

Summary
Mother–daughter centriole disengagement, the necessary first step in centriole duplication, involves Plk1 activity in early mitosis and separase activity after APC/C activity mediates securin degradation. Plk1 activity is thought to be essential and sufficient for centriole disengagement with separase activity playing a supporting but non-essential role. In separase null cells, however, centriole disengagement is substantially delayed. The ability of APC/C activity alone to mediate centriole disengagement has not been directly tested. We investigate the interrelationship between Plk1 and APC/C activities in disengaging centrioles in S or G2 HeLa and RPE1 cells, cell types that do not reduplicate centrioles when arrested in S phase. Knockdown of the interphase APC/C inhibitor Emi1 leads to centriole disengagement and reduplication of the mother centrioles, though this is slow. Strong inhibition of Plk1 activity, if any, during S does not block centriole disengagement and mother centriole reduplication in Emi1 depleted cells. Centriole disengagement depends on APC/C–Cdh1 activity, not APC/C–Cdc20 activity. Also, Plk1 and APC/C–Cdh1 activities can independently promote centriole disengagement in G2 arrested cells. Thus, Plk1 and APC/C–Cdh1 activities are independent but slow pathways for centriole disengagement. By having two slow mechanisms for disengagement working together, the cell ensures that centrioles will not prematurely separate in late G2 or early mitosis, thereby risking multipolar spindle assembly, but rather disengage in a timely fashion only late in mitosis.
Key words: APC/C, Centriole, Disengagement, Plk1
Introduction
The centrosome is the primary microtubule organizing center (MTOC) of the interphase cell and in mitosis the centrosomes act in a dominant fashion to control spindle polarity. Since centriole pairs collect the pericentriolar material that forms the MTOC, the duplication of the centrosome as a whole is determined by the duplication and separation of centriole pairs (Sluder and Rieder, 1985).
Centriole duplication starts with the functional separation, or disengagement, of mother from daughter centrioles. This was first reported as the disorientation of the orthogonal arrangement of mother–daughter centrioles during late G1 (Kuriyama and Borisy, 1981). Later live cell studies of HeLa cells revealed that unduplicated mother and daughter centrioles inherited from mitosis are often spatially separate and show independent movements early in G1 (Piel et al., 2000). In a pathfinding study Tsou and Stearns reported that the disengagement of mother–daughter centrioles occurs in anaphase, and is dependent upon the proteolytic activity of separase which also cleaves the centromeric cohesin complexes that hold sister chromatids together in metaphase (Tsou and Stearns, 2006). Reports of cohesins at centrosomes (Wong et al., 2006) and functional evidence that their cleavage leads to centriole disengagement (Schöckel et al., 2011) reinforce the notion of a common mechanism for centriole and chromosome disengagement. Separase becomes active when its inhibitor securin is targeted for degradation by anaphase-promoting complex/cyclosome (APC/C) activity at the metaphase–anaphase transition. Centriole disengagement is a necessary prerequisite for later centriole duplication when the daughter cells enter S phase (Tsou and Stearns, 2006). This model received strong support from the finding that laser ablation of daughter centrioles in S phase cells allows mother centrioles to assemble new daughter centrioles (Loncarek et al., 2008). Thus, S phase is constitutively permissive for centriole duplication and a later study showed that prolonged G2 also is permissive for repeated centriole reduplication (Lončarek et al., 2010). Thus, the centrosome intrinsic block to centrosome duplication that prevents centriole re-duplication during S and G2 (Wong and Stearns, 2003) is based in the engagement of daughter centrioles to their mothers thereby preventing them from duplicating again.
Separase is not the only player in centriole disengagement; Plk1 activity also promotes disengagement. Centriole duplication occurs in separase null HCT116 cells and is dependent upon Plk1 activity during late G2 or early mitosis. However, without separase activity centriole disengagement is delayed into late G1 or S phases (Tsou et al., 2009). Without Plk1 activity, separase null cells driven out of mitosis did not show centriole disengagement or duplication by the time they entered S phase.
Even though separase activity is not absolutely required for centriole disengagement, a number of observations have led to the belief that Plk1 activity is necessary for centriole disengagement and duplication (Wang et al., 2011; reviewed by Mardin and Schiebel, 2012). For example, siRNA depletion of Plk1 prevented centriole reduplication in S phase arrested U2OS cells (Liu and Erikson, 2002). Additionally, the disengagement of isolated mammalian centrioles in Xenopus egg extracts depends upon ongoing Plk1 mediated phosphorylation of centriolar cohesin subunits allowing them to be cleaved by separase (Schöckel et al., 2011). Centriole disengagement and reduplication during G2 arrest is also dependent upon Plk1 activity (Lončarek et al., 2010).
Whether or not APC/C activity alone, without Plk1 activity, can mediate centriole disengagement in live cells is uncertain and has not been directly tested. The possibility that APC/C activity alone can disengage centrioles is suggested by the report that knockdown of Evi5, which stabilizes the APC/C inhibitor Emi1 in interphase, leads to an incidence of extra centrosomes/spindle poles in mitotic human cells (Eldridge et al., 2006). However, the basis for this was not clear and was interpreted to possibly result from spindle abnormalities and consequent mitotic defects. On the other hand, after siRNA depletion of Emi1 in cycling HeLa cells, only 10% showed more than two centrosomes as seen by gamma tubulin foci (Lončarek et al., 2010). This was interpreted to indicate that APC/C activity alone is not sufficient to disengage centrioles.
We have further investigated the interrelationship between APC/C and Plk1 activities in the control of centriole disengagement in live cells. In particular, we were interested in testing whether if Plk1 activity and APC/C activity represent two pathways that can independently cause centriole disjoining or alternatively if Plk1 activity is required with APC/C activity playing a supporting but not essential role, as currently thought. To avoid investigating centriole disengagement against the complicated regulatory landscape of cells going through mitosis, we used S phase arrested HeLa and RPE1 cells, which normally do not disjoin or reduplicate centrioles during prolonged S phase. This phase of the cell cycle is constitutively permissive for procentriole assembly (Loncarek et al., 2008).
Results
We used HeLa and RPE1 cells stably expressing low levels of GFP-centrin 1 to tag the centrioles. Centriole duplication is normal in these cells (Piel et al., 2000; LaTerra et al., 2005). When arrested in S phase with thymidine or aphidicolin, our HeLa cells exhibit a less than 2.5% incidence of extra centrioles after 72 hours.
