

Abstract
Transposases (Tnps) are enzymes that participate in the movement of insertion sequences (ISs) within and between genomes. Genes that encode Tnps are amongst the most abundant and widely distributed genes in nature. However, they are difficult to predict bioinformatically and given the increasing availability of prokaryotic genomes and metagenomes, it is incumbent to develop rapid, high quality automatic annotation of ISs. This need prompted us to develop a web service, termed TnpPred for Tnp discovery. It provides better sensitivity and specificity for Tnp predictions than given by currently available programs as determined by ROC analysis. TnpPred should be useful for improving genome annotation. The TnpPred web service is freely available for noncommercial use.
1. Introduction
Insertion sequences (ISs) are small, mobile DNA elements that usually contain a gene encoding a transposase that catalyzes the movement of the ISs from one part of the genome to another. ISs are found in nearly all prokaryotes [1, 2], sometimes at very high frequency per genome and are among the most abundant genes in nature [3]. They play a major role in lateral gene transfer, genome organization, and genome evolution [4]. Many ISs are bounded by short terminal inverted repeats (IRs) and some generate short direct repeats (DRs) when they integrate into the genome. ISs are classified into 19 families based on amino acid sequence similarity of the transposases, DNA sequence similarity including respective IRs and DRs and, in some cases, supported by phylogenetic profiling [5, 6].
Given the increasing availability of prokaryotic genomes and metagenomes, it is incumbent to develop rapid, high quality automatic annotation of ISs. Unfortunately, currently transposases of many ISs are incorrectly annotated as having other functions or are identified as “hypothetical.” In addition, their annotation is exacerbated by the presence of numerous partial ISs scattered in most genomes, representing the remains of once active ISs.
Recently, the web application ISsaga was released, providing high quality ISs annotation [7], based on information available from curated ISs families present in the ISfinder database [5]. One advantage of the ISsaga pipeline is that it combines IS (DNA) and transposase (protein) sequence searches for the prediction of complete and partial ISs. The DNA and protein sequence searches are based on a suite of BLAST programs (BLASTN, BLASTX, and BLASTP) [8, 9]. IScan is another application that makes use of BLAST to scan whole genomes for ISs and includes in its prediction pipeline searches for transposases and inverted and direct repeats [10]. However, it is widely acknowledged that sequence-sequence comparison as carried out by this BLAST suite is inferior to profile-sequence comparisons such as Profile Hidden Markov Models (HMMs) when searching for remote homologies [11]. Recognizing this advantage, HMMs have been generated for transposases belonging to 19 of the 23 characterized families of ISs (PFAM database [12] and ACLAME database [13]). An additional bioinformatic resource for IS prediction is the Superfamily database [14] of structural and functional annotation of genomes based on a library of HMM profiles derived from structural domains in SCOP database [15]. Currently, Superfamily hosts 6 HMM profiles from domains belonging to two prokaryotic families of transposases, mu bacteriophage transposase, and IS200. A third HMM profile in Superfamily recognizes the eukaryotic Hermes transposase.
Since existing bioinformatic resources for predicting transposases via HMMs are limited to less than 60% of the IS families, we have developed a web service, termed TnpPred, that provides HMM profiles for transposases of the remaining ~40% of the IS families. In addition, newly available sequence information and manual curation allowed us to generate new HMM profiles for the ~60% of IS families for which HMM profiles already exist, that, with the exception of two cases, are as sensitive or in some cases more sensitive than those currently available in the PFAM database.
2. Materials and Methods
Transposase sequences were obtained from the ISFinder website [5]. The sequences were then manually curated using Blastp against RefSeq database [16], and several HMM profiles were developed for each IS family using multiple sequence alignments generated by ClustalW [17] and HMMer software [18], version 2.3.2. TnpPred was programmed in HTML [19] and Cascading Style Sheets, CSS [20], complying with the World Wide Web Consortium, W3C (http://www.w3.org/), guidelines. Compliance with these guidelines facilitates the accessibility of Mobilomics from any browser in any available operative system. Evaluation of the sensitivity and specificity of the HMM models was done by ROC analysis [21].
