

Summary
Remodeling of the actin cytoskeleton is required for vasopressin (VP)-induced aquaporin 2 (AQP2) trafficking. Here, we asked whether VP and forskolin (FK)-mediated F-actin depolymerization depends on AQP2 expression. Using various MDCK and LLC-PK1 cell lines with different AQP2 expression levels, we performed F-actin quantification and immunofluorescence staining after VP/FK treatment. In MDCK cells, in which AQP2 is delivered apically, VP/FK mediated F-actin depolymerization was significantly correlated with AQP2 expression levels. A decrease of apical membrane associated F-actin was observed upon VP/FK treatment in AQP2 transfected, but not in untransfected cells. There was no change in basolateral actin staining under these conditions. In LLC-PK1 cells, which deliver AQP2 basolaterally, a significant VP/FK mediated decrease in F-actin was also detected only in AQP2 transfected cells. This depolymerization response to VP/FK was significantly reduced by siRNA knockdown of AQP2. By immunofluorescence, an inverse relationship between plasma membrane AQP2 and membrane-associated F-actin was observed after VP/FK treatment again only in AQP2 transfected cells. This is the first report showing that VP/FK mediated F-actin depolymerization is dependent on AQP2 protein expression in renal epithelial cells, and that this is not dependent on the polarity of AQP2 membrane insertion.
Keywords: AQP2, Vasopressin, Actin cytoskeleton
Introduction
AQP2 is expressed in renal collecting duct principal cells and is critical for water reabsorption and urine concentration (Brown, 2003; Deen and Knoers, 1998; Fushimi et al., 1997; Fushimi et al., 1993; Katsura et al., 1997; Knepper et al., 1997; Nielsen et al., 2002). Vasopressin (VP) binding to the vasopressin type 2 receptor (V2R) triggers AQP2 plasma membrane accumulation via intracellular cAMP elevation, PKA activation (Katsura et al., 1997; Klussmann et al., 1999; Kuwahara et al., 1995) and AQP2 phosphorylation pattern changes (Fenton et al., 2008; Fushimi et al., 1997; Hoffert et al., 2008; Hoffert et al., 2007; Katsura et al., 1997; Moeller et al., 2009; Nishimoto et al., 1999). Phosphorylation increases AQP2 membrane accumulation by inhibiting its clathrin-mediated endocytosis (Bouley et al., 2006; Brown et al., 2008; Lu et al., 2007; Moeller et al., 2010; Sun et al., 2002), but the role of phosphorylation in the exocytotic process remains to be clarified. It is also well established that the filamentous structure of actin is affected by vasopressin stimulation (Hays et al., 1993). In rat kidney inner medullary collecting duct cells and in toad bladder epithelial cells, filamentous actin (F-actin) depolymerization after arginine vasopressin (AVP) treatment was reported 20 years ago (Ding et al., 1991; Simon et al., 1993).
Since the discovery of aquaporins, several studies have shown that VP mediated AQP2 plasma membrane accumulation involves F-actin depolymerization. Cytochalasin D, an F-actin depolymerizing agent, induces AQP2 plasma membrane accumulation in rabbit collecting duct CD8 cells (Tamma et al., 2001). Inhibition of RhoA, which stimulates actin polymerization in its active form, by Clostridium difficile toxin B, or inhibition of Rho kinase, an important Rho downstream effector, by Y-27632 both result in AQP2 plasma membrane accumulation (Klussmann et al., 2001; Tamma et al., 2001). The adenylyl cyclase activator forskolin (FK), which also stimulates AQP2 membrane accumulation, increases RhoA phosphorylation and binding to RhoGDI, thus stabilizing the inactive form of RhoA in CD8 cells (Tamma et al., 2003). From the same group, it was reported that moesin, an ERM protein, that mediates cross-linking between F-actin and trans-membrane proteins is involved in FK mediated F-actin depolymerization in CD8 cells (Tamma et al., 2005). We recently showed that simvastatin induces AQP2 plasma membrane accumulation through RhoA inactivation (Li et al., 2011). Taken together, these data indicate that F-actin depolymerization is crucial molecular step for AQP2 plasma membrane accumulation.
In addition, AQP2 interactions with the actin regulating proteins, SPA-1, and tropomyosin 5b, have been identified. SPA-1 is a GTPase activating protein (GAP) for Rap1. FK mediated AQP2 apical accumulation required the Rap1GAP activity of SPA-1 in MDCK cells (Noda et al., 2004). Importantly, the finding in MDCK cells that phosphorylation of AQP2 decreases its affinity for globular actin (G-actin) but increases its binding to the actin stabilizing protein tropomyosin 5b (Noda et al., 2008), suggests a “catalytic” function for AQP2 in VP- mediating F-actin depolymerization.
Recently, we established an exocytosis assay system by expressing soluble secreted (ss) YFP in LLC-PK1 cells. In cells transfected with AQP2, ssYFP was localized in many (but not all) AQP2 containing vesicles. When these vesicles fuse with the plasma membrane, ssYFP is released into the culture medium. The released ssYFP can be quantified by measuring its fluorescence (Nunes et al., 2008). Interestingly, a vasopressin and forskolin (VP/FK) mediated burst of ssYFP exocytosis was observed only in LLC-PK1 cells stably transfected with AQP2, but not in AQP2 untransfected LLC-PK1 cells (Nunes et al., 2008). This finding supports the notion that AQP2 in some way “catalyzes” the effect of VP/FK on cells–In this case, the stimulation of exocytosis.
Overall, these findings have changed the concept of VP-mediated AQP2 plasma membrane accumulation from “AQP2 accumulates in plasma membranes as a consequence of F-actin depolymerization” to “AQP2 accumulates in the plasma membrane after catalyzing F-actin re-organization”. However, a definitive study investigating whether AQP2 is indeed required for VP-mediated F-actin depolymerization has not so far been performed.
In the present study, therefore, we aimed to fill this knowledge gap by investigating whether or not the VP/FK mediated F-actin depolymerization is affected by AQP2 expression levels both in MDCK cells and in LLC-PK1 cells. These are both well established renal epithelial cell lines that have been extensively used for AQP2 intracellular translocation studies. However, they accumulate AQP2 in different plasma membrane domains upon VP stimulation – apical in the case of MDCK (Deen et al., 1997), and basolateral in the case of LLC-PK1 (Brown, 2003), providing an opportunity to assess any potential differences in their VP-induced actin depolymerization response.