Emi1 depletion leads to centriole disengagement and reduplication in S phase
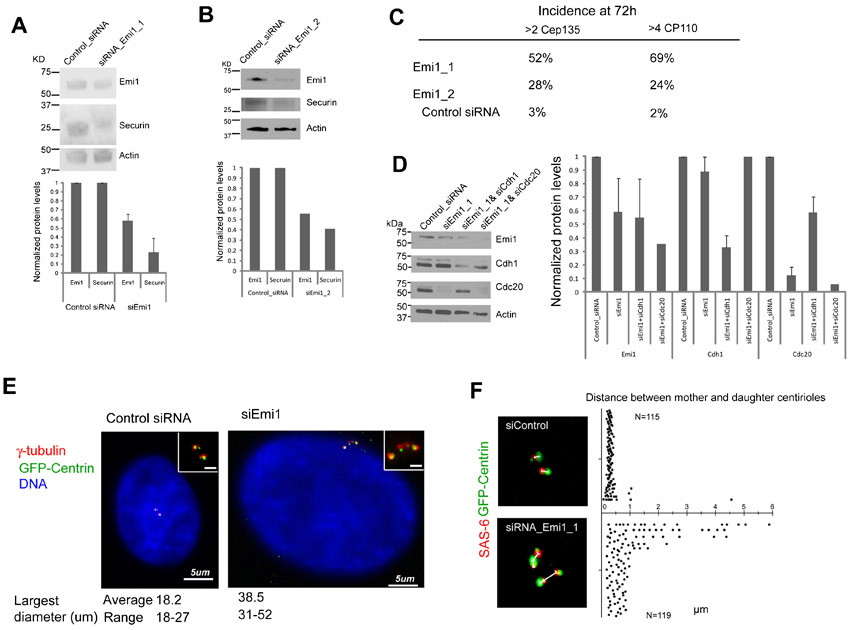
We first determined if APC/C activity can disengage centrioles during S phase. Asynchronous cultures were treated with thymidine to arrest them in S phase, and 16 hours later the interphase APC/C inhibitor Emi1 was knocked down using siRNA constructs previously shown to be effective (Di Fiore and Pines, 2007). In 3 experiments, transfection with Emi1_1 siRNA resulted in a mean 58% reduction of Emi1 protein levels and a mean 77% reduction in securin protein levels when assayed 48 hours after transfection in whole populations of transfected plus untransfected cells (supplementary material Fig. S1A). Functional efficacy of our Emi1 knockdowns was confirmed by evidence of DNA re-replication in asynchronous cultures as previously reported (Di Fiore and Pines, 2007; Machida and Dutta, 2007; Lončarek et al., 2010). This was seen at 72 hours after transfection by increases in nuclear size (supplementary material Fig. S1E) and >60 clearly separate CREST positive nuclear spots in the enlarged nuclei (not shown).
To assay for centriole disengagement/reduplication we fixed cultures 48 and 72 hours after transfection and immunostained for a number of recognized centriolar proteins and modifications to centriolar microtubules. Since daughter centriole assembly requires disengagement, the spatial separation of centrin foci associated with a number of centriolar proteins and the assembly of supernumerary centrioles are evidence of mother daughter centriole disengagement in S phase. We determined the incidence of cells containing elevated numbers of centriolar protein immunoreactive spots associated with bright GFP-centrin foci relative to S phase arrested cultures transfected with control siRNA constructs. As shown in Fig. 1A–D we found a marked increase in the incidence of cells with elevated numbers of Cep135 (cartwheel protein found in centrioles), poly-glutamylated tubulin (a characteristic modification of centriolar microtubules), and pericentriolar gamma tubulin foci in Emi1 knockdown cells relative to control cells. Furthermore, we found a similar increase in the incidence of cells with elevated numbers of CP110 (cap protein on both mother and daughter centrioles) foci and a lower, but still elevated, incidence of cells with elevated numbers of acetylated tubulin foci (modification of centriolar microtubules) and SAS-6 (cartwheel protein found in daughter centrioles) foci after Emi1 knockdown. The incidence of supernumerary acetylated tubulin foci may be limited by the time-dependent nature of tubulin acetylation and the incidence of cells with elevated numbers of SAS-6 foci may be limited by this protein being targeted, perhaps slowly, for degradation by APC/C activity (Strnad et al., 2007). In addition we immunostained Emi1 knockdown cells for C-Nap1 and found an increase in the incidence of cells with supernumerary immunoreactive spots colocalized with centrin foci relative to cells transfected with control siRNA. More than two C-Nap1 spots in S phase cells is indicative of centriole disengagement. Lastly, we immunostained Emi1 knockdown cells for Cep170, a marker for the oldest mature mother centriole (Guarguaglini et al., 2005). Almost all cells showed one Cep170 patch associated with a pair of centrin foci containing one larger and one smaller centrin spots in both control and Emi1 knockdown cells (Fig. 1D). The ranges in number of centrin spots associated with the various centriolar proteins for control and Emi1 knockdown cells are shown in Table 1.
Fig. 1. Emi1 knockdown in S phase leads to centriole disengagement and mother centriole reduplication.

(A) Diagram of the experimental protocol. (B) Incidence of cells with >2 Cep135 or >2 poly-glutamylated tubulin spots associated with centrin foci at 48 hours (blue bars) and 72 hours (brown bars). Representative images of the Cep135, poly-glutamylated tubulin, and GFP-centrin signals are shown for each condition. (C) Incidence of cells with >4 CP110, >2 SAS-6 or >2 acetylated tubulin spots associated with centrin foci at 48 hours (blue bars) and 72 hours (brown bars). Representative images are shown below the histograms. (D) Incidence of cells with, >2 gamma tubulin, >2 C-Nap1 spots, or >1 Cep170 patches associated with centrin foci at 48 hours (blue bars). (E) Emi1 knockdown without prior S phase arrest leads to centriole disengagement and mother centriole reduplication. Mitotic cells were collected by shake off were transfected Emi1_1 or control siRNA constructs 1.5 hours later according to the protocol of Lončarek et al. (Lončarek et al., 2010). The experimental protocol is outlined at the top of this portion of the figure. Histograms show the incidence of cells with >2 spots of the indicated proteins associated with centrin foci. Representative images of the indicated protein signals are shown below for each condition. Images are maximum intensity point projections from Z series images. Scale bars = 1 µm. All histogram bars indicate the average from 3 experiments with >200 cells counted for each condition. Error bars are one standard deviation.
Table 1. Range for the number of immunoreactive spots per HeLa cell for the indicated centriolar proteins associated with centrin foci under the experimental conditions listed on the left.

