstructed to pcDNA3.1 E-cadherin in our laboratory (Cancer Biology Medical Center, Tongji Hospital) and the empty vector pcDNA3.1 was preserved. Cells were harvested after 48 h and protein levels were measured using western blotting. E-cadherin siRNA sequences were as follows: forward, 5′-CAGACAAAGACCAGGACU-AdTdT-3′; reverse, 3′-dTdT GUCUGUUUCUGGUCCUGAU-5′.
Wound healing assay
Exponentially growing cells were detached from culture plates by trypsinization and seeded into 6-well plates (1×105 cells/well in complete media) with 10% FBS in DMEM. The plates were maintained at 37°C in 5% CO2 at saturated humidity in an incubator overnight. The following day, the confluent cell monolayers were converged and scrape-wounded using a micropipette tip. Floating cells were removed after three washes with serum-free medium, while scratched cells were observed under an inverted phase microscope to identify the scratch widths. Cells were cultured in serum-free culture media and images were captured under phase contrast microscopy every 12 h after wounding. For evaluation of ‘wound closure’, five randomly selected points along each wound were marked, and the horizontal distance of the migrating cells from the initial wound was measured. Each experiment was performed twice or in triplicate. Data collected were as the mean ± SD. The measurements were obtained by measuring the area of the wound using Image J software (available from the National Institute of Health website at http://imagej.nih.gov/ij/download/).
Invasion assay
Transwell chambers with polycarbonate membrane filters with 24-well inserts, 6.5 mm diameter and 8 μm pore size (Corning Life Sciences, Corning, NY, USA) were coated with 1.5 mg/ml Matrigel™ (BD Biosciences, Franklin Lakes, NJ, USA). The lower chamber was filled with 600 μl RPMI-1640 containing 20% FBS and mouse embryonic fibroblast cell line (NIH 3T3) supernatant, acting as a chemotactic factor (CF), or just DMEM with 1% fetal calf serum (FCS). A total of 1×104 cells/200 μl were seeded into the upper compartment and incubated for 48 h at 37°C and 5% CO2. Following removal of the non-migratory cells from the upper surface of the filter, invasive cells that had migrated through to the lower surface of the filter were fixed in 2.5% (v/v) glutaraldehyde for 15 min and stained with crystal violet for 10 min. The number of invasive cells were calculated by counting at least three randomly chosen visual fields at 100-fold magnification (Leica, Solms, Germany). Three independent experiments were conducted for each set.
Solid-phase adhesion assay
Adhesion was determined using an MTT assay. Exponentially growing cells were detached from culture plates by trypsinization. After washing, cells were resuspended in serum-free medium. Equal numbers of cells (4×104 cells/well) were seeded into 96-well plates precoated with 1 μg/ml FN (Sigma), which proved to be the most evident in pre-experiments (data not shown). All FN-coated wells were compared with cells seeded in 1% (w/v) bovine serum albumin (BSA). After 1 h of incubation at 37°C, the plate was immersed in PBS containing 1 mmol/l MgCl2 to remove non-adherent cells. The adhered cells were then measured using an MTT assay at 490 nm wavelength. Subsequently, 30 μl of MTT (5 mg/ml) was added 4 h prior to the end of the incubation period. Following incubation, the samples were centrifuged at 2200 × g for 5 min. Once the supernatant was removed, 150 μl of dimethyl sulphoxide (DMSO) was added to resolve the crystals and the optical density (OD) values of each well was measured at 490 nm with a microplate reader after 15 min. The OD values reflect the proportion of cells adhered to the FN-coated 96-well plate. The rate of adhesion was calculated using the following equation: (the OD value of test/the OD value of control) × 100. All experiments were conducted four times and repeated twice.
Confocal microscopy imaging
For immunofluorescence cytoskeleton staining, 1×104 cells were seeded on glass coverslips (13 mm diameter). Once the coverslips were fixated with 4% paraformaldehyde (Merck, Haar, Germany) in PBS, cell membranes were permeabilized with 0.2% (v/v) Triton X-100 in PBS, and non-specific binding sites were blocked with 5% (w/v) BSA in PBS for 30 min at 37°C. Treatment with the first antibody (α-tublin, 1:50) was conducted at 4°C overnight. Subsequently, the cells were washed three times with cold PBS and incubated with Cy3-conjugated IgG (1:50 dilution) in PBS for 30 min at room temperature. Nuclei were stained for 5 min at room temperature with DAPI (2 μg/ml methanol). Cells were then rinsed with PBS and were observed under confocal microscopy (Olympus, Tokyo, Japan).