Results
Validation of the F-actin quantification assay
The F-actin quantification assay used here has been applied previously to several mammalian cells types, for example, CD8 cells (Tamma et al., 2005). However the adequacy of the assay for MDCK cells or LLC-PK1 cells was not reported. Therefore, we determined the adequacy of the assay for our MDCK and LLC-PK1 cells using latrunculin B (1 µM for 30 minutes), which directly binds to G-actin and inhibits actin polymerization (Wakatsuki et al., 2001). In F-actin quantification assays, a striking and significant 50% decrease in F-actin was found both in MDCK and LLC-PK1 cells after latrunculin B treatment (Fig. 1). F-actin staining by rhodamine phalloidin confirmed that the filamentous structure of actin was greatly disrupted by latrunculin B in both cell types (supplementary material Fig. S1). This major effect of latrunculin B on F-actin was identical in cells with or without AQP2 transfection. We conclude that the F-actin quantification assay is suitable for use in the cell lines that we are studying here.
Fig. 1. Validation of F-actin quantification assay in MDCK and in LLC-PK1 cells.
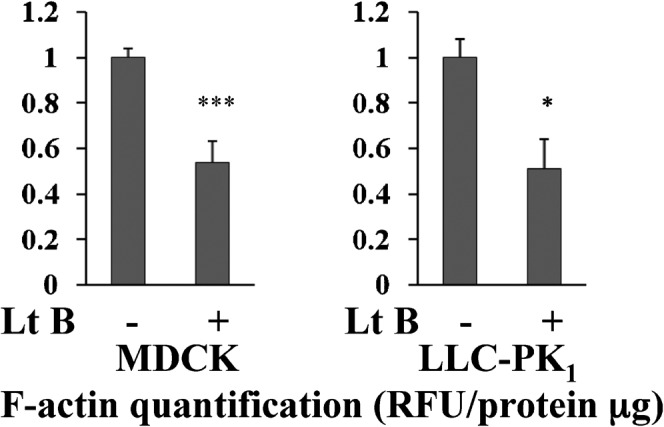
MDCK and LLC-PK1 cells were treated with 1 µM latrunculin B for 30 minutes, then subjected to the rhodamine phalloidin based F-actin quantification assay. A 50% significant reduction in F-actin content was quantified both in MDCK cells (to 0.54±0.09, mean ± s.d., n = 5, P<0.001) and in LLC-PK1 cells (to 0.51±0.13, mean ± s.d., n = 3, P<0.05). These results demonstrate that the F-actin quantification assay is suitable for MDCK cells and LLC-PK1 cells.
AQP2 expression level in different MDCK and LLC-PK1 cell lines
For the purpose of the present study to test whether the level of AQP2 expression affects VP/FK mediated F-actin depolymerization, we used 5 cell lines in our experiments; WT-MDCK, MDCK cells stably expressing a low level of rat AQP2 (low-AQP2 MDCK), a higher level of AQP2 (high-AQP2 MDCK), WT LLC-PK1, and LLC-PK1 cells stably transfected with rat AQP2 (LLC-PK1-AQP2).
Western blotting was performed to examine AQP2 expression levels in these cell lines (Fig. 2). In untransfected LLC-PK1 cells, no signal was detected at 25kD, but this band was strongly detected in LLC-PK1-AQP2 cells (Fig. 2, B). High-AQP2 MDCK showed about 2 times more AQP2 signal (2.14±0.01, n = 3, P<0.05) compared to low-AQP2 MDCK cells (Fig. 2, A and supplementary material Fig. S2). In contrast to WT LLC-PK1 cells, WT-MDCK cells showed a faint signal at the same molecular weight as AQP2 suggesting endogenous AQP2 expression (Fig. 2, A). This finding was examined in more detail below.
Fig. 2. AQP2 expression levels in MDCK and LLC-PK1 cell lines.

Three MDCK and two LLC-PK1 cell lines were subjected to semi quantitative western blotting analysis to determine AQP2 expression levels. In MDCK cells (Fig. 2A–left panels), high AQP2 expression cells showed about 2 times higher AQP2 expression than low AQP2 expression cells. A faint AQP2 signal was also detected in WT MDCK cells. In LLC-PK1 cells (Fig. 2B–right panels), WT (untransfected) cells showed no AQP2 signal, whereas, AQP2 transfected cells showed a strong AQP2 signal. 5 µg protein was loaded in each lane. GAPDH is shown as a loading control.
WT MDCK cells express low levels of endogenous AQP2
To test further whether endogenous AQP2 expression was indeed present in WT-MDCK cells, we picked up 3 colonies from WT-MDCK cells, to examine the possibility that a small number of AQP2-MDCK cells contaminated the WT-MDCK cell pool. We divided cells from these 3 selected clones into 3 groups, cultured in DMEM (FBS 10%) containing 500 µg/ml geneticin (group 1), DMEM without geneticin (group 2), and cultured in DMEM with 500 µg/ml geneticin after transfection with an empty pcDNA 3.1 vector to confer geneticin resistance (group 3). All of the cells in group 1 died within 10 days, whereas some of cells in group 3 survived and became confluent. This shows that the 3 WT-MDCK clones were geneticin sensitive, that they were not contaminated with geneticin resistant AQP2 transfected cells. Next we determined whether AQP2 signals could be detected or not in these 6 clones (3 geneticin sensitive, 3 resistant) by western blotting. In all clones, a faint 25 kD was detected by the AQP2 C-17 antibody (sc9882) (supplementary material Fig. S3, A). In addition, an antibody specific for the pS261 phosphorylated form of AQP2 was applied. A strong band was detected in high-AQP2 MDCK cells, but faint bands were also detected in the 6 clones from untransfected MDCK cells (supplementary material Fig. S3, A). The specificity of the pS261 AQP2 antibody was confirmed in our AQP2 transfected cells, and the pS261 AQP2 signal was strikingly decreased by VP/FK stimulation as previously shown (supplementary material Fig. S3, B) (Tamma et al., 2011). Furthermore, the faint signals detected by polyclonal AQP2 antibody in WT-MDCK cells were significantly increased after overnight incubation with dDAVP (10 nM) (supplementary material Fig. S3, C). These data suggest that AQP2 is endogenously expressed in our untransfected WT-MDCK cells, that the basal expression level is very low, and that expression can be increased by activation AQP2 gene transcription with VP.
VP/FK mediated F-actin depolymerization is correlated with AQP2 expression levels in MDCK cells
To quantify changes in cellular F-actin content mediated by VP/FK, MDCK cells were treated with or without VP/FK (AVP 20 nM and FK 50 µM) for the indicated times. To test whether AQP2 expression level affects VP/FK mediated F-actin depolymerization, we used clones in which AQP2 expression levels were different, as shown in Fig. 2.
