

Summary
Obesity is one of the most serious health problems of the 21st century. It is associated with highly increased risk of type 2 diabetes, high blood pressure, cardiovascular disease as well as several cancers. The expansion of the fat tissue needs the differentiation of preadipocytes to adipocytes, a process called adipogenesis. Dysfunction of adipogenesis is a hallmark of obesity and delineation of underlying mechanisms has high priority for identifying targets for pharmacological intervention. Here we investigate the impact of the COP9 signalosome (CSN), a regulator of cullin-RING ubiquitin ligases (CRLs), and of C/EBP homologous protein (CHOP) on the differentiation of LiSa-2 preadipocytes. CHOP induced by piceatannol or by permanent overexpression in LiSa-2 cells blocks adipocyte differentiation as characterized by inhibited fat droplet formation and vascular endothelial growth factor (VEGF) production. Knockdown of the CSN by permanent downregulation of CSN1 in LiSa-2 cells elevates CHOP and retards adipogenesis. The effect of the CSN knockdown on CHOP stability can be explained by the protection of the CRL component Keap1 by the CSN associated ubiquitin-specific protease 15 (USP15). Pulldowns and glycerol gradients reveal that CHOP interacts with a supercomplex consisting of the CSN, cullin 3 and Keap1. Transient knockdown of Keap1 increases CHOP steady state level and retards its degradation. We conclude that CHOP stability is controlled by a CSN-CRL3Keap1 complex, which is crucial for adipogenesis. Our data show that CHOP is a distinguished target for pharmacological intervention of obesity.
Keywords: Adipogenesis, CHOP, COP9 signalosome, Cullin 3, Keap1
Introduction
The incidence of obesity is increasing at an alarming rate worldwide. Dysfunction of adipogenesis is a hallmark of obesity. Therefore, understanding adipogenesis, the adipocyte differentiation from fibroblast-like preadipocytes to lipid-loaded adipocytes, is relevant for identifying targets for pharmacological interventions. The process of adipogenesis is regulated by an elaborate network of transcription factors that coordinate the expression of hundreds of proteins responsible for establishing obesity. In the centre of that network are the two master regulators of adipocyte differentiation, the peroxisome proliferator-activated receptor γ (PPAR-γ) and CCAAT/enhancer binding protein α (C/EBPα) (Farmer, 2006). PPAR-γ is a nuclear hormone receptor, essential for adipogenesis (Koppen and Kalkhoven, 2010; White and Stephens, 2010). C/EBP proteins are a family of transcription factors with different functions. C/EBPα is considered a primary transcription factor that mediates adipogenesis. Early in the differentiation program C/EBPβ is expressed, which is the transcriptional activator of PPAR-γ and C/EBPα. Its activity is delayed by CHOP (also called growth arrest-DNA damage-induced 153, GADD153), a dominant negative form of C/EBP family members (Li et al., 2006) that blocks adipogenesis (Batchvarova et al., 1995). Interestingly, CHOP protein is degraded by the ubiquitin (Ub) proteasome system (UPS) and proteasome inhibitors affect adipocyte differentiation (Li et al., 2006).
The CSN is a regulator of the UPS in all eukaryotic cells (Kato and Yoneda-Kato, 2009; Schmaler and Dubiel, 2010; Wei et al., 2008). This function of the CSN is predominantly executed by influencing the activity of cullin-RING Ub ligases (CRLs). The CSN forms supercomplexes with CRLs and removes the Ub-like protein Nedd8 from cullins. CSN-mediated deneddylation (Cope et al., 2002) inhibits CRL activity (Deshaies and Joazeiro, 2009; Duda et al., 2008; Saha and Deshaies, 2008). CRLs specifically recognize substrates of the UPS by their substrate recognition subunit (SRS). For example, SRSs of cullin 1 (Cul1)-containing complexes, CRL1s, are F-box proteins such as Skp2 or β-TrCP, whereas cullin 3 (Cul3) ligases (CRL3s) possess BTB proteins such as Keap1 for substrate recognition (Deshaies and Joazeiro, 2009). The CSN is associated with Ub-specific protease 15 (USP15) that protects CRL components from autoubiquitination and degradation (Hetfeld et al., 2005; Schweitzer et al., 2007; Wee et al., 2005; Zhou et al., 2003). CSN associated kinases phosphorylate substrates of the UPS which modulates protein stability (Hannß and Dubiel, 2011).