Immunohistochemistry
Tissues samples of normal prostate as well as localized and metastatic prostate cancer were obtained from the Department of Pathology at Tongji Hospital Affiliated to Huazhong University of Science and Technology (Hubei, China). Immunohistochemical staining for E-cadherin and MTA1 expression were conducted following standard procedures. The 5 μm paraffin sections cut on poly-L-lysine-coated microscopy slides were deparaffinized and rehydrated in graded alcohol. The sections were heated in 0.01 M citrate buffer (pH 6.0) in a 85–95°C microwave oven (3 times for 5 min each) and the non-specific binding sites were blocked with goat serum. Sections were incubated overnight at 4°C with rabbit monoclonal anti-human E-cadherin (1:500 dilution) and goat polyclonal anti-human MTA1 (1:100 dilution) and then washed with PBS. Biotinylated goat anti-rabbit immunoglobulin (Dako, Kyoto, Japan) was then added to the sections for 30 min at room temperature. Peroxidase-conjugated avidin (Dako) was applied once the sections had been washed with PBS. Peroxidase activity was detected by exposure of the sections to the solution of 0.05% 3,3′-diaminobenzidine (DAB) and 0.01% H2O2 in Tris-HCl buffer (DAB solution) for 3–6 min at room temperature. Finally, the slides were counterstained with hematoxylin. Negative control slides were processed similarly, but were incubated with PBS instead of primary antibody.
Statistical analysis
Data were analyzed using the ANOVA test. Statistical analysis was conducted using SPSS version 13.0 software. P<0.05 was considered to indicate a statistically significant difference. The correlation between the indicators was analyzed using Spearman correlation analysis.
Results
Silencing E-cadherin upregulates MTA1 expression
As shown in Fig. 1A, E-cadherin was present in two prostate cancer cell lines, but at different expression levels. The protein expression levels of E-cadherin in 2B4 cells were over 2.5- to 3-fold higher compared with those in 1E8 cells. Since the 2B4 cells expressed the highest levels of endogenous E-cadherin, we selected this cell line to investigate the effects of heterologous E-cadherin expression on the cellular biological properties of prostate carcinoma cells in vitro. Western blot analysis demonstrated that E-cadherin siRNA is able to specifically downregulate E-cadherin expression and significantly increase MTA1 expression (Fig. 1B).
Figure 1.
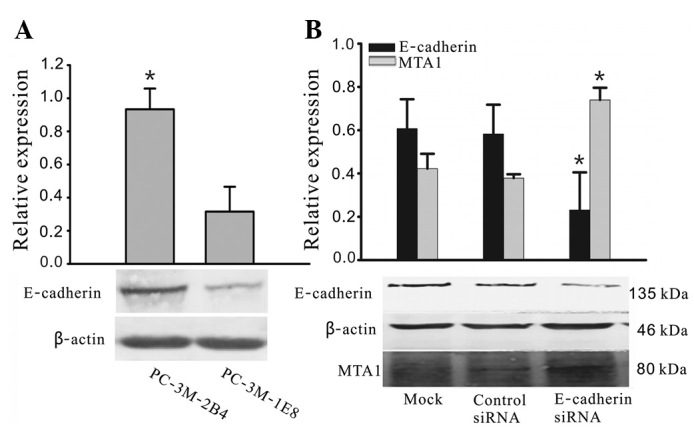
Expression of E-cadherin in prostate carcinoma cell lines. (A) Protein expression levels of E-cadherin in 2B4 cells with poor metastatic ability and 1E8 cells with high metastatic ability were analyzed by western blot analysis using specific antibodies. Expression levels were normalized against β-actin expression and quantified by bars in three independent experiments. *P<0.05 compared with 1E8. (B) Cells were treated with E-cadherin siRNA or negative control siRNA for 48 h, and expression of E-cadherin and MTA1 was evaluated by western blot analysis. Expression levels were normalized against β-actin expression and experiments were repeated three times. E-cadherin siRNA decreased E-cadherin expression and increased MTA1 expression. The quantification of the band intensities is shown. *P<0.05 compared with non-treated cells (mock) and negative control siRNA (control siRNA) transfected cells. MTA1, metastasis-associated gene 1; siRNA, small interfering RNA.