3. Results and Discussion
3.1. Validation of TnpPred HMM Profiles
In order to evaluate the sensitivity and selectivity of the TnpPred HMM profiles for predicting transposases, the HMMs were subjected to ROC curve analysis [21]. This analysis compared the performance of TnpPred HMMs derived from our study with those derived from Pfam and assessed their ability to identify transposases in a database of known, curated transposases (known positives database) versus a database of sequences devoid of known transposases (known negatives database). The known positives transposase database was constructed by amalgamating the database of ISfinder [5] with transposases from RefSeq. The known negatives transposase database was made with all sequences from Swiss-Prot Database [22, 23] after the elimination of all entries tagged as transposases, insertion sequences, resolvases, recombinases, and integrases. The ROC curves for 19 IS families are provided in supplementary file 1, see Supplementary Material available online at doi:10.1155/2012/678761 and are available for downloading at http://www.mobilomics.cl/.
TnpPRED HMMs have equal or superior selectivity, sensitivity, and cutoff e-value scores compared to those derived from Pfam HMM profiles for the prediction of Tnps belonging to 17 families of ISs (Table 1, marked with an asterick). In the two remaining cases, namely, Transposase mut of IS Family IS256 and Transposase 7 of IS Family Tn3, Pfam HMM profile outperforms TnpPRED HMMs in at least one performance criterion. In the case of IS family IS256, Pfam's Transposase mut outperforms TnpPRED HMM Profile in its selectivity and sensitivity. In the case of Tn3, Pfam's Profile Transposase 7 outperforms TnpPRED in selectivity but not sensitivity (Table 1). For these reasons, the Pfam HMM profile for predicting IS256 transposase members has been incorporated into the profiles used for the TnpPRED prediction web service, whereas both the Pfam HMM profiles and TnpPRED HMM profiles are used to predict Tn3 IS family members (Table 1, marked with asterisk).
Table 1.
A comparison of the selectivities, sensitivities, and cutoff e-values derived from TnpPred versus the corresponding Pfam HMM profiles for 19 IS families.
| IS family | Pfam | TnpPred | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HMM | Selectivity | Sensitivity | Cutoff1 | HMM | Selectivity | Sensitivity | Cutoff1 | ||
| Transposase_27 | 96.4% | 83.2% | 1.2E + 04 | Combined* | 100.0% | 95.4% | 2.0E − 06 | ||
| 1 | IS1 | — | — | — | — | ORF1* | 99.9% | 100.0% | 32E − 34 |
| — | — | — | — | ORF2* | 99.4% | 100.0% | 3.9E − 40 | ||
|
| |||||||||
| 2 | IS110 | Transposase_9 | 95.3% | 94.2% | 7.0E + 03 | ORF1* | 100.0% | 100.0% | 1.3E − 63 |
| Transposase_20 | 100.0% | 99.1% | 1.2E − 06 | ||||||
|
| |||||||||
| 3 | IS1380 | — | — | — | — | ORF1* | 100.0% | 100.0% | 2.3E − 224 |
|
| |||||||||
| 4 | IS200/IS605 | Transposase_17 | 100.0% | 100.0% | 3.9E − 21 | ORF1* | 100.0% | 100.0% | 4.2E − 79 |
|
| |||||||||
| — | — | — | — | Combined* | 93.9% | 96.0% | l.8E − 09 | ||
| 5 | IS21 | — | — | — | — | ORF1* | 100.0% | 93.7% | 84E − 10 |
| IstB_N | 72.8% | 79.3% | 6.7E + 04 | ORF2* | 100.0% | 100.0% | 2.0E − 06 | ||