Similar results were obtained when Emi1 was knocked down with an independent siRNA construct (Emi1_2) (Di Fiore and Pines, 2007) (supplementary material Fig. S1B) though the incidence of elevated numbers of centrioles was less than that observed in cells transfected with the Emi1_1 construct perhaps due to a lower reduction in securin levels (supplementary material Fig. S1C).
The colocalization of a number of centriole specific proteins or tubulin modifications with the extra centrin spots and the spatial separation of the centrin/centriolar protein spots (supplementary material Fig. S1F) indicates that knockdown of Emi1 and consequent activation of the APC/C during interphase leads to disengagement of mother from daughter centrioles and the repeated assembly of daughter centrioles that then disengage from their mothers. That extra centrioles appeared to be daughters is consistent with the report that, with the exception of certain cell types such as CHO and U2OS, daughter centrioles when separate from their mothers do not mature and duplicate again when cells are in S phase (Lončarek et al., 2010).
Lončarek et al. reported that for cycling HeLa cells arrested in S phase by Emi1 knockdown (Di Fiore and Pines, 2007; Machida and Dutta, 2007), 90% of the cells contained only two centrosomes (with two centrioles apiece) as detected by gamma tubulin staining (Lončarek et al., 2010). The remaining 10% showed elevated centrosome numbers. This was interpreted to show that Emi1 knockdown does not lead to centriole disengagement or reduplication. To explore the basis for the difference between our results and theirs, mitotic cells were collected by shake off and transfected Emi1_1 or control siRNA constructs 1.5 hours later according to the protocol of Lončarek et al. (Lončarek et al., 2010). Cultures were fixed 72 hours after transfection and immunostained for SAS-6, Cep135, or gamma tubulin. We counted centrioles in cells with markedly enlarged nuclei (indicative of arrest in S phase). We found that 25% of the cells showed more than 2 gamma tubulin foci associated with centrin spots, 38% contained >2 Cep135 spots, and 22% had >2 SAS-6 spots (Fig. 1E). These results reveal that our findings with S phase arrested cells were not an artifact of the means used to arrest the cells in S or their being in S before Emi1 was knocked down. We propose that the difference in results of the two groups is quantitative rather than qualitative.
To test if our results were specific to HeLa cells, we knocked down Emi1 in S phase arrested RPE1 cells – immortalized untransformed human cells. We observed a marked increase in the incidence of elevated centriole number relative to cells transfected with the control siRNA construct though the incidence of extra centrioles in the population was lower than that found in HeLa cells (supplementary material Fig. S2A–E). The reason why RPE1 cells show a lower incidence of centriole disengagement/reduplication is not fully known but may reflect the less efficient knockdown of Emi1 in this cell type (Di Fiore and Pines, 2007).
APC/C–Cdh1, not APC/C–cdc20, activity disengages centrioles
To determine whether APC/C–Cdh1 and/or Cdc20 were responsible for centriole disengagement, we co-depleted Emi1 and either Cdh1 or Cdc20 in S phase cells (Fig. 2A–C). 48 hours after transfection cell populations were fixed and immunostained for Cep135, Cp110, or SAS-6. Co-depletion of Emi1 plus Cdh1 greatly diminished the incidence of centriole disengagement and mother centriole reduplication relative to knockdown of Emi1 alone (Fig. 2A–C). Co-depletion of Emi1 plus Cdc20 allowed disengagement and mother centriole reduplication at levels only slightly less than knockdown of Emi1 alone. These observations reveal that primarily APC/C–Cdh1 rather than APC/C–Cdc20 activity promotes centriole disengagement during S phase arrest.
Fig. 2. After Emi1 knockdown in S phase, APC/C–Cdh1, not APC/C–cdc20, activity mediates centriole disengagement.
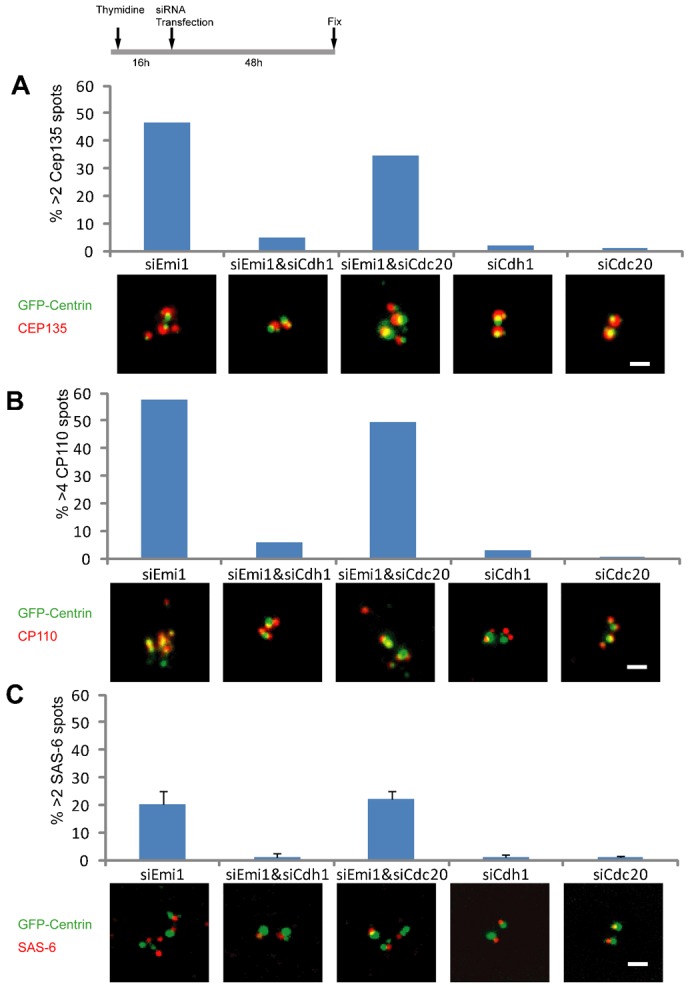
The experimental conditions are indicated in the protocol diagram at the top of the figure. (A) Incidence of cells with >2 Cep135 spots associated with centrin foci after transfection with the indicated constructs. Representative images of the Cep135 and GFP-centrin signals are shown for each experimental condition. (B) Incidence of cells with >4 CP110 spots associated with centrin foci after transfection with the indicated constructs. (C) Incidence of cells with >2 SAS-6 spots associated with centrin foci after transfection with the indicated constructs. Representative images for each condition are shown. Images are maximum intensity point projections from Z series images. Scale bars = 1 µm. In panels A and B histogram bars indicate the average from 2 experiments with >200 cells counted for each condition. In panel C histogram bars indicate the average from 3 experiments. Error bars are one standard deviation.