Silencing E-cadherin induces migration ability of 2B4 cells
To determine whether E-cadherin affects the migration ability of 2B4 cells, a wound healing assay was conducted. The wound healing ability of cells reflects their movement and migration on the surface on which they are anchored to for growth. At 0, 12 and 24 h after wounding, transfected E-cadherin siRNA cells demonstrated an increased level of wound healing compared with the mock and control siRNA cells (Fig. 2). At 24 h after wounding, the wound of the E-cadherin siRNA transfected group had healed, while that of the cells from the control siRNA and mock groups had not closed (P<0.01 compared with the control cells).
Figure 2.

Downregulation of E-cadherin expression increases migration of prostate carcinoma cells. The motility ability was analyzed using a wound healing assay after transfection with E-cadherin siRNA in 2B4 cells for 48 h. Wound closure of scrape-wounded cell monolayers over a 24 h culture period was assessed. Migration rate (mean ± SD; n=3) and degree of wound closure were assessed by measuring the distance between wound edges at 6 h time intervals. (A) Representative images (magnification, x10) at 0, 12 and 24 h after wounding from one of three independent experiments are shown. (B) Percentage of wound closure corresponds to the distance between wound edges in at least three randomly chosen regions (mean ± SD) normalized to 100% wound closure for E-cadherin siRNA-transfected cells. For each individual experiment, control immunoblots were conducted to ensure E-cadherin inhibition by siRNAs treatments (data not shown). *P<0.001 between cells transfected with E-cadherin siRNA and mock or control siRNA cells. siRNA, small interfering RNA.
Silencing E-cadherin alters the malignant phenotype of 2B4 cells
The 2B4 cells were transfected with siRNA against E-cadherin for 48 h and a solid-phase adhesion assay was used to detect the alteration in cell adhesion ability. For the cells with a similar concentration of surface-coated FN, silencing of E-cadherin by siRNA significantly inhibited the adhesion process compared with the mock and control siRNA treated cells (Fig. 3A). 2B4 cells were transfected with siRNA against E-cadherin for 48 h and a Matrigel-coated transwell model was used to detect the alteration in cell invasion ability. The number of cells at the lower side of the membrane reflects the invasion ability of the cells being investigated. The number of cells that invaded to the lower side of the membrane were 320.22±12.50, 293.56±18.32 and 521.13±21.67 for the mock, control siRNA- treated and E-cadherin-siRNA-treated cells, respectively. Compared with the mock and control-siRNA treated group, the invasion ability of the cells treated with E-cadherin siRNA was significantly greater (Fig. 3B and = C). Each assay was repeated three times. The decreased adhesion ability as well as an increased invasion and migration ability, influences a change in the architecture and composition of the cytoskeleton. To examine whether the inhibition of E-cadherin expression affects the subcellular distribution and organization of the cytokeratin filaments, immunofluorescence analyses were conducted using E-cadherin siRNA-transfected, control-transfected siRNA and mock cells. In the E-cadherin siRNA-transfected cells we observed an increased elongated spindle-shape morphology with extended pseudopodia between adjacent cells and an increase in cellular polarity strength compared with the control siRNA and mock cells (Fig. 3D). These results suggest that E-cadherin plays a key role in controling the malignant phenotypes of 2B4 cells, including the adhesion ability, invasion ability and cytoskeleton organization.
Figure 3.

Downregulation of E-cadherin affects cell adhesion, invasion ability and cytoskeleton construction. Cells were transfected with E-cadherin siRNA or negative control siRNA for 48 h, and the adhesive ability was examined by MTT assay. All experiments were repeated three times. (A) The adhesion ability to solid phase was significantly downregulated in cells treated with E-cadherin siRNA. *P<0.05 compared with non-treated cells (mock) and negative control siRNA transfected cells. (B) Invasion ability was assessed using the Matrigel™-coated transwell system. 8×103 cells/well were placed in a Matrigel-coated Boyden chamber and allowed to invade for 24 h. Graph reveals the quantification of invasive cells from the lower side of the transwell inserts. *P<0.05 compared with non-treated cells (mock) and negative control siRNA transfected cells. (C) Representative images of invasion experiments. (D) The altered structure (600-fold) of the cytoskeleton was detected using confocal microscopy. Red fluorescent staining represents the α-tublin and blue staining represents the nucleus. siRNA, small interfering RNA; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
E-cadherin regulates MTA1 expression
The 2B4 cells were treated with E-cadherin siRNA and full length E-cadherin plasmid for various time periods between 24 and 48 h, and western blot analyis was used to detect E-cadherin and MTA1 protein expression. As demonstrated in Fig. 4A, 48 h after treatment with E-cadherin siRNA, E-cadherin expression was significantly downregulated, while MTA1 expression was significantly upregulated. Additionally, following empty vector pCMV-SPORT6 transfection, MTA1 expression was significantly downregulated and E-cadherin was significantly upregulated 48 h after treatment (Fig. 4B). All experiments were repeated three times.