Recently we have shown that the CSN has a role in adipogenesis (Rogalla et al., 2010). Inhibitors of CSN associated kinases such as curcumin and curcumin-like substances block the production of VEGF in tumor cells (Braumann et al., 2008; Pollmann et al., 2001) as well as in adipocytes (Rogalla et al., 2010). Inhibition of adipocyte VEGF production is one explanation for the fact that obesity is downregulated by curcumin (Aggarwal, 2010; Alappat and Awad, 2010; Ejaz et al., 2009) or resveratrol (Andersen et al., 2010; Baile et al., 2011). Here we show that the curcumin-like compound piceatannol induces CHOP in LiSa-2 preadipocytes, which blocks adipocyte differentiation. Permanent overexpression of Flag-CHOP in LiSa-2 cells inhibits VEGF production and lipid droplet formation. Permanent downregulation of the CSN in LiSa-2 cells stabilizes CHOP and retards adipogenesis. CHOP degradation is controlled by a supercomplex containing the CSN, Cul3 and Keap1.
Results
Piceatannol induces CHOP and blocks adipogenesis
Piceatannol is a naturally occurring stilbene and belongs to curcumin-like compounds (Braumann et al., 2008; Füllbeck et al., 2005). Our microarray data indicated that 50 µM piceatannol induced the expression of CHOP mRNA approximately 8-fold in HeLa cells after 6 h of treatment, which was even more effective than curcumin (GEO accession: GSM472500). A substantial increase of CHOP protein was detected by Western blotting in LiSa-2 preadipocytes and in differentiated adipocytes upon piceatannol treatment for 6 h. After 24 h CHOP almost completely disappeared to its original low level (Fig. 1A). Piceatannol just like curcumin (Rogalla et al., 2010) significantly reduced the VEGF production by LiSa-2 cells already after 2 h of treatment (Fig. 1B). Images in Fig. 1C clearly demonstrate that in contrast to the control with DMSO, preadipocytes did not differentiate to adipocytes in the presence of 50 µM piceatannol. Confluent LiSa-2 cells were treated with insulin, cortisol and triiodthyronine to initiate differentiation, which was monitored by a protocol described before (Wabitsch et al., 2000). As shown by Oil Red O (ORO) staining lipid droplets accumulated significantly in DMSO control cells but not in the presence of piceatannol where we found apoptotic cells after 22 days (Fig. 1C).
Fig. 1. Piceatannol induces CHOP and inhibits differentiation of LiSa-2 preadipocytes.
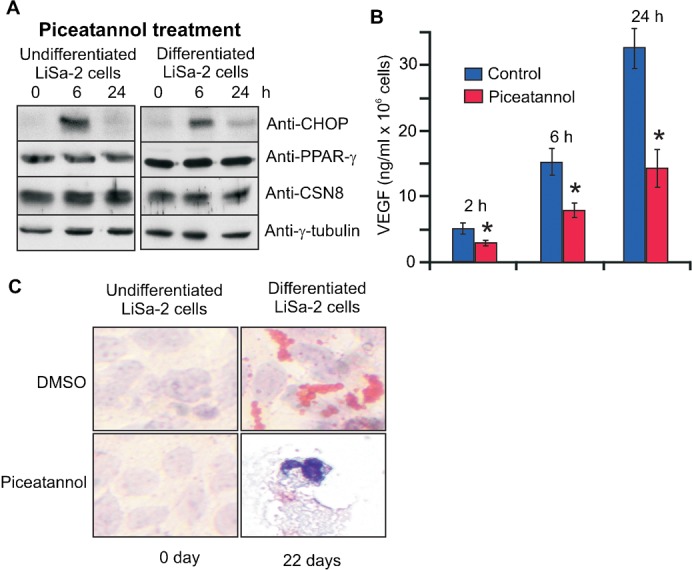
(A) Undifferentiated LiSa-2 cells and LiSa-2 cells after 22 days of differentiation were treated with 50 µM piceatannol and Western blots were performed. (B) VEGF concentration of LiSa-2 cell supernatants from 106 cells was determined by ELISA. VEGF production was significantly reduced by 50 µM piceatannol after 2, 6 and 24 h (*P<0.0001, n = 8). (C) The formation of lipid droplets was visualized by ORO staining and nuclei by hemotoxylin using the Adipogenic differentiation kit in undifferentiated LiSa-2 cells and after 22 days of differentiation without (DMSO) or with piceatannol treatment. After 22 days in the presence of piceatannol apoptotic cells were observed. Microscopy was performed at 200-fold magnification.