Plk1 activity is not required for APC/C–Cdh1 mediated centriole disengagement
Plk1 activity during S phase is low, but perhaps not zero (Golsteyn et al., 1995). Thus, we tested if strong inhibition of Plk1 activity affected APC/C–Cdh1 mediated centriole disengagement during S. We knocked down Emi1 in S phase cells and simultaneously treated with 200 nM BI2536 which should block Plk1 activity (IC50: 0.83 nM) and largely block the activities of Plk2 and Plk3 (IC50s: 3.5 nM and 9.0 nM respectively) (Lénárt et al., 2007). Functional efficacy of this drug at this concentration is supported by observations that 100 nM BI2536 completely blocked centriole reduplication in G2 arrested cells (Lončarek et al., 2010) and 200 nM blocked centriole disengagement and duplication in separase null cells driven out of mitosis (Tsou et al., 2009).
Forty-eight hours after transfection and Plk inhibitor application the incidence of cells with supernumerary centrin spots associated with Cep135, CP110, gamma tubulin, and SAS-6 was equivalent to their incidence with Emi1 knockdown alone (Fig. 3A–D). The ranges in number of centrin spots associated with the various centriolar proteins for Emi1 knockdown cells with and without BI2536 application are shown in Table 1. Similar results were obtained for RPE1 cells (supplementary material Fig. S2E) though the incidence of cells with extra centrioles was lower than that observed for HeLa cells. These results indicate that centriole disengagement and mother centriole reduplication are not dependent upon Plk1 activity when Emi1 is knocked down in S phase. Interestingly, inhibition of Plk1 activity did not diminish the incidence of cells with >2 gamma tubulin clouds associated with centrin spots relative to that found when Emi1 alone was knocked down (Fig. 3C). This indicates that acquisition of gamma tubulin around daughter centrioles is not entirely dependent upon Plk1 activity, contrary to what was expected based on a previous report (Wang et al., 2011).
Fig. 3. Plk1 activity is not required for APC/C–Cdh1 mediated centriole disengagement during S.
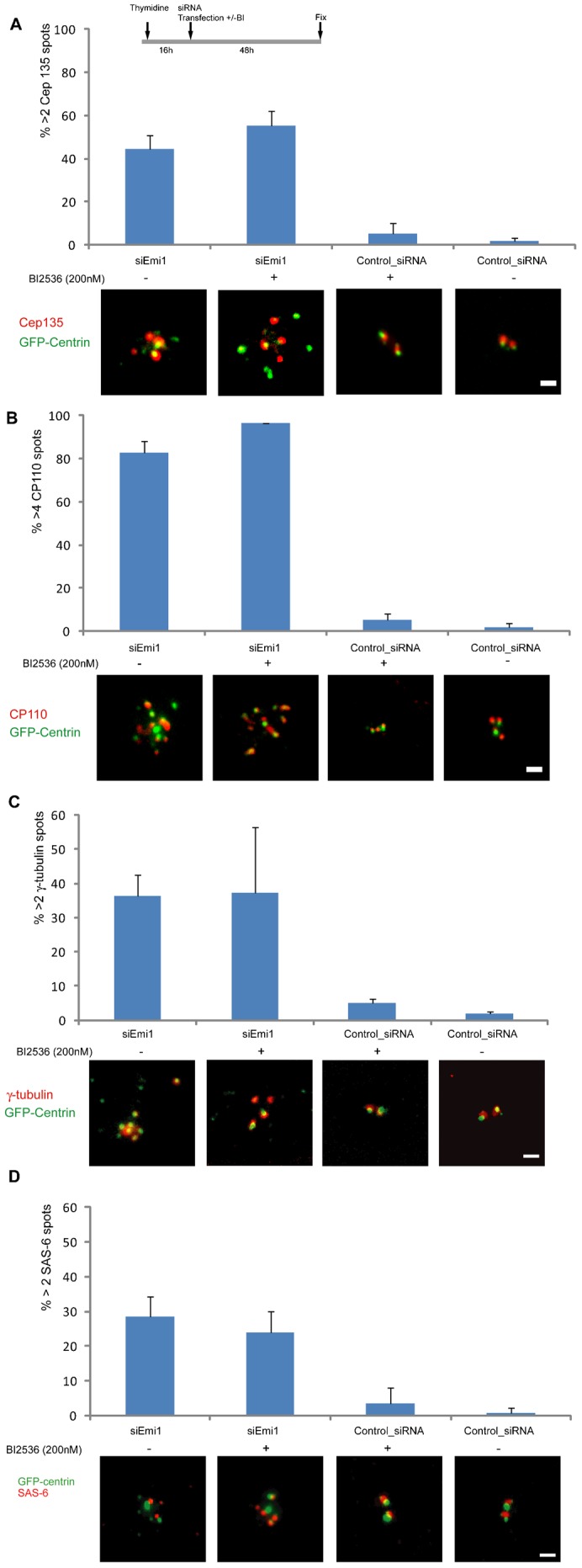
The experimental conditions are indicated in the protocol diagram at the top of the figure. (A) Incidence of cells with >2 Cep135 spots associated with centrin foci after transfection with and without Plk inhibitor. Representative images of the Cep135 and the GFP-centrin signals are shown for each condition. (B) Incidence of cells with >4 CP110 spots associated with centrin foci under the same range of conditions. Representative images are shown below. (C) Incidence of cells with >2 gamma tubulin spots associated with centrin foci under the conditions indicated along with representative images. (D) Incidence of cells with >2 SAS-6 spots associated with centrin foci under the conditions indicated. Representative images are maximum intensity point projections from Z series of images. Scale bars = 1 µm. In all panels histogram bars indicate the average from 3 experiments with >200 cells counted for each condition. Error bars are one standard deviation.
APC/C–Cdh1 and Plk1 activities can independently disengage centrioles during G2 arrest
Since significant Plk1 activity is not expected in S phase and normally rises in G2, we used HeLa cells arrested in G2 with the Cdk1 inhibitor RO-3306. By knocking down Cdh1 or inhibiting Plk1 activity during G2 we wanted to test whether Plk1 and APC/C activities can independently promote centriole disengagement in this phase of the cell cycle. Although centriole disengagement/reduplication are reported to occur in a Plk1 activity dependent fashion in G2 (Lončarek et al., 2010), we re-examined this issue because that Plk1 phosphorylates Emi1 to target it for SCF-Trcp1 mediated destruction (Guardavaccaro et al., 2003; Margottin-Goguet et al., 2003; Hansen et al., 2004; Moshe et al., 2004). Thus, inhibition of Plk1 activity starting before cells reach G2 (Lončarek et al., 2010) should also block Emi1 destruction thereby inhibiting APC/C activity; the resulting lack of centriole disengagement in the absence of Plk1 and APC/C activity would thus be expected (Tsou et al., 2009).