Figure 4.
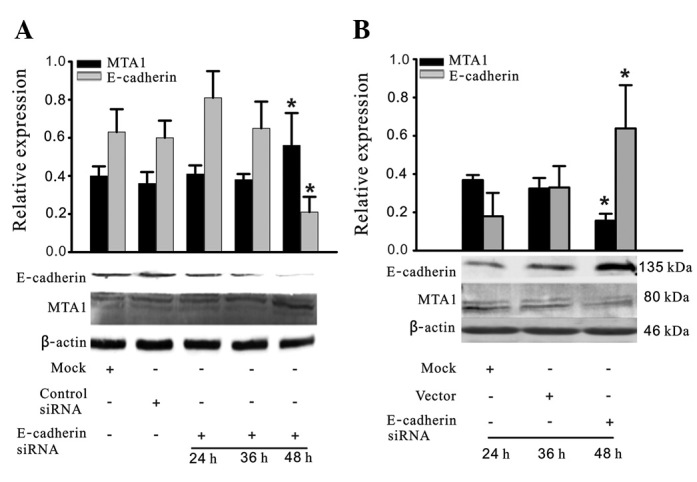
E-cadherin regulates MTA1 expression. (A) Cells were transfected with E-cadherin siRNA or negative control siRNA for 24, 36 or 48 h. Western blot analysis demonstrated that E-cadherin siRNA increased MTA1 expression at 48 h. (B) Cells were transfected with E-cadherin full length plasmid for 24, 36 or 48 h. Western blot analysis demonstrated that MTA1 expression was decreased at 48 h. The changes were quantified in the upper panels. *P<0.05 compared with negative control siRNA transfection. All experiments were repeated three times. MTA1, metastasis-associated gene 1; siRNA, small interfering RNA.
Orientation of E-cadherin and MTA1 protein in prostate tissue
E-cadherin expression was detected predominantly in the cytoplasm and membrane of the prostate cancer tissues. MTA1 protein positive signaling was detected in the cytoplasm or nucleus of cancer cells. A representative result of the immunohistochemistry for MTA1 and E-cadherin in prostate cancer is shown in Fig. 5. The samples probed with E-cadherin antibody demonstrated 2+ or 3+ membrane staining of the epithelial cells. Cytoplasmic staining was 0− or 1+. The positive staining in the prostate lesions had distinct circumferential E-cadherin immunoreactivity of high intensity, which revealed that E-cadherin is a cell membrane protein.
Figure 5.

Expression of E-cadherin and MTA1 in prostate cancer tissues. Typical results for E-cadherin and MTA1 staining determined by immunohistochemistry was observed in BPH, localized prostate cancer and metastatic prostate cancer. Magnification, x200. MTA1, metastasis-associated gene 1; BPH, benign prostatic hyperplasia.
Correlation between expression of E-cadherin and MTA1 in prostate carcinoma tissue
To further assess whether E-cadherin expression levels correlate with the expression of MTA1 in prostate cancer metastasis, we analyzed the results for E-cadherin and MTA1 immunoreactivity. Intact E-cadherin immunostaining was observed in 70% (14/20) of benign prostatic hyperplasia (BPH), 53.6% (15/28) of primary cancer and 16.7% (2/12) of metastatic lesion tissues. MTA1 expression levels in BPH, primary cancer and metastatic lesion tissues were 30 (6/20), 75 (21/28) and 25% (3/12), respectively. E-cadherin expression was decreased in the prostate cancer tissues compared with the BPH tissues (P<0.001), while MTA expression was increased in the prostate cancer tissues compared with the BPH tissues (P<0.05; Table I). Furthermore, we analyzed the correlation between the expression levels of E-cadherin and MTA1 in the tumor group. As shown in Table II, we observed a statistically significant correlation between E-cadherin and MTA1 immunoreactivity in the prostate cancer group (P<0.05; rs=−0.434, respectively).
Table I.