LiSa-2 cells permanently overexpressing CHOP do not differentiate
We established Flag-CHOP-LiSa-2 cells which permanently express Flag-CHOP (Fig. 2A). Interestingly, this caused a slight reduction of CSN subunit steady state levels. However, an elevation of neddylated versus deneddylated Cul1 was not observed. In contrary, deneddylated Cul1 and Cul3 increased. PPAR-γ was clearly reduced (Fig. 2A).
Fig. 2. Permanent overexpression of CHOP prevents differentiation of LiSa-2 cells.
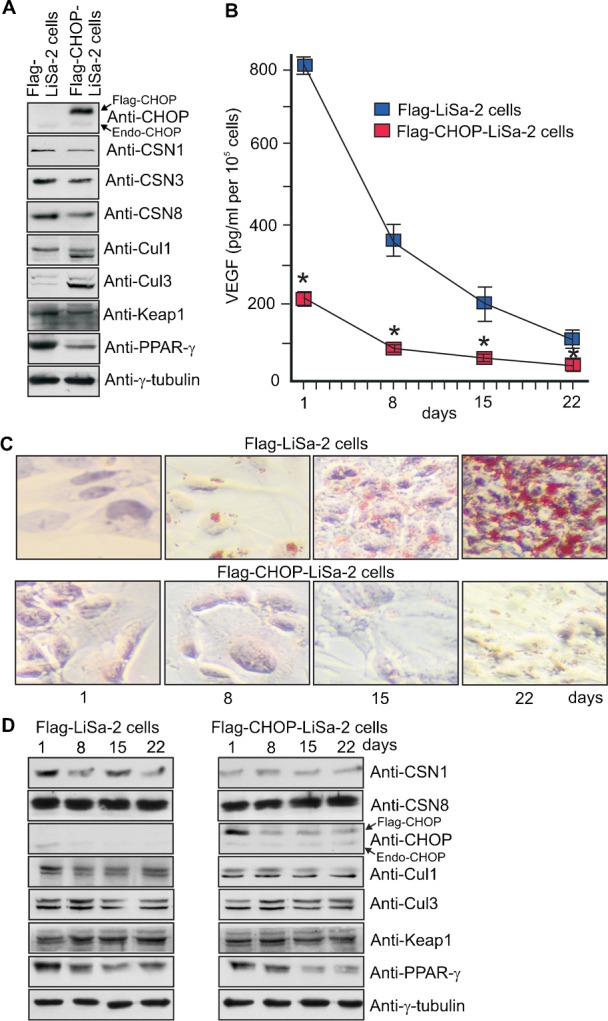
(A) The Western blot compares lysates from Flag-LiSa-2 control cells and Flag-CHOP-LiSa-2 cells permanently expressing Flag-CHOP. (B) Flag-LiSa-2 cells and Flag-CHOP-LiSa-2 cells were treated with a hormone cocktail to induce differentiation. After days 1, 8, 15 and 22 the VEGF concentration was measured in the supernatants of 105 cells (*P<0.0001, n = 6). (C) Lipid formation was analyzed by ORO staining using the Adipogenic differentiation kit after days 1, 8, 15 and 22 upon stimulation of differentiation in Flag-CHOP-LiSa-2 cells and in control cells. Nuclei appear in blue. Microscopy was performed at 200-fold magnification. (D) Dynamics of selected regulatory proteins at days 1, 8, 15 and 22 after stimulation of differentiation analyzed by Western blotting. Flag-CHOP and endogenous CHOP are indicated.
The ability of differentiating preadipocytes to produce VEGF and other adipokines is a major hallmark of cell vitality and proper adipocyte differentiation (Christiaens and Lijnen, 2010; Vona-Davis and Rose, 2009). After induction of differentiation by hormones the VEGF production in Flag-CHOP-LiSa-2 cells was significantly blocked and declined below 25% compared to control at day 1, 8, 15 as well as 22 (Fig. 2B). Moreover, ORO stain revealed that there was no detectable accumulation of lipid droplets in Flag-CHOP-LiSa-2 cells (Fig. 2C). The dynamics of Flag-CHOP revealed that it was high in preadipocytes and declined during differentiation (Fig. 2D). There was a reduction of CSN1, which however had no impact on Cul1 or Cul3 neddylation. After induction with hormones in Flag-CHOP-LiSa-2 cells Keap1 was mostly unchanged whereas PPAR-γ was reduced throughout the 22-day period (Fig. 2D). In summary, these data demonstrate that permanent overexpression of CHOP is sufficient to block adipocyte differentiation.