We shook off mitotic cells and 3 hours later continuously treated them with 5 µM RO-3306 to arrest the population in G2. Some cultures were transfected with siRNA for Cdh1 an hour and a half after shake off and others were treated with 200 nM BI2536 six hours after shake off (Fig. 4A, protocol diagram). 49.5 hours after shake off, we fixed the cultures and immunostained for SAS-6 and Cep135. For cultures treated with RO-3306 alone we observed a 55.4% incidence of cells with >2 SAS-6 spots associated with centrin foci (Fig. 4A, first bar) and a similar incidence of cells with >2 Cep135 spots (Fig. 4B, first bar). Inhibition of Plk1 activity with 200 nM BI2536 almost eliminated the incidence of >2 SAS-6 and >2 Cep135 spots (Fig. 4A,B, second bars). These observations are in accord with those of Lončarek et al. (Lončarek et al., 2010).
Fig. 4. APC/C–Cdh1 and Plk1 activities can independently disengage centrioles during G2.
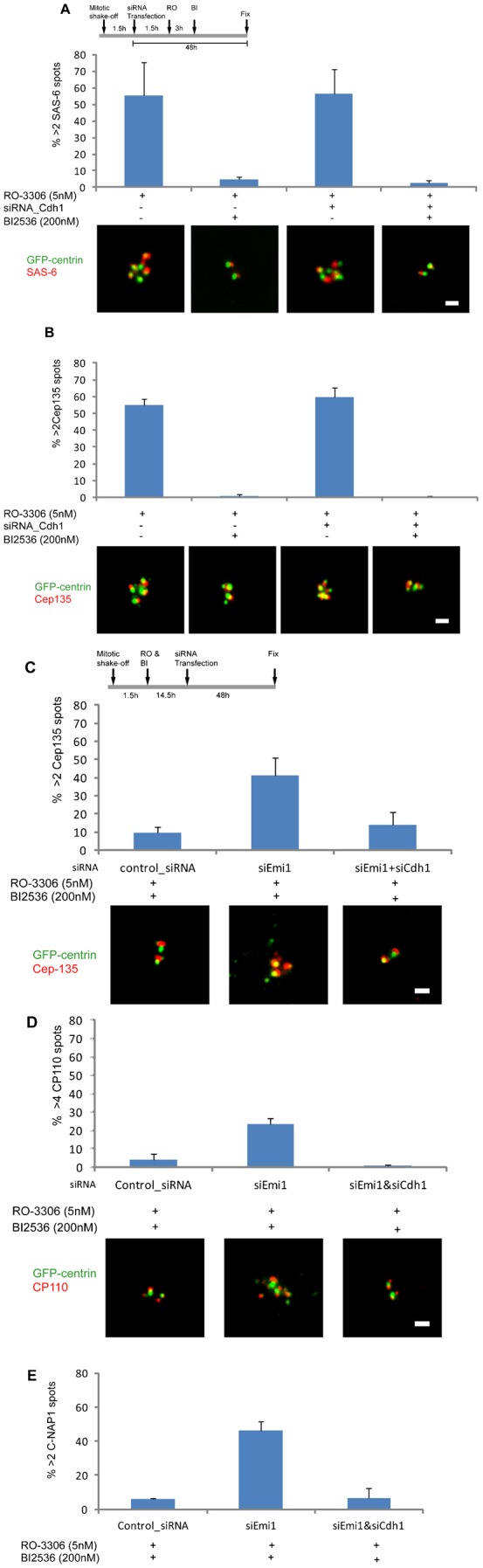
(A,B) Incidence of cells with >2 SAS-6 or >2 Cep135 spots associated with centrin foci under the conditions indicated in the protocol diagram at the top of panel A. Representative images of the SAS-6, Cep135 and the GFP-centrin signals are shown for each condition. (C–E) Incidence of cells with >2 Cep135, >4 CP110, or >2 C-Nap1 spots associated with centrin foci under the conditions indicated in the protocol diagram at the top of panel C. Representative images are shown for each condition. Images are maximum intensity point projections from Z series of images. Scale bars = 1 µm. Histogram bars indicate the average from 3 experiments with >200 cells counted for each condition. Error bars are one standard deviation.
Cultures transfected with siRNA to Cdh1 showed a 54% incidence of cells with >2 SAS-6 spots associated with centrin foci (Fig. 4A, third bar) and a similar incidence of cells with >2 Cep135 spots (Fig. 4B, third bar). These observations indicate that Plk1 activity in the absence of APC/C–Cdh1 activity is sufficient to disengage centrioles during G2. Knockdown of Cdh1 plus inhibition of Plk1 activity almost completely blocked centriole disengagement/reduplication (Fig. 4A,B, fourth bars).
To test if APC/C activity alone can disengage centrioles during G2, we shook of mitotic cells and 1.5 hours later treated them with RO-3306 plus 200 nM BI2536 to arrest the daughter cells in G2 and inhibit Plk1 activity. 16 hours after shake off we transfected to knock down Emi1 and 48 hours later fixed the cultures. These cultures exhibited a 42% incidence of cells with supernumerary Cep135 spots associated with centrin foci, a 27% incidence of extra CP110 spots, and a 46% incidence of extra C-Nap1 spots (Fig. 4C–E). When we co-knocked down Emi1 and Cdh1, the incidences of extra Cep135, CP110, and C-Nap1 spots were greatly reduced (Fig. 4C–E). These observations indicate that APC/C–Cdh1 activity alone can promote centriole disengagement during G2.
Discussion
Centriole disengagement must be tightly controlled to prevent it from occurring during G2 or early mitosis, something that can lead to spindle multipolarity and genomic instability through chromosome missegregation. Perhaps more pernicious, transient spindle multipolarity can lead to one or more merotelically attached chromosomes that lag in anaphase/telophase (Ganem et al., 2009). These laggards may become micronuclei which do not show complete DNA replication in the following cell cycle. When the daughter cell with a micronucleus enters mitosis, chromosome condensation leads to chromosome damage (Crasta et al., 2012).