| IstB | 76.6% | 79.5% | 2.6E + 04 | 100.0% | 100.0% | 2.0E − 06 | |||
|
| |||||||||
| 6 | IS256 | Transposase_mut* | 100.0% | 100.0% | 8.7E − 01 | ORF1 | 99.4% | 98.8% | 3.1E − 55 |
|
| |||||||||
| — | — | — | — | Combined* | 99.5% | 81.8% | 2.7E + 02 | ||
| IS3_IS150 | — | — | — | — | ORF1* | 90.3% | 69.7% | 5.4E + 03 | |
| — | — | — | — | ORF2 | 100.0% | 100.0% | 2.4E − 114 | ||
| Transposase_8 | 80.1% | 78.8% | 2.5E + 04 | Combined* | 100.0% | 100.0% | 5.7E − 06 | ||
| IS3_IS2 | — | — | — | — | ORF1* | 100.0% | 90.0% | 1.2E − 83 | |
| — | — | — | — | ORF2* | 100.0% | 100.0% | 1.3E − 223 | ||
| — | — | — | — | Combined* | 98.6% | 89.6% | 3.3E + 02 | ||
| 7 | IS3_IS3 | — | — | — | — | ORF1* | 93.6% | 76.5% | 2.5E − 11 |
| — | — | — | — | ORF2* | 100.0% | 100.0% | 7.9E − 140 | ||
| — | — | — | — | Combined* | 99.9% | 100.0% | 3.9E − 04 | ||
| IS3_IS407 | — | — | — | — | ORF1* | 99.7% | 95.8% | 3.0E − 44 | |
| — | — | — | — | ORF2* | 100.0% | 100.0% | 3.5E − 135 | ||
| — | — | — | — | Combined* | 100.0% | 91.4% | 5.4E − 98 | ||
| IS3_IS51 | — | — | — | — | ORF1* | 87.4% | 74.3% | 5.5E − 24 | |
| — | — | — | — | ORF2 | 100.0% | 100.0% | 8.1E − 205 | ||
|
| |||||||||
| 8 | IS30 | — | — | — | — | ORF1* | 100.0% | 100.0% | 1.7E − 127 |
|
| |||||||||
| 9 | IS4 | Transposase_11 | 99.0% | 96.0% | 9.5E + 02 | ORF1* | 100.0% | 96.1% | 1.3E − 01 |
| Transposase_Tn5 | 51.8% | 58.9% | 1.1E + 05 | ||||||
|
| |||||||||
| 10 | IS481 | Mu-transpos_C | 67.7% | 54.0% | 6.7E + 04 | ORF1* | 99.9% | 100.0% | 4.0E − 84 |
|
| |||||||||
| IS5_IS1031 | — | — | — | — | ORF1* | 100.0% | 100.0% | 1.2E − 162 | |
| IS5_IS427 | — | — | — | — | Combined* | 99.7% | 97.7% | 6.7E − 11 | |
| 11 | IS5_IS5 | Transposase_33 | 54.6% | 60.4% | 1.1E + 05 | ORF1* | 100.0% | 100.0% | 4.2E − 48 |
| IS5_IS903 | — | — | — | — | ORF1* | 100.0% | 100.0% | 7.3E − 155 | |
| IS5_ISH1 | — | — | — | — | ORF1* | 100.0% | 100.0% | 1.5E − 216 | |
| IS5_ISL2 | — | — | — | ORF1* | 100.0% | 100.0% | 2.6E − 129 | ||
|
| |||||||||
| 12 | IS6 | — | — | — | — | ORF1* | 100.0% | 100.0% | 4.1E − 65 |
|
| |||||||||
| 13 | IS630 | Transposase_14 | 52.8% | 68.2% | 9.8E + 04 | ORF1* | 98.4% | 97.7% | 2.7E − 93 |
|
| |||||||||
| Transposase_34 | 89.9% | 73.1% | 2.4E − 35 | Combined | 85.6% | 79.0% | 1.5E + 04 | ||
| 14 | IS66 | — | — | — | — | ORF1* | 97.8% | 82.6% | 1.4E − 32 |
| — | — | — | — | ORF2* | 94.0% | 88.4% | 3.4E + 02 | ||
| — | — | — | — | ORF3* | 100.0% | 88.8% | 1.7E − 299 | ||
|
| |||||||||
| 15 | IS91 | Transposase_32 | 100.0% | 100.0% | 7.9E − 16 | ORF1* | 100.0% | 100.0% | 4.1E − 216 |
|
| |||||||||
| 16 | IS982 | — | — | — | — | ORF1* | 99.3% | 99.2% | 3.5E − 102 |
|
| |||||||||
| 17 | ISAs1 | — | — | — | — | ORF1* | 100.0% | 100.0% | 4.1E − 205 |
|
| |||||||||
| 18 | ISL3 | Transposase_12 | 100.0% | 99.0% | 4.7E − 31 | ORF1* | 100.0% | 99.0% | 5.0E − 95 |
|
| |||||||||
| 19 | Tn3 | Transposase_7* | 100.0% | 63.3% | 1.7E − 156 | ORF1* | 94.8% | 68.3% | 0.0E + 00 |
1Cutoff e-values are derived from ROC charts for each model (Supplementary File 1); *indicates the HMM that was selected for incorporation into the TnpPred web service.