In all AQP2 transfected cells, the F-actin content was significantly decreased at both 20 minutes and 60 minutes after VP/FK addition compared to untransfected cells (Fig. 3, A), However, a smaller but significant effect of VP/FK on F-actin content was also observed in WT MDCK cells at 20 minutes, but not at 60 minutes. As shown in Fig. 2 and supplementary material Fig. S3, a low level of AQP2 was endogenously expressed in the WT MDCK cells, which could explain this small effect.
Fig. 3. VP/FK mediated F-actin depolymerization is correlated with AQP2 expression levels in MDCK cells.

(A) MDCK cells were treated with or without VP/FK (20 nM AVP and 50 µM FK) for indicated times, then F-actin was quantified using the F-actin quantification assay shown in Fig. 1. In higher AQP2 expressing cells (high-AQP2 MDCK, right panel), F-actin was significantly decreased by VP/FK at both 20 minutes and 60 minutes. (n = 5, *** P<0.001). In lower AQP2 expressing cells (low-AQP2 MDCK, center panel), F-actin was also significantly decreased, but to a lesser extent, by VP/FK at both 20 minutes and 60 minutes. (n = 5, * P<0.05, *** P<0.001). In WT MDCK cells, F-actin was slightly but significantly decreased by VP/FK at 20 minutes, but was not decreased at 60 minutes. (n = 5, ** P<0.01, NS = not significant). These figures are representative of three independent experiments. (B) F-actin content in each MDCK cell line at the same VP/FK time points was compared using data from Fig. 3A. At both 20 minutes and 60 minutes, higher AQP2 expression is associated with a lower F-actin content (n = 5, * P<0.05, ** P<0.01, *** P<0.001). These figures are representative of three independent experiments.
Next, we quantified the F-actin content of AQP2 transfected MDCK cells compared to untransfected MDCK cells at the same VP/FK time points, at 20 minutes and at 60 minutes (Fig. 3, B). F-actin content was significantly lower in AQP2 transfected cells than in untransfected cells at both time points. Furthermore, the F-actin content of high-AQP2 MDCK cells was significantly lower than low-AQP2 MDCK cells at 20 minutes and at 60 minutes VP/FK treatment (Fig. 3, B). We confirmed that the β-actin expression level was not affected by VP/FK treatment in high AQP2-MDCK cells, in which the VP/FK mediated F-actin decrease was the most striking (supplementary material Fig. S4, A). Taken together, these data demonstrate that VP/FK mediated F-actin depolymerization is correlated with AQP2 expression levels in these cells.
Apical F-actin content is decreased by VP/FK in AQP2-MDCK cells, but not in WT MDCK cells
We monitored F-actin staining and AQP2 translocation after VP/FK treatment by confocal immunofluorescence microscopy in MDCK cells. In high-AQP2 MDCK cells, AQP2 was located intracellularly and sub-apically under non-stimulated conditions (Fig. 4, A, panel a), After VP/FK treatment (AVP 20 nM and FK 50 µM for 60 minutes), AQP2 accumulated at the level of apical plasma membrane (Fig. 4, A, compare panel b and c to panel a). The intensity of apical F-actin signal was selectively and obviously decreased by VP/FK treatment when examining the z-plane sections (Fig. 4A, compare panel e and f to panel d); in contrast, the intensity of the lateral F-actin signal was not changed by VP/FK (Fig. 4, A, panel d and e). The VP/FK mediated selective apical F-actin decrease was not observed in WT MDCK cells (Fig. 4, A, see panel j and k, compare panel k to panel e). The apical and basolateral fluorescence intensities were compared using ImageJ (Fig. 4, B). In high-AQP2 MDCK cells, a significant 60% decrease of apical F-actin signal was detected, whereas, the basolateral F-actin signal was not significantly decreased after 60 minutes VP/FK treatment (Fig. 4, B). In WT-MDCK cells, no significant decrease of the F-actin signal was detected either in apical or basolateral cellular domains (Fig. 4, B). These results are in accordance with the results of the F-actin quantification assay shown in Fig. 3.
Fig. 4. Apical F-actin content is decreased by VP/FK in AQP2-MDCK cells, but not in WT MDCK cells.

(A) MDCK cells were stained with AQP2 antibody (green) and rhodamine phalloidin (red) with or without VP/FK treatment (AVP 20 nM and FK 50 µM for 60 minutes). The larger panels represent confocal sections through the middle or apical regions of the cells as indicated. The smaller horizontal strips at the bottom of each panel are Z-sections that clearly show apical and basolateral cell membranes for direct comparison of their respective staining intensities. In high AQP2-MDCK cells, intracellular and subapical localization of AQP2 was detected under non-stimulated conditions (panels a, g), with well defined cortical F-actin staining both apically and basolaterally (panels d, g). Upon stimulation by VP/FK, AQP2 was strongly accumulated in the apical plasma membrane, and intracellular AQP2 signals could not be detected (panels b, c and h, i). A selective decrease of the apical F-actin was evident (panels e, f and h, i), but the basolateral F-actin staining was not decreased by VP/FK (panels d, e). A VP/FK-induced (same concentration and incubation time) selective apical F-actin signal decrease was not detectable in WT-MDCK cells (panels j, k). The images are representative of three independent experiments. Scale bars, 10 µm. (B) F-actin fluorescence in MDCK cells was analyzed using ImageJ. All images were acquired under identical microscope parameters within the linear range of intensity. No thresholding was applied to the image prior to quantification. A region of interest (ROI) was drawn over both apical and basolateral membranes for quantification. Background intensities were evaluated from an ROI drawn over the nucleus, and were subtracted from the membrane ROI intensities. In high AQP2-MDCK cells, a significant 60% decrease of apical F-actin fluorescence was observed after VP/FK treatment (1.00±0.05 to 0.41±0.02). In contrast, basolateral F-actin fluorescence was not significantly decreased by VP/FK treatment (1.00±0.07 to 1.07±0.07) (right graphs). The selective VP/FK mediated apical F-actin signal decrease was not detected in WT (untransfected) MDCK cells (apical: 1.00±0.04 to 1.07±0.04, basolateral: 1.00±0.04 to 1.07±0.05) (left graphs). Quantification shows the means of more than 20 cells taken from three independent experiments (means ± s.e., *** P<0.001, NS = not significant).
VP/FK induces a decrease in F-actin in AQP2 expressing LLC-PK1 cells, but not in WT LLC-PK1 cells
Next, we performed F-actin quantification assays using WT LLC-PK1 cells and LLC-PK1-AQP2 cells. Cells were treated with or without VP/FK (LVP 20 nM, FK 10 µM) for the indicated times, then subjected to F-actin quantification. In LLC-PK1-AQP2 cells, the F-actin content was significantly decreased after VP/FK treatment compared to untreated cells. In contrast, a VP/FK mediated F-actin decrease was not detected in WT LLC-PK1 cells. We confirmed that β-actin expression levels were not affected by VP/FK treatment in LLC-PK1-AQP2 cells, in which a significant F-actin decrease was found. (supplementary material Fig. S4, B).