Expression of E-cadherin and MTA1 in prostate carcinoma tissue.
| E-cadherin
|
MTA1
|
||||||
|---|---|---|---|---|---|---|---|
| Group | No. of cases | − | + | P-value | − | + | P-value |
| BPH | 20 | 6 | 14 | 0.044 | 14 | 6 | 0.028 |
| Tumor | 40 | 23 | 17 | 16 | 24 | ||
| Primary cancer | 28 | 13 | 15 | 0.030 | 7 | 21 | 0.003 |
| Metastatic lesion | 12 | 10 | 2 | 9 | 3 | ||
MTA1, metastasis-associated gene 1; BPH, benign prostatic hyperplasia.
Table II.
Correlation between immunohistochemically detected expression of MTA1 and E-cadherin in prostate cancer.
| MTA1
|
|||||
|---|---|---|---|---|---|
| E-cadherin | − | + | Total | P-value | rs |
| − | 5 | 18 | 23 | 0.005 | −0.434 |
| + | 11 | 6 | 17 | ||
| Total | 16 | 24 | 40 | ||
MTA1, metastasis-associated gene 1.
Discussion
Cancer metastasis is a complex and multi-step process that commonly leads to the mortality of cancer patients. Cell adhesion molecules play an important role in the development of recurrent, invasive and distant metastasis in the process of cancer progression (6). E-cadherin is a key cell-to-cell adhesion molecule, which plays a significant role in the invasion and metastasis of tumor cells (10). In the majority of, if not all, cancers of epithelial origin, the loss of cell-to-cell adhesion mediated by E-cadherin may occur concomitantly with the progression towards tumor malignancy (10).
The majority of previous studies indicate that E-cadherin has a close correlation with metastasis and invasion of a number of tumors, including ovarian (13), breast (34,35), pancreatic (36), gastric (37) and prostate cancer (38,39). With regard to prostate cancer, E-cadherin expression in the cancer cells at metastasis in lymph node sites is lower than in the primary prostate cancer (38). Studies have suggested that prostate cancer cells with low expression of E-cadherin are more invasive (40); therefore, the absence of E-cadherin expression in prostate cancer predicts the potential of metastasis to the bone compared with prostate cancer that does express E-cadherin (41). A recent study suggested that upregulating E-cadherin expression by valproic acid inhibits prostate cancer cell migration (42). The effects of E-cadherin on the malignant phenotype of prostate cancer cells and possible molecular mechanisms are not entirely understood.
MTA1 is a highly expressed with high potential to metastasize in a number of cancer types (22–24). Earlier studies have demonstrated an inverse correlation between the levels of MTA1 and E-cadherin, and identified that increased MTA1 expression is associated with increased invasiveness and reduced expression of E-cadherin in tumor cells (23,43). However, studies with regard to the expression and correlation of E-cadherin and MTA1 in prostate cancer have rarely been reported. In this study, we demonstrate that the loss of E-cadherin is able to induce metastasis of prostate cancer cells through upregulated MTA1 expression.
Downregulated expression of E-cadherin in prostate cancer and upregulated expression of MTA1 is consistent with previous studies (38,41,44). Therefore, it was hypothesized that there is a certain inherent connection between E-cadherin and MTA1, and their coordination may lead to tumor cell invasion and metastasis. E-cadherin may be upregulated by MTA1 siRNA in melanoma cells, which was also confirmed in our previous study in cervical cancer cells (29,30). MTA1 overexpression resulted in the downregulation of E-cadherin expression in ovarian cancer (23).
In the present study, E-cadherin protein was markedly expressed in 2B4 (poorly-metastatic prostate adenocarcinoma cell lines) cells and weakly expressed in 1E8 (highly-metastatic prostate adenocarcinoma cell lines) cells. To clarify the molecular characteristics of E-cadherin, E-cadherin siRNA was transfected into 2B4 cells for 48 h. Our results revealed that MTA1 expression was increased when E-cadherin expression was decreased. E-cadherin downregulation is one of the principal events during EMT and a frequently reported phenomena in embryonic development and cancer progression (45,46). MTA1 has been suggested to determine EMT phenotype, mainly through the downregulated expression of E-cadherin, which leads to EMT (28). Developing cancer cells acquire migratory features concomitant with a loss of E-cadherin expression during carcinogenesis (13,47). We identified that downregulation of E-cadherin expression resulted in the promotion of 2B4 cell migration and invasion, and the inhibition of adhesion capability via upregulated MTA1 in vitro.