3.2. Comparison of TnpPred with ISsaga
To assess the predictive power of ISsaga, it was used to predict Tnps in the genomes of Acaryochloris marina MBIC11017 and Stenotrophomonas maltophilia K279a [7]. We have also annotated these two genomes in order to compare the Tnps predictions of TnpPred with those of ISsaga and to evaluate the types of additional IS predictions offered by TnpPred. In the genome of Acaryochloris marina MBIC11017, ISsaga predicts a total of 272 Tnps for 17 IS families or subfamilies, whereas TnpPred HMM profiles predict a total of 266 Tnps for the same 17 IS families (Table 2). Summing the predictions for both ISsaga and TnpPred gives a total of 293 unique Tnps. In the case of S. maltophilia K279a, ISsaga predicts a total of 39 Tnps from 9 IS families, whereas TnpPred predicts a total of 47 Tnps for 10 IS families (Table 2) summing to 53 unique Tnps.
Table 2.
Summary of Tnp predictions by TnpPred compared to ISsaga.
| Organism |
Acaryochloris marina
MBIC11017 |
Stenotrophomonas maltophilia
K279a |
|---|---|---|
| Kingdom | Bacteria | Bacteria |
| Class | Acaryochloris | Gammaproteobacteria |
| Date | May 27, 10 | July 9, 10 |
| Accession number | NC_009925 | NC_010943 |
| % G + C | 47.3% | 66.3% |
| Length (Mbp) | 6.5 | 4.9 |
| Confirmed total | 244 | 46 |
| Class A* | 214 | 42 |
| Class B* | 30 | 4 |
| Class C* | 22 | 1 |
| Not found by TnpPred | 27 | 6 |
| Number of IS families TnpPred | 17 | 10 |
| Total: not found + TnpPred | 293 | 53 |
| Number of IS families ISsaga | 17 | 9 |
| Total TnpPred | 266 | 47 |
| Total ISsaga | 272 | 39 |
∗See Figure 1 for the definition of classes.
Inspection of the Tnps predicted by TnpPred but not by ISsaga revealed three broad classes of novel predictions as outlined in Figure 1. In class (a), TnpPred provides a family prediction for a gene previously annotated only as “transposase”; in class (b), TnpPred adds information to a gene previously annotated as “hypothetical” or with “no known function” and in class (c), TnpPred predicts a Tnp in a DNA sequence where no gene had previously been annotated.
Figure 1.
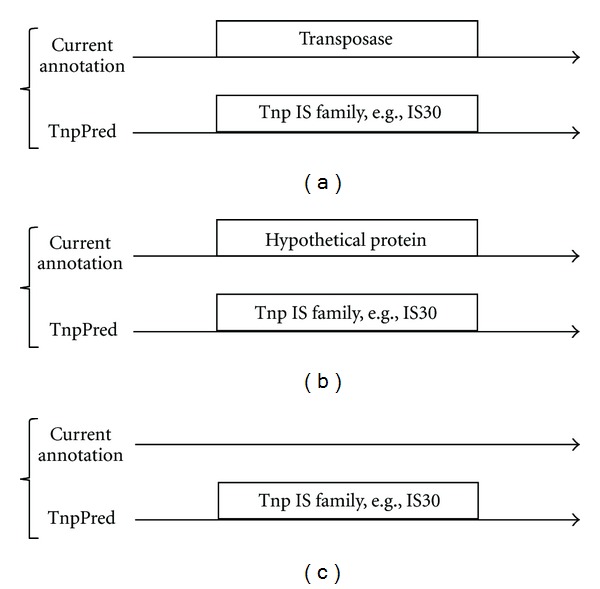
Classes of improvement of gene annotation using TnpPred. (a) Additional information such as “family classifiaction” is provided for a previously annotated transposase, (b) prediction of a transposase where a previously hypothetical gene had been annotated, (c) prediction of a transposase where no prior annotation existed.