AQP2 membrane accumulation and F-actin membrane localization are inversely related in LLC-PK1-AQP2 cells
In LLC-PK1-AQP2 cells, AQP2 was located intracellularly under non-stimulated conditions, and accumulated in lateral plasma membranes after VP/FK treatment (Fig. 6, compare b to a). F-actin signals were markedly, but heterogeneously decreased after VP/FK treatment (Fig. 6, compare e to d). Interestingly, F-actin signals and AQP2 signals after VP/FK treatment showed an inverse relationship like puzzle pieces (Fig. 6, see b, e and h, more remarkable at the high magnification shown in c, f and i), suggesting that VP/FK mediated F-actin depolymerization occurred together with AQP2 plasma membrane accumulation. The selective F-actin decrease mediated by VP/FK was not observed in WT LLC-PK1 cells (Fig. 6, see j and k). These results were in accordance with F-actin quantification assay shown in Fig. 5 as well as MDCK experiments.
Fig. 6. AQP2 membrane accumulation and F-actin membrane localization are inversely related in LLC-PK1-AQP2 cells.

LLC-PK1 cells were stained with AQP2 antibody (green) and rhodamine phalloidin (red) with or without VP/FK treatment (LVP 20 nM and FK 10 µM for 20 minutes). AQP2 was located intracellularly (panels a, g) with well defined basolateral cortical F-actin under non-stimulated conditions (panels d, g). After VP/FK treatment, AQP2 accumulated in lateral plasma membranes (panels b, c) where F-actin depolymerization was clearly detectable in a heterogeneous pattern (panels e, f). There was a clear inverse relationship between the intensity of F-actin staining and AQP2 membrane accumulation (panels b, e, and h). This inverse relationship between AQP2 and F-actin after VP/FK treatment is more clearly appreciated at high magnification (panels c, f, and i). The VP/FK-mediated (same concentration and incubation time) selective decrease of F-actin signal was not observed in untransfected LLC-PK1 cells (panels j, k). The images are representative of three independent experiments. Scale bars, 10 µm.
Fig. 5. VP/FK induces a decrease in F-actin in AQP2 expressing LLC-PK1 cells, but not in WT LLC-PK1 cells.
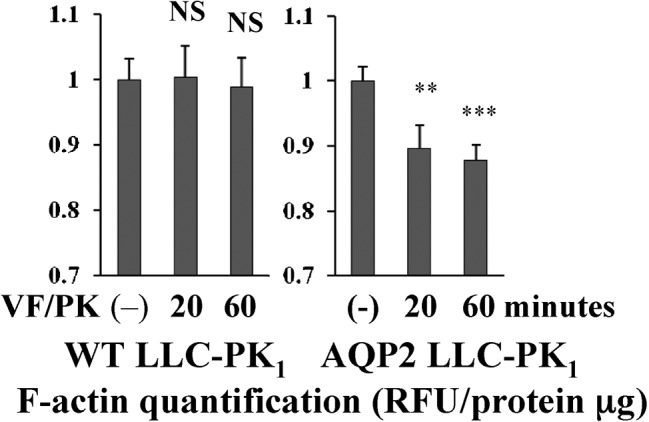
LLC-PK1 cells were treated with or without VP/FK (LVP 20 nM and FK 10 µM for indicated times), then subjected to F-actin quantification assay. In AQP2 expressing LLC-PK1 cells (LLC-PK1-AQP2), F-actin content was decreased by VP/FK at both 20 minutes and 60 minutes. In WT LLC-PK1 cells, F-actin content was not decreased by VP/FK at either 20 minutes and 60 minutes (n = 5, ** P<0.01, *** P<0.001, NS = not significant). These figures are representative of three independent experiments.
AQP2 knockdown blunts the VP/FK mediated F-actin decrease in LLC-PK1-AQP2 cells
Contrary to MDCK cells, we tested only one AQP2 transfected LLC-PK1 clone. Because of the possibility that F-actin organization in this LLC-PK1-AQP2 clone could be especially susceptible to VP/FK stimulation even if AQP2 was not expressed, we performed siRNA-mediated AQP2 knockdown in this cell line (Fig. 7). AQP2 expression was strikingly decreased 48 hours after AQP2 siRNA transfection (Fig. 7, A) and the VP/FK mediated F-actin depolymerization rate was significantly blunted by AQP2 knockdown (Fig. 7, B). Note that F-actin depolymerization was not totally suppressed compared to untransfected cells, reflecting the continued expression of small amount of AQP2 even after siRNA-mediated knockdown. These data further support the necessity of AQP2 expression for VP/FK mediated F-actin depolymerization.
Fig. 7. AQP2 knock down blunts the VP/FK mediated F-actin decrease in LLC-PK1-AQP2 cells.

(A) A highly significant 80% decrease in AQP2 protein expression was achieved by transfection of 80 nM AQP2 siRNA into LLC-PK1-AQP2 cells. 5 µg protein was loaded in each lane. After AQP2 detection, the same PVDF membrane was stripped, reprobed with GAPDH antibody as a loading control. Band intensity was analyzed using ImageJ (Mean ± s.d., n = 3, *** P< 0.001). (B) In AQP2 knockdown cells, F-actin content was still decreased by VP/FK treatment (black bars), but the decrease was significantly blunted compared to cells without siRNA induced AQP2 knockdown (compare black bars to gray bars, mean ± s.d., n = 5, ** P< 0.01). The figure is representative of three independent experiments.
Intracellular cAMP measurement in MDCK and LLC-PK1 cell lines
Finally, we tested whether the VP/FK induced increase in intracellular cAMP concentration was similar among MDCK cell lines and LLC-PK1 cell lines (supplementary material Fig. S5). In MDCK cells, VP/FK increased cAMP in all cell lines to a similar degree (supplementary material Fig. S5, left graph). In both WT LLC-PK1 and LLC-PK1-AQP2 cells, cAMP concentrations were not significantly different after VP/FK treatment (supplementary material Fig. S5, right graph). These data show that all cell clones used in this study react similarly to VP/FK treatment. The blunted VP/FK mediated F-actin depolymerization found in untransfected MDCK cells and the lack of VP/FK mediated F-actin depolymerization in untransfected LLC-PK1 cells is not due to impaired sensitivity to VP/FK.
Discussion
In the present study, we show that F-actin depolymerization mediated by VP/FK stimulation is correlated with AQP2 expression levels, and that F-actin depolymerization is closely associated with AQP2 plasma membrane accumulation in both MDCK cells and LLC-PK1 cells, both, well established renal epithelial cell lines.