We investigated the interrelationship between APC/C and Plk1 activities in centriole disengagement, the necessary first step in centriole duplication that normally occurs during late mitosis. Previous work had shown that Plk1 activity alone can disengage centrioles during G2 and mitosis (Tsou et al., 2009; Lončarek et al., 2010). However, the ability of APC/C activity alone to disengage centrioles had not been tested. We found that APC/C–Cdh1, not APC/C–Cdc20, activity alone is sufficient to disengage centrioles during S phase and G2. In S this was seen by the assembly of multiple new centrioles as well as their spatial separation from each other and from the original mother centrioles. The assembly of multiple daughter centrioles indicates that there were repeated cycles of disengagement. Our observations that many of the centrin foci were associated with SAS-6 and there was only one Cep170 patch associated with a brighter centrin focus in each cell indicates that the new centrioles are daughters (Guarguaglini et al., 2005; Strnad et al., 2007). This is consistent with findings that laser ablation of the daughter centriole (a definitive form of disengagement) allows the mother to assemble new daughter centrioles in S phase arrested cells and that daughter centrioles are not capable of becoming mothers during S phase (Lončarek et al., 2010).
Plk1 activity by itself is also sufficient to disengage centrioles without APC/C activity, as first shown in separase null HCT116 cells driven out of mitosis (Tsou et al., 2009) and later supported by the finding that centriole reduplication during G2 arrest is Plk1 dependent (Lončarek et al., 2010). We confirmed the ability of Plk1 activity alone to disengage G2 centrioles using our experimental system. Knockdown of Cdh1, which blocked disengagement/reduplication of centrioles during S phase, did not do so when cells were arrested in G2, at a time when Plk1 becomes active. However, when Cdh1 was knocked down and Plk1 inhibited, centrioles did not disengage.
A number of studies led to the understanding that Plk1 activity is essential for centriole disengagement whether or not APC/C activity was present (Liu and Erikson, 2002; Tsou et al., 2009; Lončarek et al., 2010; Schöckel et al., 2011; Wang et al., 2011; reviewed by Mardin and Schiebel, 2012). It was proposed that Plk1 activity was necessary to “prime” centriole associated cohesin subunits for separase mediated cleavage (Tsou et al., 2009; Schöckel et al., 2011). Significant levels of Plk1 activity during S phase are not expected, but nevertheless we found that strong inhibition of Plk1 (and Plk2 and Plk3) activities, which is sufficient to block centriole reduplication during G2, did not inhibit APC/C–Cdh1 mediated centriole disengagement during S phase. However, we should be clear that our results do not speak against the importance of Plk1 “priming” of centriolar cohesin subunits for prompt cleavage in anaphase; rather our findings reveal that this is not absolutely necessary in living cells if enough time is allowed.
Thus, Plk1 and APC/C–Cdh1 activities are redundant pathways each of which can disengage and thereby “license” centrioles for duplication. One can ask why the cell uses two pathways that work cooperatively to disjoin centrioles and what advantage that might provide. Plk1 activity during late G2 and mitosis is slow to disengage centrioles; in separase null cells driven out of mitosis centrioles do not disengage until sometime in G1 (Tsou et al., 2009). We found that APC/C activity alone is also slow in disengaging centrioles (Fig. 5) with the incidence of cells with disengaged/reduplicated centrioles increasing steadily until at least 72 hours after siRNA transfection when the experiments were terminated. Given the obvious problems with disengagement occurring too early (spindle multipolarity) or too late (possibly incomplete centriole duplication in the next cell cycle), the cooperative action of Plk1 and APC/C activities ensure the timeliness and fidelity of disengagement in the short time-window of the cell finishing mitosis. Indeed, there may be an advantage for the cell to use two relatively slow mechanisms to disengage centrioles. If Plk1 or APC/C activities, working singly, were efficient in releasing the links between centrioles the cell could be subject to the risk that when Plk1 activity rises in G2/early mitosis and Emi1 begins to be degraded as early as 20 minutes before nuclear envelope breakdown (Di Fiore and Pines, 2007), centrioles could prematurely separate leading to an incidence of spindle multipolarity. Such a phenomenon can be dramatically seen when rapidly cycling sea urchin zygotes are held in prometaphase by a variety of methods. During prolonged prometaphase, the centriole pairs split leading to spindle multipolarity before anaphase onset (Sluder and Begg, 1985; Sluder and Rieder, 1985). Additionally, by having the completion of disengagement be linked to APC/C–Cdh1 activity and not sensitive to APC/C–Cdc20 activity, centriole disengagement is pushed off to late mitosis.
Fig. 5. Incidence of HeLa cells with >2 Cep135 spots associated with centrin foci as a function of time after siRNA transfection to knock down Emi1 during S phase.
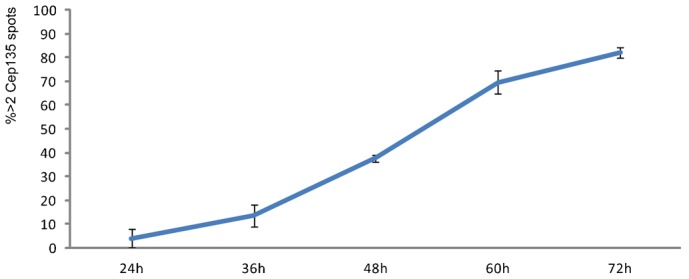
The experimental protocol is outlined in Fig. 1A with the exception that fixation times ranged from 24 to 72 hours after transfection. Each data-point is the mean of three independent experiments (>200 cells counted for each time-point of each experiment) and the error bars are one standard deviation.
Our results also revealed that many of the extra centrioles were associated with SAS-6 even 48–72 hours after the start of Emi1 knockdown. Given reports that APC/C–Cdh1 activity targets SAS-6 for proteasomal degradation in late anaphase (Strnad et al., 2007; Puklowski et al., 2011), persistence of this protein was at first glance unexpected. Nevertheless, this cartwheel protein of daughter centrioles must be stable for some time both in daughter centrioles and in the cellular subunit pool in the presence of APC/C–Cdh1 activity, because SAS-6 is required for procentriole formation (Strnad et al., 2007). We would not have seen supernumerary daughter centrioles had it been completely degraded. Persistence of SAS-6 after Emi1 knockdown may be due to APC/C–Cdh1 mediated degradation of the F-box protein FBXW5, part of an SCF complex that targets SAS-6 for proteasomal degradation (Puklowski et al., 2011). Perhaps APC/C–Cdh1 activity alone targets SAS-6 for degradation but does so slowly. This is consistent with our finding that the incidence of extra SAS-6 foci was consistently lower than the incidence of extra Cep135 and CP110 foci.