Knockdown of CSN1 prevents LiSa-2 cell differentiation
LiSa-2 cells were established which permanently express specific siRNA against CSN1. Our siCSN1-LiSa-2 cells were analyzed by Western blotting. Similar as in siCSN1-HeLa cells (Leppert et al., 2011) the protein steady state level of CSN1 was permanently downregulated to approximately 40% as compared to control cells (Fig. 3A). This is accompanied with a reduction of the entire CSN complex as it has been shown before in siCSN1-HeLa cells (Leppert et al., 2011). Downregulation of the CSN caused an approximately 2-fold increase of endogenous CHOP protein in siCSN1-LiSa-2 cells, demonstrating a reciprocal correlation between the CSN and CHOP. Knockdown of the CSN increased the ratio of neddylated versus deneddylated Cul1, which was not observed for Cul3. The BTB protein Keap1 and the transcriptional regulator PPAR-γ were reduced in siCSN1-LiSa-2 cells as compared to siGFP-LiSa-2 control cells (Fig. 3A). In siCSN1-LiSa-2 cells the VEGF production was reduced to less than 20% compared to control values at day 1, 8, 15 as well as 22 (Fig. 3B). In contrast to control cells siCSN1-LiSa-2 cells did not form detectable lipid droplets during 22 days (Fig. 3C). The steady state levels of CSN1 and CSN8 remained downregulated over 22 days in siCSN1-LiSa-2 cells. As consequence CHOP protein increased significantly at day 8 and 15. The ratio of neddylated/deneddylated Cul1 increased during the process, whereas neddylation of Cul3 seemed not to be affected. There is a decrease of Keap1 as well as of PPAR-γ in siCSN1-LiSa-2 cells as compared to normally differentiating control cells (Fig. 3D). Summing up, downregulation of the CSN led to a block of LiSa-2 cell differentiation.
Fig. 3. Downregulation of CSN1 prevents differentiation of LiSa-2 cells.
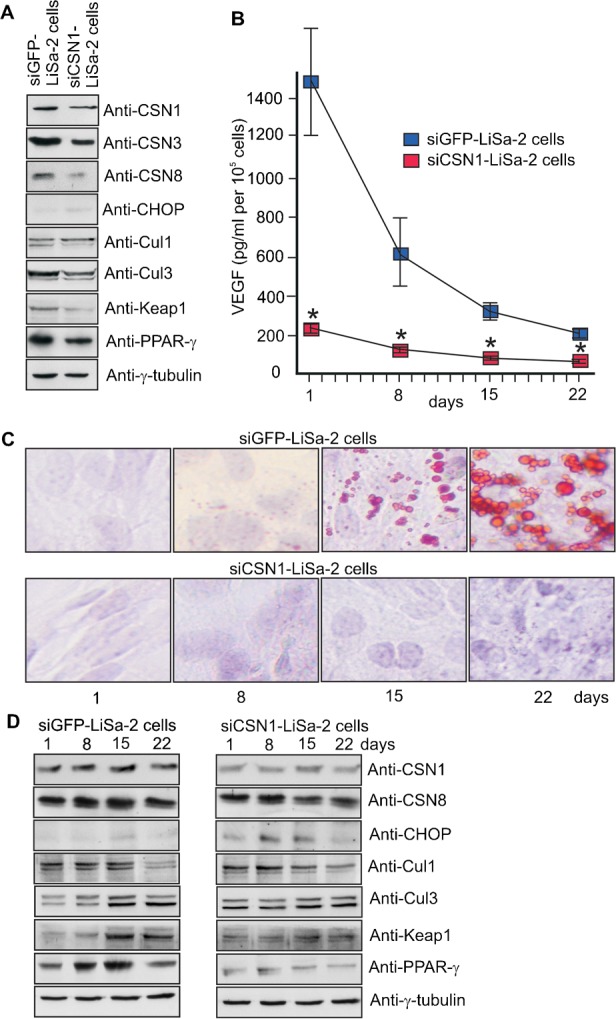
(A) The Western blot compares lysates from siGFP-LiSa-2 control cells and siCSN1-LiSa-2 cells permanently expressing siRNA against CSN1. (B) siGFP-LiSa-2 cells and siCSN1-LiSa-2 cells were treated with a hormone cocktail to induce differentiation. After days 1, 8, 15 and 22 the VEGF concentration was measured in the supernatants of 105 cells (*P<0.0001, n = 6). (C) Lipid formation was analyzed by ORO staining using the Adipogenic differentiation kit after days 1, 8, 15 and 22 upon stimulation of differentiation. After these conditions nuclei are blue. Microscopy was performed at 200-fold magnification. (D) Dynamics of selected regulatory proteins during differentiation at days 1, 8, 15 and 22 analyzed by Western blotting.