Compared to latrunculin B mediated ‘global’ F-actin depolymerization, the extent of VP/FK mediated F-actin depolymerization was relatively lower both in MDCK and LLC-PK1 cells. However, as shown by immunofluorescence staining, VP/FK mediated F-actin depolymerization occurred ‘locally’ and was closely related to AQP2 plasma membrane accumulation. We believe, therefore, the smaller (about 10%) decrease in total cellular F-actin measured after VP/FK treatment probably is an underestimate of the local actin changes that occur in the vicinity of AQP2 target membranes. Interestingly, similar changes were seen in both cell types, independently of the polarity of membrane insertion (apical in MDCK versus basolateral in LLC-PK1) of AQP2. This is consistent with the idea that AQP2 itself is influencing local actin polymerization at or close to its site of membrane accumulation, since no depolymerization was seen in the absence of AQP2.
In MDCK cells, F-actin depolymerization rates were clearly related to AQP2 expression levels at the same VP/FK time point. However, a smaller F-actin depolymerization effect was also observed in untransfected MDCK cells. We found subsequently that very low AQP2 signals are detected even in untransfected MDCK cells. The signal was also revealed by another AQP2 antibody (pS261-AQP2 antibody), and was significantly increased by overnight exposure to dDAVP (10 nM), consistent with previous data showing the presence of cAMP responsive element in the AQP2 gene promoter (Hozawa et al., 1996; Matsumura et al., 1997; Yasui et al., 1997). So far, there is little or no evidence that VP affects post-translational regulation of AQP2 abundance. Because the effect of VP/FK in these WT MDCK cells was reduced compared to AQP2 transfected cells, these results further support the concept that AQP2 is required for VP/FK mediated F-actin depolymerization.
Our present results also provide a potential explanation of our previous data using an ssYFP exocytosis assay in LLC-PK1 cells, as described above (Nunes et al., 2008). We found that VP/FK-induced a rapid burst of ssYFP exocytosis in transfected LLC-PK1-AQP2 cells but not in untransfected LLC-PK1 cells (Nunes et al., 2008). Since actin remodeling is necessary for AQP2 trafficking, we conclude that VP/FK mediated F-actin depolymerization is dependent on AQP2 expression, and is necessary for VP-induced exocytosis in LLC-PK1 cells.
Previous pioneering findings performed prior to the discovery of AQP2 also showed that AVP treatment depolymerizes F-actin in rat inner medullary collecting duct cells (Simon et al., 1993) and in toad bladder epithelial cells (Ding et al., 1991). Toad bladder epithelial cells also express an aquaporin, known as AQP-TB which is 44% identical to rat-AQP2 and may also have a similar effect on the actin cytoskeleton (Siner et al., 1996). Furthermore, Holmgren et al reported that the AVP-induced actin depolymerization was restricted to the apical pole of the cell, where water channels (now identified as AQP2) are inserted (Holmgren et al., 1992). This effect was believed to be due to the VP-induced removal of an apical actin “barrier” that impeded the access of water-channel transporting vesicles to the plasma membrane (Ding et al., 1991).
Our current results also support the notion that AQP2 catalyses F-actin depolymerization by FK via interaction with actin regulating proteins, including SPA-1 and tropomyosin 5b (Noda et al., 2004; Noda et al., 2008). The effect of RhoA or ERM proteins on F-actin depolymerization might also require the presence of AQP2, but this has not yet been examined. In the view of vast array of actin regulatory proteins (Saarikangas et al., 2010), including Rho GEFs, GAPs, GDIs (Ridley, 2006), it is likely that the interaction AQP2 with tropomyosin-5b and SPA-1 are only the tip of a vast iceberg of actin signaling complexes that involve AQP2. Indeed, interactions between AQP2 containing vesicles and actin-related proteins, including ARP2/3 and gelsolin, which interacts with PI(4,5)P2, were identified in isolated rat inner medullary collecting ducts (Barile et al., 2005).
The molecular mechanisms by which AQP2 interacts with many of these actin regulating proteins have yet to be investigated, but deeper understanding of the cell physiology of AQP2/actin associations might contribute to the development of new strategies for treatment of water balance disorders including nephrogenic diabetes insipidus (NDI) and congestive heart failure (CHF). Indeed, we recently showed that statins induce the plasma membrane accumulation of AQP2 and increase urinary concentration in Brattleboro rats by inhibiting RhoA. This results in actin depolymerization, and inhibits endocytosis, thus allowing increased AQP2 membrane accumulation to occur (Li et al., 2011). Our present results show a clear inverse relationship between the presence of F-actin close to the plasma membrane, and AQP2 membrane accumulation. This could reflect local “hotspots” of either accumulation of AQP2 due to impaired endocytosis or increased exocytosis when actin is depolymerized, or the reverse situation (inhibition of exocytosis and stimulation of endocytosis) in the membrane regions where F-actin is abundant. Since our prior data indicate that AQP2 is required for the VP-induced exocytosis of ssYFP in LLC-PK1 cells, it is likely that, as discussed earlier, AQP2/actin association and signaling processes have multiple cellular effects on the AQP2 recycling pathway.
Materials and Methods
Reagents
Lysine vasopressin (LVP), arginine vasopressin (AVP), deamino-Cys1, D-Arg8 vasopressin (dDAVP), forskolin (FK), latrunculin B (LtB), rhodamine phalloidin (Phalloidin Tetramethylrhodamine B isothiocyanate) were purchased from Sigma Aldrich (MO). Goat AQP2 antibody (sc9882, Santa Cruz, CA), mouse anti GAPDH antibody (AM4300, Ambion, CA), mouse anti β-actin antibody (A5441, Sigma Aldrich, MO), donkey anti goat antibody FITC conjugated (Jackson ImmunoResearch, PA), donkey anti goat antibody HRP conjugated (Santa Cruz, CA), donkey anti rabbit antibody HRP conjugated (Santa Cruz, CA), donkey anti mouse antibody HRP conjugated (Santa Cruz, CA) were used following the manufacturer's protocol.
Cell culture
MDCK and LLC-PK1 cells stably expressing rat AQP2 were generated as described previously (Lu et al., 2004; Yui et al., 2009). WT MDCK cells and WT LLC-PK1 cells were parental cells of these AQP2 transfected clones, respectively. The cells were cultured in DMEM containing 10% FBS in a 5% CO2 incubator at 37°C under neomycin selection for transfected cells (500 µg/ml), passaged using TrypLE (GIBCO-BRL). 24 hours before experiments, culturing DMEM was replaced with fresh FBS 10% DMEM without antibiotics. The adenylyl cyclase activator forskolin (FK) was used in combination with the V2R agonist vasopressin (VP) to ensure that a maximum response was elicited in the cells.