The increased adhesion ability and increased invasion and migration ability is accompanied by a change in the architecture and composition of the cytoskeleton. EMT is a key step toward cancer metastasis, and E-cadherin is regarded as a main indicator of the epithelial/mesenchymal phenotype switching (19). E-cadherin loss is suggestive of EMT, and tumor cell invasion and metastasis are associated with EMT (10,48). Cell changes, including morphological and gene expression alterations, are necessary for EMT (49,50). In the present study, cells acquired an elongated spindle-shape morphology with extended pseudopodia between adjacent cells due to a decrease in E-cadherin and an increase in cellular polarity strength. Our results indicate that E-cadherin may play an important role in the cellular polarity of prostate cancer cells. We identified that the protein level of MTA1 was upregulated when E-cadherin was decreased. We also identified that expression of E-cadherin regulates MTA1 expression in 2B4 cells treated with E-cadherin siRNA or full length E-cadherin plasmid at different times (24–48 h). Our data suggest that E-cadherin regulates MTA1 in a time-dependent manner.
To further investigate the expression levels of and correlation between E-cadherin and MTA1 in prostate cancer, we examined BPH, carcinoma in situ and metastatic carcinoma tissues using immunohistochemical staining. Our results demonstrated that there is an inverse correlation between protein expression of E-cadherin and MTA1 in prostate cancer. Loss of E-cadherin expression in 57.5% of prostate cancer tissues in association with the increase of MTA expression in 60% of tissues, suggests that the two proteins are closely related to prostate cancer progression. This result suggests the possibility that E-cadherin and MTA1 act together as prognosis predictors of metastasis and progression of prostate cancer. A combined testing strategy for detecting MTA1 and E-cadherin may be sufficient for selecting high-risk patients with metastasis.
We revealed that loss of E-cadherin-induced MTA1 expression in prostate cancer 2B4 cells promotes the migration and invasion of 2B4 cells. Our results provide a new insight into the mechanisms of E-cadherin regulation of MTA1 in prostate cancer, and suggests that E-cadherin may combine with MTA1 and alter the malignant phenotype of prostate cancer. A combined testing strategy for detecting MTA1 and E-cadherin may be sufficient for selecting high-risk patients with metastasis. E-cadherin and MTA1 may be potential powerful targets for the treatment of various types of cancer.
Acknowledgments
This study was supported by grants from the Major State Basic Research Development Program of China (973 Program, 2009; No. CB521808) and the National Science Foundation of China (Nos. 30700895, 30770913, 81001006, 30901587 and 81101971).
References
- 1.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 2.Boyle P, Ferlay J. Cancer incidence and mortality in Europe, 2004. Ann Oncol. 2005;16:481–488. doi: 10.1093/annonc/mdi098. [DOI] [PubMed] [Google Scholar]
- 3.Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- 4.Logothetis CJ, Lin SH. Osteoblasts in prostate cancer metastasis to bone. Nat Rev Cancer. 2005;5:21–28. doi: 10.1038/nrc1528. [DOI] [PubMed] [Google Scholar]
- 5.Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. Lancet. 2007;369:1742–1757. doi: 10.1016/S0140-6736(07)60781-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okegawa T, Pong RC, Li Y, Hsieh JT. The role of cell adhesion molecule in cancer progression and its application in cancer therapy. Acta Biochim Pol. 2004;51:445–457. [PubMed] [Google Scholar]
- 7.Bogenrieder T, Herlyn M. Axis of evil: molecular mechanisms of cancer metastasis. Oncogene. 2003;22:6524–6536. doi: 10.1038/sj.onc.1206757. [DOI] [PubMed] [Google Scholar]
- 8.Cavallaro U, Dejana E. Adhesion molecule signalling: not always a sticky business. Nat Rev Mol Cell Biol. 2011;12:189–197. doi: 10.1038/nrm3068. [DOI] [PubMed] [Google Scholar]
- 9.Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548–558. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 10.Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4:118–132. doi: 10.1038/nrc1276. [DOI] [PubMed] [Google Scholar]