Degradation of CHOP is mediated by the CSN-CRL3Keap1 supercomplex
Immunoprecipitations with anti-CSN7 antibody from LiSa-2 cell lysates revealed CHOP in the precipitate. On the other hand, the CSN was co-immunoprecipitated with the anti-CHOP antibody (Fig. 4B). In addition, Cul3 was identified in the precipitate indicating that a CSN-CRL3 supercomplex might control CHOP ubiquitination. Human CHOP complete cDNA (Fig. 4A) was cloned into Flag-vector. Interestingly, transient overexpression of Flag-CHOP in LiSa-2 cells led to a downregulation of CSN subunits (Fig. 4C). Also PPAR-γ was clearly reduced. Flag-CHOP pulldowns confirmed that CHOP occurs in supercomplexes containing the CSN and Cul3 but not Cul1 (Fig. 4D). Co-expression of Xpress-His-CSN1 and Flag-CHOP and subsequent pulldown with His-beads demonstrated binding of CHOP to the CSN subunit CSN1 (Fig. 4E).
Fig. 4. CHOP occurs in complexes together with the CSN and with Cul3.
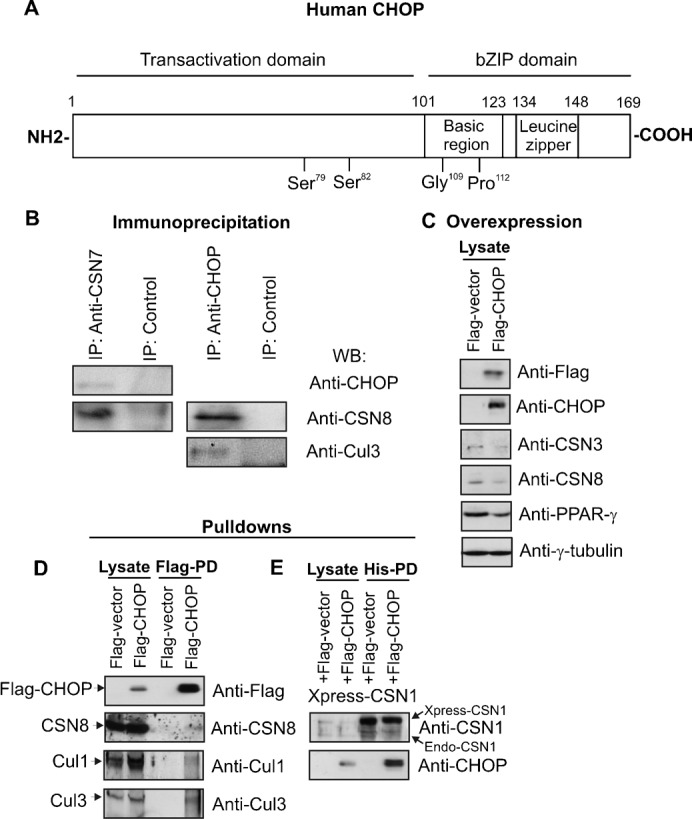
(A) Functional domains of the CHOP protein. (B) The immunoprecipitation with the anti-CSN7 antibody revealed CHOP in the precipitate and the anti-CHOP antibody co-precipitated CSN8 and Cul3 as shown by Western blots. (C) Transient overexpression of Flag-CHOP in LiSa-2 cells led to slight reduction of CSN3 and CSN8 and a decrease of PPAR-γ. (D) Flag-CHOP was transiently overexpressed in LiSa-2 cells and then pulled down by Flag beads (Flag-PD) as described in Materials and Methods. CSN8 and Cul3 but not Cul1 were detected in the precipitate by Western blotting. (E) Xpress-His-CSN1 and Flag-CHOP were co-expressed in LiSa-2 cells. Pulldown of Xpress-His-CSN1 with His beads revealed a co-precipitation of Flag-CHOP.