In A. marina MBIC11017, an example of a class (a) annotation improvement is YP_001515477.1, annotated in ISsaga as “transposase” and in TnpPred as “transposase Family IS630”; a class (b) annotation improvement is YP_001516695.1, annotated as “hypothetical protein” in ISsaga and as “IS5 sub-family ISL2” in TnpPred, and a class (c) annotation improvement is a sequence not annotated in ISsaga (coordinates 5666475..5666933 +strand) and as “IS200/IS605” in TnpPred (supplementary files 2 and 4). Similar examples exist for S. maltophilia K279a (supplementary files 3 and 5).
However, TnpPred failed to detect 27 out of 293 predicted Tnps in A. marina MBIC11017 and 6 Tnps out of 53 in S. maltophilia K279a. There are several possible reasons for this: (i) some sequences in ISsaga fall below the accepted e value cutoff for TnpPred, (ii) some sequences are incorrectly annotated in ISsaga because it uses the NCBI nr data base via BLAST to predict Tnps and some of these Tnps are incorrectly annotated in NCBI, and (iii) ISsaga has predictions for four new families of Tnps [5] that were not available when TnpPred was developed. These new families will be incorporated into a future update of TnpPred.
3.3. Additional Discussion
TnpPred is able to detect fragments or pseudogenes of Tnps if the relevant sequence has an e-value lower than the accepted evalue cutoff score specified by the HMM. It is often useful to be able to detect such “molecular fossils” because they can aid in the prediction of genes and gene islands, including pathogenicity islands, that may have been horizontally transferred [24, 25].
4. Conclusions
TnpPred is a web service that supplements and extends currently available programs and HMM Profiles for the prediction of 19 prokaryotic transposase families. In a comparison of the sensitivity and selectivity by ROC analysis of the HMMs used by TnpPred versus those used by Pfam HMMs, the TnpPred predictions of the 19 families outperformed Pfam in all but two cases. The ability of TnpPred to predict Tnps in whole genomes was compared to the currently available ISsaga annotations for A. marina MBIC11017 and S. maltophilia K279a. TnpPred successfully predicted 266 Tnps out of 293 for A. marina and 47 Tnps out of 53 for S. maltophilia. In addition, TnPred predicted additional loci for Tnps in both genomes that were not recognized by ISsaga and improved the prediction of several Tnps by the assignment of a Family designation to Tnps that were only identified by the general term “Tnps” in ISsaga. Therefore, it is proposed that TnPred could be a useful aid to predict Tnps in microbial genomes.
5. Website and FTP
The TnpPred web service of Tnp IS family HMM prediction for aminoacid sequences and the HMM Profiles for 19 Tnp IS families can be accessed at http://www.mobilomics.cl/.
Supplementary Material
The Supplementary Materials include the following files: Excel file showing each IS amily HMM profile ROC curve, sensitivity and selectivity. Acaryochloris marina MBIC11017 uid58167 TnpPred annotation in genbank format (.gbk). Stenotrophomonas maltophilia K279a TnpPred annotation in genbank format (.gbk). Acaryochloris marina MBIC11017 uid58167 full report of TnpPred predictions. NC_009925.gbk Chromosome. Stenotrophomonas maltophilia K279a full report of TnpPred predictions. NC_010943.gbk Chromosome.
Acknowledgment
This work was supported by Fondecyt (1090451).