- 11.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Berx G, van Roy F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb Perspect Biol. 2009;1:a003129. doi: 10.1101/cshperspect.a003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sawada K, Mitra AK, Radjabi AR, et al. Loss of E-cadherin promotes ovarian cancer metastasis via alpha 5-integrin, which is a therapeutic target. Cancer Res. 2008;68:2329–2339. doi: 10.1158/0008-5472.CAN-07-5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68:3645–3654. doi: 10.1158/0008-5472.CAN-07-2938. [DOI] [PubMed] [Google Scholar]
- 15.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28:15–33. doi: 10.1007/s10555-008-9169-0. [DOI] [PubMed] [Google Scholar]
- 17.Moreno-Bueno G, Portillo F, Cano A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene. 2008;27:6958–6969. doi: 10.1038/onc.2008.346. [DOI] [PubMed] [Google Scholar]
- 18.Gravdal K, Halvorsen OJ, Haukaas SA, Akslen LA. A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clin Cancer Res. 2007;13:7003–7011. doi: 10.1158/1078-0432.CCR-07-1263. [DOI] [PubMed] [Google Scholar]
- 19.Liu YN, Liu Y, Lee HJ, Hsu YH, Chen JH. Activated androgen receptor downregulates E-cadherin gene expression and promotes tumor metastasis. Mol Cell Biol. 2008;28:7096–7108. doi: 10.1128/MCB.00449-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toh Y, Pencil SD, Nicolson GL. A novel candidate metastasis-associated gene, mta1, differentially expressed in highly metastatic mammary adenocarcinoma cell lines. cDNA cloning, expression, and protein analyses. J Biol Chem. 1994;269:22958–22963. [PubMed] [Google Scholar]
- 21.Manavathi B, Peng S, Rayala SK, et al. Repression of Six3 by a corepressor regulates rhodopsin expression. Proc Natl Acad Sci USA. 2007;104:13128–13133. doi: 10.1073/pnas.0705878104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qian H, Lu N, Xue L, et al. Reduced MTA1 expression by RNAi inhibits in vitro invasion and migration of esophageal squamous cell carcinoma cell line. Clin Exp Metastasis. 2005;22:653–662. doi: 10.1007/s10585-006-9005-2. [DOI] [PubMed] [Google Scholar]
- 23.Dannenmann C, Shabani N, Friese K, Jeschke U, Mylonas I, Bruning A. The metastasis-associated gene MTA1 is upregulated in advanced ovarian cancer, represses ERbeta, and enhances expression of oncogenic cytokine GRO. Cancer Biol Ther. 2008;7:1460–1467. doi: 10.4161/cbt.7.9.6427. [DOI] [PubMed] [Google Scholar]
- 24.Ryu SH, Chung YH, Lee H, et al. Metastatic tumor antigen 1 is closely associated with frequent postoperative recurrence and poor survival in patients with hepatocellular carcinoma. Hepatology. 2008;47:929–936. doi: 10.1002/hep.22124. [DOI] [PubMed] [Google Scholar]
- 25.Nicolson GL, Nawa A, Toh Y, Taniguchi S, Nishimori K, Moustafa A. Tumor metastasis-associated human MTA1 gene and its MTA1 protein product: role in epithelial cancer cell invasion, proliferation and nuclear regulation. Clin Exp Metastasis. 2003;20:19–24. doi: 10.1023/a:1022534217769. [DOI] [PubMed] [Google Scholar]
- 26.Toh Y, Nicolson GL. The role of the MTA family and their encoded proteins in human cancers: molecular functions and clinical implications. Clin Exp Metastasis. 2009;26:215–227. doi: 10.1007/s10585-008-9233-8. [DOI] [PubMed] [Google Scholar]
- 27.Pakala SB, Singh K, Reddy SD, et al. TGF-β1 signaling targets metastasis-associated protein 1, a new effector in epithelial cells. Oncogene. 30:2230–2241. doi: 10.1038/onc.2010.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Radaelli E, Damonte P, Cardiff RD. Epithelial-mesenchymal transition in mouse mammary tumorigenesis. Future Oncol. 2009;5:1113–1127. doi: 10.2217/fon.09.93. [DOI] [PubMed] [Google Scholar]
- 29.Qian H, Yu J, Li Y, et al. RNA interference of metastasis-associated gene 1 inhibits metastasis of B16F10 melanoma cells in a C57BL/6 mouse model. Biol Cell. 2007;99:573–581. doi: 10.1042/bc20060130. [DOI] [PubMed] [Google Scholar]
- 30.Rao Y, Wang H, Fan L, Chen G. Silencing MTA1 by RNAi reverses adhesion, migration and invasiveness of cervical cancer cells (SiHa) via altered expression of p53, and E-cadherin/β-catenin complex. J Huazhong Univ Sci Technolog Med Sci. 2011;31:1–9. doi: 10.1007/s11596-011-0141-9. [DOI] [PubMed] [Google Scholar]