Western blotting
Cells were grown on 6 well plates for at least 3 days and lysed in RIPA buffer (500 µl for MDCK, 250 µl for LLC-PK1 cells) supplemented with a Complete Mini (1 tablet/40 ml RIPA buffer). Lysate was rotated for 30 min at 4°C and centrifuged for 10 min at 10,000 rpm. Protein concentration in each sample was determined using a BCA kit (PIERCE). Samples were diluted in RIPA buffer and 35 µl NuPage SDS sample buffer to a protein concentration of 50 µg in 100 µl loading solution. 10 µl reduced samples (5 µg protein) per lane were run on a NuPage 1.0-mm 15 well 4–12% Bis-Tris gel for ∼90 min at 80 V, transferred onto PVDF membrane (LC2002, Invitrogen, CA) for 90 minutes at 100 V. The transferred PVDF membranes were incubated in 5% skimmed milk in 0.05% PBST at 4°C overnight, then incubated with primary antibody, diluted 1:10,000 in 0.05% PBST for 1h (AQP2 polyclonal antibody, GAPDH antibody, and β-actin antibody), or overnight (pS261 AQP2 antibody) at 4°C. Then PVDF membranes were washed 4 times for 15 min in 0.05% PBST, incubated with HRP conjugated secondary antibody diluted 1:10,000 for 30 min, washed in 0.05% PBST 4 times for 15 min. Signals were visualized using Western Lightning ECL and Biomax XAR film. For loading controls, antibodies on PVDF membranes were stripped in stripping buffer (0.2M glycine, 0.05% Tween 20, pH was adjusted to 2.5 by adding HCl) for 1 h, were blocked in 5% skimmed milk/PBST again for 15 minutes, then reprobed with mouse GAPDH antibody (diluted 1:10,000). Band intensities were analyzed by ImageJ.
Immunocytochemistry
8×104 cells were plated on coverslips (for MDCK cells) in 24 wells or Transwell filters (Costar 3460, for LLC-PK1 cells) in 12 wells. After 2 days for MDCK cells and 3 days for LLC-PK1 cells, polarized monolayers were formed. MDCK cells were treated with 50 µM indomethacin overnight (Deen et al., 1997) before experiments to reduce endogenous cAMP levels. Serum containing DMEM was replaced with pre-warmed, serum and antibiotic-free DMEM for 2 hours, then VP/FK (AVP 20 nM and FK 50 µM for MDCK cells, LVP 20 nM and FK 10 µM for LLC-PK1 cells, final concentration) or latrunculin B (1 µM, final concentration) were added to the medium. After incubation for various times, cells were fixed with 4% paraformaldehyde at 37°C for 10 minutes. Cells were washed in PBS 3 times, permeabilized with 0.1% Triton-X/PBS for 10 minutes, washed in PBS, blocked in 1% BSA/PBS for 30 min at room temperature. AQP2 antibody was diluted 1:1,000 in 1%BSA/PBS, incubated overnight at 4°C. Cells were washed in PBS (10 min, 3 times), incubated with 1:1,000 diluted donkey anti goat IgG, FITC conjugated antibody for 3 hours at room temperature, then washed in PBS (10 min, 3 times). Finally, cells were incubated with 20 nM rhodamine-phalloidin diluted in cold PBS for 20 min on ice. Cells were washed in PBS (10 min, 3 times), then mounted on glass slides with Vectashield mounting medium (Vector Labs. Burlingame. CA).
F-actin quantification assay
8×104 cells were plated on 24 well plates, and incubated in normal DMEM for 2 days (LLC-PK1) or 3 days (MDCK). MDCK cells were treated with 50 µM indomethacin overnight prior to use (Deen et al., 1997). DMEM was replaced with HBSS and cells were incubated for 2 hours. After treatment with VP/FK or Latrunculin B as above for various times, HBSS buffer was removed and cells were incubated in binding buffer containing phalloidin (20 mM KH2PO4, 10 mM PIPES, 5 mM EGTA, 2 mM MgCl2, 4% PFA, 0.1% TritonX-100, 250 nM rhodamine phalloidin) for 15 min at room temperature. Negative controls to estimate background autofluorescence were prepared using binding buffer lacking rhodamine-phalloidin. Cells treated with binding buffer were washed 4 times in PBS, incubated in 300 µl (in each well) methanol overnight at −20°C to extract bound rhodamine-phalloidin. The extracted rhodamine-phalloidin fluorescence was read using a Beckman DTX-880 multi-plate reader (excitation 535 nm, emission 595 nm). F-actin content values were expressed as relative fluorescence after subtraction of the negative-control values. Data was adjusted for protein concentration. Each fluorescence value was expressed as relative fluorescence unit (RFU)/protein (µg) and data were analyzed using a two-tailed Student's t-test.
RNA interference
Duplex siRNAs for AQP2 knockdown (5′-CUGCCAUCCUCCAUGAGAU-3′, 5′-AUCUCAUGGAGGAUGGCAG-3′) were purchased from Sigma Aldrich, and diluted in distilled water to 20 µM. 105 LLC-PK1 cells were plated on 6 well plates and incubated overnight. Next day, DMEM was replaced with serum free medium. 80 nM AQP2 siRNA (per well in 1 ml serum free DMEM) was transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). 6 h after adding siRNA, serum free DMEM was replaced with 10% FBS DMEM. 48 hours after transfection, cells were lysed in 200 µl RIPA buffer and subjected to western blotting analysis, or cells were subjected to F-actin quantification assay as described above. Negative controls were prepared using Lipofectamine 2000 without AQP2 siRNA.
Intracellular cAMP measurement
Cells were plated on 96 well plates, were treated with or without VP/FK as above, then analyzed using an ELISA based cAMP Biotrak enzyme immunoassay (Amersham Biosciences) according to the manufacturer's protocol. MDCK cells were treated with 50 µM indomethacin overnight before exposure to VP/FK. Absorbance values at 450 nm were measured using the DTX880 plate reader. The results were adjusted by cell number, and analyzed by a two-tailed Student's t-test.
Supplementary Material
Acknowledgments
This work was supported by NIH grant DK38452 (DB, RB), DK75940, and a Gottschalk research grant (American Society of Nephrology). We thank Dr S. Uchida and Dr S. Sasaki (Tokyo Medical and Dental University) for their providing us MDCK clones. We thank Dr M. Knepper (NIH) for providing the pS261 AQP2 polyclonal antibody.