We also found that daughter centrioles are able to acquire small gamma tubulin clouds around them during S even when Plk1 activity, if any, is strongly suppressed by BI2536. This indicates that acquisition of gamma tubulin by daughter centrioles is not entirely dependent upon Plk1 activity as previously proposed (Wang et al., 2011). That the number of gamma tubulin foci associated with centrin spots was less than the extra Cep135 and CP110 foci suggests, however, that gamma tubulin acquisition may be relatively slow in the absence of Plk1 activity. Also, the fact that daughter centrioles do not duplicate during S despite presence of gamma tubulin on some of them suggests that reproductive maturation does not simply result from the acquisition of pericentriolar gamma tubulin but rather depends on some other Plk1 dependent event (Lončarek et al., 2010; Wang et al., 2011).
Materials and Methods
Cell culture and drug treatment
HeLa cells expressing low levels of GFP-Centrin1 (La Terra et al., 2005) were cultured in DME medium supplemented with 10% FBS and 1% Penicillin–Streptomycin cocktail (Life Technologies, Grand Island, NY). RPE-1 cells expressing GFP-Centrin1 were cultured in F12/DME (1:1) medium supplemented with 10% FBS and 1% Penicillin–Streptomycin cocktail (Life Technologies, Grand Island, NY). Cells were arrested in S phase with 1 mM thymidine, or in G2 with 5 nM RO3306 (Calbiochem, La Jolla, CA). Plk1 activity was inhibited with 200 nM BI2536 (ChemieTek, Indianapolis, IN). Protocols for cell collection, siRNA transfection, drug treatments, and fixation times are shown diagrammatically at the top of each figure and described in the text and figure legends.
RNAi
The siRNA constructs used were: siEmi1_1, sequence GATTGTGATCTCTTATT AA (Di Fiore and Pines, 2007); siEmi1_2, sequence ACTTGCTGCCAGTTCTTCA (Di Fiore and Pines, 2007); siCdh1 (Brummelkamp et al., 2002); and siCdc20 (Wolthuis et al., 2008). All were synthesized by Dharmacon (Lafayette, CO). Control siRNA constructs used: siluciferase (GL2, Dharmacon Lafayette, CO) and FITC conjugated scrambled oligonucleotides (Santa Cruz Biotechnology Inc., Santa Cruz, CA). siRNA constructs were transfected at a final concentration of 10 nM according to the manufacturer's instructions using Oligofectamine (Life Technologies, Grand Island, NY) for HeLa cells and RNAi MAX (Life Technologies, Grand Island, NY) for RPE-1 cells. In all experiments the transfecting agent was removed by changing to fresh medium 16 hours after initial application.
Immunofluorescence
Cells were fixed in ice-cold methanol for >5 minutes. The primary antibodies used were: SAS-6 (Santa Cruz Biotechnology Inc., Santa Cruz, CA) at 1:100 dilution; Cep170 (Life Technologies, Grand Island, NY) at 1:500; Cep135 (Abcam, Cambridge, MA) at 1:500; CP110 (gift from Harold Fisk ) at 1:100; gamma tubulin (Santa Cruz Biotechnology Inc., Santa Cruz, CA) at 1:200; acetylated tubulin (Qiagen Inc., Valencia, CA) at 1:200; poly-glutamylated tubulin (GT335 clone, ENZO Life Science, Farmingdale, NY) at 1:2000; CREST serum (ImmunoVision, Springdale, AR) at 1:500. Secondary antibodies conjugated to AlexaFluor 568 or 647 (Life Technologies, Grand Island, NY) were used at 1:1000 dilutions.
Cell preparations were observed with a Leica DMR microscope equipped for phase contrast and epifluorescence. A 100× NA 1.3 objective lens was used to collect Z stacks (0.2 µm steps). Image series were deconvolved using SlideBook 5.0 software (Intelligent Imaging Innovations, Denver, CO). Distances between mother and daughter centrioles were measured using ImageJ software (National Institute of Health, Bethesda, MD) and plotted in a graph using GraphPad Prism for Windows version. 5.04 (GraphPad Software Inc., La Jolla, CA).
Immunoblotting
Cells were lysed with (50 mM Tris, 150 nM NaCl, 1% NP-40, 5% Glycerol) supplimented with protease inhibitor mix (Sigma–Aldrich, St. Louis, MO). Anti-Emi1 antibody was used at 1:100. Anti-Cdh1, Cdc20 and Securin antibodies were used at 1:1000. All antibodies above are from Santa Cruz Biotechnology Inc., Santa Cruz, CA. Band intensities were quantified using ImageJ software. Protein levels were normalized to the actin loading control bands in the same lanes of the same gels.
Supplementary Material
Acknowledgments
We thank Drs Yumi Uetake, Paramasivam Murugan, Elizabeth Luna, Jadranka Lončarek and Alexey Khodjakov for useful suggestions and discussions. We are grateful to Harold Fisk for the gift of anti-CP110 antibody. This work was supported by National Institutes of Health grant [GM 30758 to G.S.] and partial support from The Uehara Memorial Foundation [2008 Postdoctoral Fellowship for T.H.].
Footnotes
Competing interests: The authors have no competing interests to declare.