- 31.Hunt NC, Douglas-Jones AG, Jasani B, Morgan JM, Pignatelli M. Loss of E-cadherin expression associated with lymph node metastases in small breast carcinomas. Virchows Arch. 1997;430:285–289. doi: 10.1007/BF01092751. [DOI] [PubMed] [Google Scholar]
- 32.Manavathi B, Kumar R. Metastasis tumor antigens, an emerging family of multifaceted master coregulators. J Biol Chem. 2007;282:1529–1533. doi: 10.1074/jbc.R600029200. [DOI] [PubMed] [Google Scholar]
- 33.Liu Y, Zheng J, Fang W, et al. Isolation and characterization of human prostate cancer cell subclones with different metastatic potential. Zhonghua Bing Li Xue Za Zhi. 1999;28:361–364. (In Chinese) [PubMed] [Google Scholar]
- 34.Chao YL, Shepard CR, Wells A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol Cancer. 2010;9:179. doi: 10.1186/1476-4598-9-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kowalski PJ, Rubin MA, Kleer CG. E-cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res. 2003;5:R217–R222. doi: 10.1186/bcr651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.von Burstin J, Eser S, Paul MC, et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroent erology. 2009;137:361–371. doi: 10.1053/j.gastro.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 37.Tang B, Peng ZH, Yu PW, Yu G, Qian F. Expression and significance of Cx43 and E-cadherin in gastric cancer and metastatic lymph nodes. Med Oncol. 2011;28:502–508. doi: 10.1007/s12032-010-9492-5. [DOI] [PubMed] [Google Scholar]
- 38.Cheng L, Nagabhushan M, Pretlow TP, Amini SB, Pretlow TG. Expression of E-cadherin in primary and metastatic prostate cancer. Am J Pathol. 1996;148:1375–1380. [PMC free article] [PubMed] [Google Scholar]
- 39.Rubin MA, Mucci NR, Figurski J, Fecko A, Pienta KJ, Day ML. E-cadherin expression in prostate cancer: a broad survey using high-density tissue microarray technology. Hum Pathol. 2001;32:690–697. doi: 10.1053/hupa.2001.25902. [DOI] [PubMed] [Google Scholar]
- 40.Mao Q, Zheng X, Yang K, et al. Suppression of migration and invasion of PC3 prostate cancer cell line via activating E-cadherin expression by small activating RNA. Cancer Invest. 2010;28:1013–1018. doi: 10.3109/07357900802620844. [DOI] [PubMed] [Google Scholar]
- 41.Pontes J, Jr, Srougi M, Borra PM, Dall’ Oglio MF, Ribeiro-Filho LA, Leite KR. E-cadherin and beta-catenin loss of expression related to bone metastasis in prostate cancer. Appl Immunohistochem Mol Morphol. 2010;18:179–184. doi: 10.1097/PAI.0b013e3181640bca. [DOI] [PubMed] [Google Scholar]
- 42.Zhang L, Wang G, Wang L, Song C, Wang X, Kang J. Valproic acid inhibits prostate cancer cell migration by up-regulating E-cadherin expression. Pharmazie. 2011;66:614–618. [PubMed] [Google Scholar]
- 43.Zhang H, Stephens LC, Kumar R. Metastasis tumor antigen family proteins during breast cancer progression and metastasis in a reliable mouse model for human breast cancer. Clin Cancer Res. 2006;12:1479–1486. doi: 10.1158/1078-0432.CCR-05-1519. [DOI] [PubMed] [Google Scholar]
- 44.Hofer MD, Kuefer R, Varambally S, et al. The role of metastasis-associated protein 1 in prostate cancer progression. Cancer Res. 2004;64:825–829. doi: 10.1158/0008-5472.can-03-2755. [DOI] [PubMed] [Google Scholar]
- 45.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 46.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 47.Richmond PJ, Karayiannakis AJ, Nagafuchi A, Kaisary AV, Pignatelli M. Aberrant E-cadherin and alpha-catenin expression in prostate cancer: correlation with patient survival. Cancer Res. 1997;57:3189–3193. [PubMed] [Google Scholar]
- 48.Di Croce L, Pelicci PG. Tumour-associated hypermethylation: silencing E-cadherin expression enhances invasion and metastasis. Eur J Cancer. 2003;39:413–414. doi: 10.1016/s0959-8049(02)00815-8. [DOI] [PubMed] [Google Scholar]
- 49.Maeda M, Johnson KR, Wheelock MJ. Cadherin switching: essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J Cell Sci. 2005;118:873–887. doi: 10.1242/jcs.01634. [DOI] [PubMed] [Google Scholar]
- 50.Christofori G. New signals from the invasive front. Nature. 2006;441:444–450. doi: 10.1038/nature04872. [DOI] [PubMed] [Google Scholar]