References
- Barile M., Pisitkun T., Yu M. J., Chou C. L., Verbalis M. J., Shen R. F., Knepper M. A. (2005). Large scale protein identification in intracellular aquaporin-2 vesicles from renal inner medullary collecting duct. Mol. Cell Proteomics 4, 1095–1106 10.1074/mcp.M500049-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouley R., Hawthorn G., Russo L. M., Lin H. Y., Ausiello D. A., Brown D. (2006). Aquaporin 2 (AQP2) and vasopressin type 2 receptor (V2R) endocytosis in kidney epithelial cells: AQP2 is located in ‘endocytosis-resistant’ membrane domains after vasopressin treatment. Biol. Cell 98, 215–232 10.1042/BC20040054 [DOI] [PubMed] [Google Scholar]
- Brown D. (2003). The ins and outs of aquaporin-2 trafficking. Am. J. Physiol. Renal Physiol. 284, F893–F901 10.1152/ajprenal.00387.2002 [DOI] [PubMed] [Google Scholar]
- Brown D., Hasler U., Nunes P., Bouley R., Lu H. A. (2008). Phosphorylation events and the modulation of aquaporin 2 cell surface expression. Curr. Opin. Nephrol. Hypertens. 17, 491–498 10.1097/MNH.0b013e3283094eb1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deen P. M., Knoers N. V. (1998). Physiology and pathophysiology of the aquaporin-2 water channel. Curr. Opin. Nephrol. Hypertens. 7, 37–42 10.1097/00041552-199801000-00007 [DOI] [PubMed] [Google Scholar]
- Deen P. M., Rijss J. P., Mulders S. M., Errington R. J., van Baal J., van Os C. H. (1997). Aquaporin-2 transfection of Madin-Darby canine kidney cells reconstitutes vasopressin-regulated transcellular osmotic water transport. J. Am. Soc. Nephrol. 8, 1493–1501 [DOI] [PubMed] [Google Scholar]
- Ding G. H., Franki N., Condeelis J., Hays R. M. (1991). Vasopressin depolymerizes F-actin in toad bladder epithelial cells. Am. J. Physiol. 260, C9–C16 [DOI] [PubMed] [Google Scholar]
- Fenton R. A., Moeller H. B., Hoffert J. D., Yu M. J., Nielsen S., Knepper M. A. (2008). Acute regulation of aquaporin-2 phosphorylation at Ser-264 by vasopressin. Proc. Natl. Acad. Sci. USA 105, 3134–3139 10.1073/pnas.0712338105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fushimi K., Uchida S., Hara Y., Hirata Y., Marumo F., Sasaki S. (1993). Cloning and expression of apical membrane water channel of rat kidney collecting tubule. Nature 361, 549–552 10.1038/361549a0 [DOI] [PubMed] [Google Scholar]
- Fushimi K., Sasaki S., Marumo F. (1997). Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J Biol. Chem 272, 14,800–14,804 10.1074/jbc.272.23.14800 [DOI] [PubMed] [Google Scholar]
- Hays R. M., Condeelis J., Gao Y., Simon H., Ding G., Franki N. (1993). The effect of vasopressin on the cytoskeleton of the epithelial cell. Pediatr. Nephrol. 7, 672–679 10.1007/BF00852577 [DOI] [PubMed] [Google Scholar]
- Hoffert J. D., Nielsen J., Yu M. J., Pisitkun T., Schleicher S. M., Nielsen S., Knepper M. A. (2007). Dynamics of aquaporin-2 serine-261 phosphorylation in response to short-term vasopressin treatment in collecting duct. Am. J. Physiol. Renal Physiol. 292, F691–F700 10.1152/ajprenal.00284.2006 [DOI] [PubMed] [Google Scholar]
- Hoffert J. D., Fenton R. A., Moeller H. B., Simons B., Tchapyjnikov D., McDill B. W., Yu M. J., Pisitkun T., Chen F., Knepper M. A. (2008). Vasopressin-stimulated increase in phosphorylation at Ser269 potentiates plasma membrane retention of aquaporin-2. J Biol. Chem 283, 24617–24627 10.1074/jbc.270.18.10384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren K., Magnusson K. E., Franki N., Hays R. M. (1992). ADH-induced depolymerization of F-actin in the toad bladder granular cell: a confocal microscope study. Am. J. Physiol. 262, C672–C677 [DOI] [PubMed] [Google Scholar]
- Hozawa S., Holtzman E. J., Ausiello D. A. (1996). cAMP motifs regulating transcription in the aquaporin 2 gene. Am. J. Physiol. 270, C1695–C1702 [DOI] [PubMed] [Google Scholar]
- Katsura T., Gustafson C. E., Ausiello D. A., Brown D. (1997). Protein kinase A phosphorylation is involved in regulated exocytosis of aquaporin-2 in transfected LLC-PK1 cells. Am. J. Physiol. 272, F817–F822 [PubMed] [Google Scholar]
- Klussmann E., Maric K., Wiesner B., Beyermann M., Rosenthal W. (1999). Protein kinase A anchoring proteins are required for vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J. Biol. Chem. 274, 4934–4938 10.1074/jbc.274.8.4934 [DOI] [PubMed] [Google Scholar]
- Klussmann E., Tamma G., Lorenz D., Wiesner B., Maric K., Hofmann F., Aktories K., Valenti G., Rosenthal W. (2001). An inhibitory role of Rho in the vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J. Biol. Chem. 276, 20451–20457 10.1074/jbc.M010270200 [DOI] [PubMed] [Google Scholar]
- Knepper M. A., Verbalis J. G., Nielsen S. (1997). Role of aquaporins in water balance disorders. Curr. Opin. Nephrol. Hypertens. 6, 367–371 [DOI] [PubMed] [Google Scholar]
- Kuwahara M., Fushimi K., Terada Y., Bai L., Marumo F., Sasaki S. (1995). cAMP-dependent phosphorylation stimulates water permeability of aquaporin-collecting duct water channel protein expressed in Xenopus oocytes. J. Biol. Chem. 270, 10384–10387 [DOI] [PubMed] [Google Scholar]
- Li W., Zhang Y., Bouley R., Chen Y., Matsuzaki T., Nunes P., Hasler U., Brown D., Lu H. A. (2011). Simvastatin Enhances Aquaporin 2 Surface Expression and Urinary Concentration in Vasopressin Deficient Brattleboro Rats through Modulation of Rho GTPase. Am. J. Physiol. Renal Physiol. 301, F309–F318 10.1152/ajprenal.00001.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H., Sun T. X., Bouley R., Blackburn K., McLaughlin M., Brown D. (2004). Inhibition of endocytosis causes phosphorylation (S256)-independent plasma membrane accumulation of AQP2. Am. J. Physiol. Renal Physiol 286, F233–F243 10.1152/ajprenal.00179.2003 [DOI] [PubMed] [Google Scholar]