References
- 1.Touchon M, Rocha EPC. Causes of insertion sequences abundance in prokaryotic genomes. Molecular Biology and Evolution. 2007;24(4):969–981. doi: 10.1093/molbev/msm014. [DOI] [PubMed] [Google Scholar]
- 2.Siguier P, Filée J, Chandler M. Insertion sequences in prokaryotic genomes. Current Opinion in Microbiology. 2006;9(5):526–531. doi: 10.1016/j.mib.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 3.Aziz RK, Breitbart M, Edwards RA. Transposases are the most abundant, most ubiquitous genes in nature. Nucleic Acids Research. 2010;38(13):4207–4217. doi: 10.1093/nar/gkq140.gkq140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De la Cruz F, Davies J. Horizontal gene transfer and the origin of species: lessons from bacteria. Trends in Microbiology. 2000;8(3):128–133. doi: 10.1016/s0966-842x(00)01703-0. [DOI] [PubMed] [Google Scholar]
- 5.Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Research. 2006;34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahillon J, Chandler M. Insertion Sequences Revisited. In Mobile DNA II. Washington, DC, USA: ASM Press; 2002. [Google Scholar]
- 7.Varani AM, Siguier P, Gourbeyre E, Charneau V, Chandler M. ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biology. 2011;12(3, article R30) doi: 10.1186/gb-2011-12-3-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altschul SF, Wootton JC, Gertz EM, et al. Protein database searches using compositionally adjusted substitution matrices. FEBS Journal. 2005;272(20):5101–5109. doi: 10.1111/j.1742-4658.2005.04945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner A, Lewis C, Bichsel M. A survey of bacterial insertion sequences using IScan. Nucleic Acids Research. 2007;35(16):5284–5293. doi: 10.1093/nar/gkm597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Madera M, Gough J. A comparison of profile hidden Markov model procedures for remote homology detection. Nucleic Acids Research. 2002;30(19):4321–4328. doi: 10.1093/nar/gkf544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finn RD, Mistry J, Tate J. The Pfam protein families database. Nucleic Acids Research. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leplae R, Lima-Mendez G, Toussaint A. ACLAME: a CLAssification of mobile genetic elements, update 2010. Nucleic Acids Research. 2009;38(1):D57–D61. doi: 10.1093/nar/gkp938.gkp938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson D, Madera M, Vogel C, Chothia C, Gough J. The SUPERFAMILY database in 2007: families and functions. Nucleic Acids Research. 2007;35(1):D308–D313. doi: 10.1093/nar/gkl910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andreeva A, Howorth D, Chandonia JM, et al. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Research. 2008;36(1):D419–D425. doi: 10.1093/nar/gkm993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pruitt KD, Tatusova T, Klimke W, Maglott DR. NCBI reference sequences: current status, policy and new initiatives. Nucleic Acids Research. 2009;37(1):D32–D36. doi: 10.1093/nar/gkn721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chenna R, Sugawara H, Koike T, et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Research. 2003;31(13):3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Durbin R, Eddy SR, Krogh A, Mitchison GJ. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge University Press; 1998. [Google Scholar]
- 19. Upgrading to TLS Within HTTP/1.1, http://tools.ietf.org/html/rfc2817.
- 20. The text/css Media Type, http://tools.ietf.org/html/rfc2318.
- 21.Fawcett T. An introduction to ROC analysis. Pattern Recognition Letters. 2006;27(8):861–874. [Google Scholar]
- 22.Boeckmann B, Bairoch A, Apweiler R, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Research. 2003;31(1):365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Apweiler R, Martin MJ, O'Donovan C, et al. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Research. 2011;39(supplement 1):D214–D219. doi: 10.1093/nar/gkq1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hooper SD, Mavromatis K, Kyrpides NC. Microbial co-habitation and lateral gene transfer: what transposases can tell us. Genome Biology. 2009;10(4, article R45) doi: 10.1186/gb-2009-10-4-r45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmitz-Esser S, Penz T, Spang A, Horn M. A bacterial genome in transition—an exceptional enrichment of IS elements but lack of evidence for recent transposition in the symbiont Amoebophilus asiaticus. BMC Evolutionary Biology. 2011;11(1, article 270) doi: 10.1186/1471-2148-11-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Supplementary Materials include the following files: Excel file showing each IS amily HMM profile ROC curve, sensitivity and selectivity. Acaryochloris marina MBIC11017 uid58167 TnpPred annotation in genbank format (.gbk). Stenotrophomonas maltophilia K279a TnpPred annotation in genbank format (.gbk). Acaryochloris marina MBIC11017 uid58167 full report of TnpPred predictions. NC_009925.gbk Chromosome. Stenotrophomonas maltophilia K279a full report of TnpPred predictions. NC_010943.gbk Chromosome.