References
- Brummelkamp T. R., Bernards R., Agami R. (2002). A system for stable expression of short interfering RNAs in mammalian cells. Science 296, 550–553 10.1126/science.1068999 [DOI] [PubMed] [Google Scholar]
- Crasta K., Ganem N. J., Dagher R., Lantermann A. B., Ivanova E. V., Pan Y., Nezi L., Protopopov A., Chowdhury D., Pellman D. (2012). DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–58 10.1038/nature10802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fiore B., Pines J. (2007). Emi1 is needed to couple DNA replication with mitosis but does not regulate activation of the mitotic APC/C. J. Cell Biol. 177, 425–437 10.1083/jcb.200611166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge A. G., Loktev A. V., Hansen D. V., Verschuren E. W., Reimann J. D., Jackson P. K. (2006). The evi5 oncogene regulates cyclin accumulation by stabilizing the anaphase-promoting complex inhibitor emi1. Cell 124, 367–380 10.1016/j.cell.2005.10.038 [DOI] [PubMed] [Google Scholar]
- Ganem N. J., Godinho S. A., Pellman D. (2009). A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278–282 10.1038/nature08136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golsteyn R. M., Mundt K. E., Fry A. M., Nigg E. A. (1995). Cell cycle regulation of the activity and subcellular localization of Plk1, a human protein kinase implicated in mitotic spindle function. J. Cell Biol. 129, 1617–1628 10.1083/jcb.129.6.1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardavaccaro D., Kudo Y., Boulaire J., Barchi M., Busino L., Donzelli M., Margottin-Goguet F., Jackson P. K., Yamasaki L., Pagano M. (2003). Control of meiotic and mitotic progression by the F box protein β-Trcp1 in vivo. Dev. Cell 4, 799–812 10.1016/S1534-5807(03)00154-0 [DOI] [PubMed] [Google Scholar]
- Guarguaglini G., Duncan P. I., Stierhof Y. D., Holmström T., Duensing S., Nigg E. A. (2005). The forkhead-associated domain protein Cep170 interacts with Polo-like kinase 1 and serves as a marker for mature centrioles. Mol. Biol. Cell 16, 1095–1107 10.1091/mbc.E04-10-0939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen D. V., Loktev A. V., Ban K. H., Jackson P. K. (2004). Plk1 regulates activation of the anaphase promoting complex by phosphorylating and triggering SCFβTrCP-dependent destruction of the APC Inhibitor Emi1. Mol. Biol. Cell 15, 5623–5634 10.1091/mbc.E04-07-0598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama R., Borisy G. G. (1981). Centriole cycle in Chinese hamster ovary cells as determined by whole-mount electron microscopy. J. Cell Biol. 91, 814–821 10.1083/jcb.91.3.814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Terra S., English C. N., Hergert P., McEwen B. F., Sluder G., Khodjakov A. (2005). The de novo centriole assembly pathway in HeLa cells: cell cycle progression and centriole assembly/maturation. J. Cell Biol. 168, 713–722 10.1083/jcb.200411126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lénárt P., Petronczki M., Steegmaier M., Di Fiore B., Lipp J. J., Hoffmann M., Rettig W. J., Kraut N., Peters J. M. (2007). The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr. Biol. 17, 304–315 10.1016/j.cub.2006.12.046 [DOI] [PubMed] [Google Scholar]
- Liu X., Erikson R. L. (2002). Activation of Cdc2/cyclin B and inhibition of centrosome amplification in cells depleted of Plk1 by siRNA. Proc. Natl. Acad. Sci. USA 99, 8672–8676 10.1073/pnas.132269599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loncarek J., Hergert P., Magidson V., Khodjakov A. (2008). Control of daughter centriole formation by the pericentriolar material. Nat. Cell Biol. 10, 322–328 10.1038/ncb1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lončarek J., Hergert P., Khodjakov A. (2010). Centriole reduplication during prolonged interphase requires procentriole maturation governed by Plk1. Curr. Biol. 20, 1277–1282 10.1016/j.cub.2010.05.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida Y. J., Dutta A. (2007). The APC/C inhibitor, Emi1, is essential for prevention of rereplication. Genes Dev. 21, 184–194 10.1101/gad.1495007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardin B. R., Schiebel E. (2012). Breaking the ties that bind: new advances in centrosome biology. J. Cell Biol. 197, 11–18 10.1083/jcb.201108006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margottin-Goguet F., Hsu J. Y., Loktev A., Hsieh H. M., Reimann J. D., Jackson P. K. (2003). Prophase destruction of Emi1 by the SCFβTrCP/Slimb ubiquitin ligase activates the anaphase promoting complex to allow progression beyond prometaphase. Dev. Cell 4, 813–826 10.1016/S1534-5807(03)00153-9 [DOI] [PubMed] [Google Scholar]
- Moshe Y., Boulaire J., Pagano M., Hershko A. (2004). Role of Polo-like kinase in the degradation of early mitotic inhibitor 1, a regulator of the anaphase promoting complex/cyclosome. Proc. Natl. Acad. Sci. USA 101, 7937–7942 10.1073/pnas.0402442101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel M., Meyer P., Khodjakov A., Rieder C. L., Bornens M. (2000). The respective contributions of the mother and daughter centrioles to centrosome activity and behavior in vertebrate cells. J. Cell Biol. 149, 317–330 10.1083/jcb.149.2.317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puklowski A., Homsi Y., Keller D., May M., Chauhan S., Kossatz U., Grünwald V., Kubicka S., Pich A., Manns M. P.et al. (2011). The SCF-FBXW5 E3-ubiquitin ligase is regulated by PLK4 and targets HsSAS-6 to control centrosome duplication. Nat. Cell Biol. 13, 1004–1009 10.1038/ncb2282 [DOI] [PubMed] [Google Scholar]
- Schöckel L., Möckel M., Mayer B., Boos D., Stemmann O. (2011). Cleavage of cohesin rings coordinates the separation of centrioles and chromatids. Nat. Cell Biol. 13, 966–972 10.1038/ncb2280 [DOI] [PubMed] [Google Scholar]
- Sluder G., Begg D. A. (1985). Experimental analysis of the reproduction of spindle poles. J. Cell Sci. 76, 35–51. [DOI] [PubMed] [Google Scholar]
- Sluder G., Rieder C. L. (1985). Centriole number and the reproductive capacity of spindle poles. J. Cell Biol. 100, 887–896 10.1083/jcb.100.3.887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strnad P., Leidel S., Vinogradova T., Euteneuer U., Khodjakov A., Gönczy P. (2007). Regulated HsSAS-6 levels ensure formation of a single procentriole per centriole during the centrosome duplication cycle. Dev. Cell 13, 203–213 10.1016/j.devcel.2007.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou M. F., Stearns T. (2006). Mechanism limiting centrosome duplication to once per cell cycle. Nature 442, 947–951 10.1038/nature04985 [DOI] [PubMed] [Google Scholar]
- Tsou M. F., Wang W. J., George K. A., Uryu K., Stearns T., Jallepalli P. V. (2009). Polo kinase and separase regulate the mitotic licensing of centriole duplication in human cells. Dev. Cell 17, 344–354 10.1016/j.devcel.2009.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. J., Soni R. K., Uryu K., Tsou M. F. (2011). The conversion of centrioles to centrosomes: essential coupling of duplication with segregation. J. Cell Biol. 193, 727–739 10.1083/jcb.201101109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolthuis R., Clay-Farrace L., van Zon W.., Yekezare M., Koop L., Ogink J., Medema R., Pines J. (2008). Cdc20 and Cks direct the spindle checkpoint-independent destruction of cyclin A. Mol. Cell 30, 290–302 10.1016/j.molcel.2008.02.027 [DOI] [PubMed] [Google Scholar]
- Wong C., Stearns T. (2003). Centrosome number is controlled by a centrosome-intrinsic block to reduplication. Nat. Cell Biol. 5, 539–544 10.1038/ncb993 [DOI] [PubMed] [Google Scholar]
- Wong R. W., Blobel G., Coutavas E. (2006). Rae1 interaction with NuMA is required for bipolar spindle formation. Proc. Natl. Acad. Sci. USA 103, 19783–19787 10.1073/pnas.0609582104 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}