- Lu H. A., Sun T. X., Matsuzaki T., Yi X. H., Eswara J., Bouley R., McKee M., Brown D. (2007). Heat shock protein 70 interacts with aquaporin-2 and regulates its trafficking. J. Biol. Chem. 282, 28721–28732 10.1074/jbc.M611101200 [DOI] [PubMed] [Google Scholar]
- Matsumura Y., Uchida S., Rai T., Sasaki S., Marumo F. (1997). Transcriptional regulation of aquaporin-2 water channel gene by cAMP. J. Am. Soc. Nephrol. 8, 861–867 [DOI] [PubMed] [Google Scholar]
- Moeller H. B., Knepper M. A., Fenton R. A. (2009). Serine 269 phosphorylated aquaporin-2 is targeted to the apical membrane of collecting duct principal cells. Kidney Int. 75, 295–303 10.1038/ki.2008.505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller H. B., Praetorius J., Rützler M. R., Fenton R. A. (2010). Phosphorylation of aquaporin-2 regulates its endocytosis and protein-protein interactions. Proc. Natl. Acad. Sci. USA 107, 424–429 10.1073/pnas.0910683107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S., Frøkiaer J., Marples D., Kwon T. H., Agre P., Knepper M. A. (2002). Aquaporins in the kidney: from molecules to medicine. Physiol. Rev. 82, 205–244 10.1152/physrev.00024.2001 [DOI] [PubMed] [Google Scholar]
- Nishimoto G., Zelenina M., Li D., Yasui M., Aperia A., Nielsen S., Nairn A. C. (1999). Arginine vasopressin stimulates phosphorylation of aquaporin-2 in rat renal tissue. Am. J. Physiol. 276, F254–259 [DOI] [PubMed] [Google Scholar]
- Noda Y., Horikawa S., Furukawa T., Hirai K., Katayama Y., Asai T., Kuwahara M., Katagiri K., Kinashi T., Hattori M. et al. (2004). Aquaporin-2 trafficking is regulated by PDZ-domain containing protein SPA-1. FEBS Lett. 568, 139–145 10.1016/j.febslet.2004.05.021 [DOI] [PubMed] [Google Scholar]
- Noda Y., Horikawa S., Kanda E., Yamashita M., Meng H., Eto K., Li Y., Kuwahara M., Hirai K., Pack C. et al. (2008). Reciprocal interaction with G-actin and tropomyosin is essential for aquaporin-2 trafficking. J. Cell Biol. 182, 587–601 10.1083/jcb.200709177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes P., Hasler U., McKee M., Lu H. A., Bouley R., Brown D. (2008). A fluorimetry-based ssYFP secretion assay to monitor vasopressin-induced exocytosis in LLC-PK1 cells expressing aquaporin-2. Am. J. Physiol. Cell Physiol. 295, C1476–C1487 10.1152/ajpcell.00344.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley A. J. (2006). Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell. Biol. 16, 522–529 10.1016/j.tcb.2006.08.006 [DOI] [PubMed] [Google Scholar]
- Saarikangas J., Zhao H., Lappalainen P. (2010). Regulation of the actin cytoskeleton-plasma membrane interplay by phosphoinositides. Physiol. Rev. 90, 259–289 10.1152/physrev.00036.2009 [DOI] [PubMed] [Google Scholar]
- Simon H., Gao Y., Franki N., Hays R. M. (1993). Vasopressin depolymerizes apical F-actin in rat inner medullary collecting duct. Am. J. Physiol. 265, C757–C762 [DOI] [PubMed] [Google Scholar]
- Siner J., Paredes A., Hosselet C., Hammond T., Strange K., Harris H. W. (1996). Cloning of an aquaporin homologue present in water channel containing endosomes of toad urinary bladder. Am. J. Physiol. 270, C372–381 [DOI] [PubMed] [Google Scholar]
- Sun T. X., Van Hoek A., Huang Y., Bouley R., McLaughlin M., Brown D. (2002). Aquaporin-2 localization in clathrin-coated pits: inhibition of endocytosis by dominant-negative dynamin. Am. J. Physiol. Renal Physiol. 282, F998–1011 10.1152/ajprenal.00257.2001 [DOI] [PubMed] [Google Scholar]
- Tamma G., Klussmann E., Maric K., Aktories K., Svelto M., Rosenthal W., Valenti G. (2001). Rho inhibits cAMP-induced translocation of aquaporin-2 into the apical membrane of renal cells. Am. J. Physiol. Renal Physiol. 281, F1092–F1101 10.1152/ajprenal.0091.2001 [DOI] [PubMed] [Google Scholar]
- Tamma G., Klussmann E., Procino G., Svelto M., Rosenthal W., Valenti G. (2003). cAMP-induced AQP2 translocation is associated with RhoA inhibition through RhoA phosphorylation and interaction with RhoGDI. J. Cell Sci. 116, 1519–1525 10.1242/jcs.00355 [DOI] [PubMed] [Google Scholar]
- Tamma G., Klussmann E., Oehlke J., Krause E., Rosenthal W., Svelto M., Valenti G. (2005). Actin remodeling requires ERM function to facilitate AQP2 apical targeting. J. Cell Sci. 118, 3623–3630 10.1242/jcs.02495 [DOI] [PubMed] [Google Scholar]
- Tamma G., Robben J. H., Trimpert C., Boone M., Deen P. M. (2011). Regulation of AQP2 localization by S256 and S261 phosphorylation and ubiquitination. Am. J. Physiol. Cell Physiol. 300, C636–C646 10.1152/ajpcell.00433.2009 [DOI] [PubMed] [Google Scholar]
- Wakatsuki T., Schwab B., Thompson N. C., Elson E. L. (2001). Effects of cytochalasin D and latrunculin B on mechanical properties of cells. J. Cell Sci. 114, 1025–1036 [DOI] [PubMed] [Google Scholar]
- Yasui M., Zelenin S. M., Celsi G., Aperia A. (1997). Adenylate cyclase-coupled vasopressin receptor activates AQP2 promoter via a dual effect on CRE and AP1 elements. Am. J. Physiol. 272, F443–F450 [DOI] [PubMed] [Google Scholar]
- Yui N., Okutsu R., Sohara E., Rai T., Ohta A., Noda Y., Sasaki S., Uchida S. (2009). FAPP2 is required for aquaporin-2 apical sorting at trans-Golgi network in polarized MDCK cells. Am. J. Physiol. Cell Physiol. 297, C1389–C1396 10.1152/ajpcell.00098.2009 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}