Immunoprecipitation of CHOP from Flag-CHOP-LiSa-2 cells revealed Keap1 as a putative SRS for CHOP in the CSN-CRL3 complex (Fig. 5A). Keap1 was also analyzed in glycerol density gradients with Flag-CHOP-LiSa-2 cell lysates (Fig. 5B). In these experiments Keap1, Cul3, the CSN and CHOP co-sedimented to the same fractions (4–8), confirming the formation of supercomplexes consisting of CSN-CRL3Keap1. To see whether CHOP degradation is influenced by Keap1 specific siRNA against Keap1 was transiently transfected into Flag-CHOP-LiSa-2 cells. Keap1 was downregulated by approximately 60% (Fig. 5C). This led to a significant increase of CHOP steady state levels as compared to the siGFP-LiSa-2 control cells at time point 0 h (Fig. 5C,D). Knockdown of Keap1 also stabilized Nrf2, a transcription factor which is known to be ubiquitinated in a Keap1-dependent manner (Taguchi et al., 2011). As shown in cycloheximide (CHX) chase experiments downregulation of Keap1 caused retardation of CHOP degradation as compared with control cells (Fig. 5C,D). Treatment with the proteasome inhibitor MG132 led to significant stabilization of Flag-CHOP. In the presence of MG132 a stronger accumulation of Ub conjugates was observed in controls contrary to Keap1 downregulation (Fig. 5C). Based on these results we concluded that CHOP degradation is mediated by CRL3Keap1.
Fig. 5. CHOP degradation is mediated by a complex containing Cul3, Keap1 and the CSN.
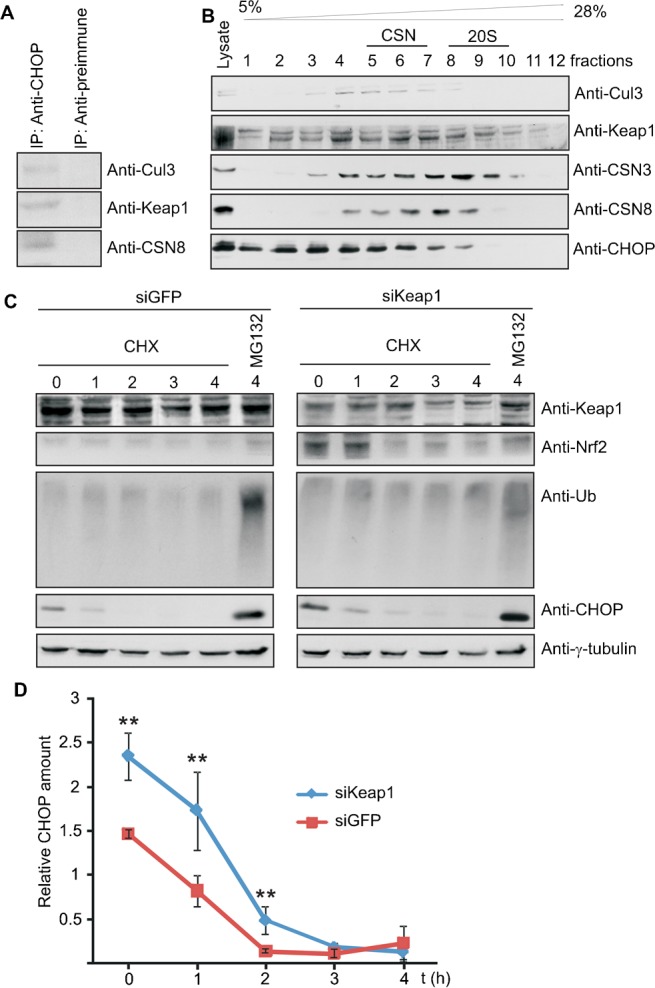
(A) The immunoprecipitate with the anti-CHOP antibody from Flag-CHOP-LiSa-2 cell lysate contained Cul3, Keap1 and the CSN as shown by Western blotting. (B) Density gradient centrifugation was performed with lysate from Flag-CHOP-LiSa-2 cells. Western blots of fractions with different density were performed with antibodies against Cul3, Keap1, CSN3, CSN8 and CHOP. The density gradient was calibrated with purified CSN (about 400 kDa, fractions 5–7) and with purified 20S proteasome (about 700 kDa, fractions 8–10). (C) siKeap1 and control siGFP were transiently transfected into Flag-CHOP-LiSa-2 cells. After transfection (48 h) CHX chase experiments were performed. Western blots were carried out with indicated antibodies. (D) CHX data as shown in Fig. 5C were quantified by densitometry, normalized against γ-tubulin and plotted as relative CHOP amount versus time. Error bars were calculated from standard deviations of three independent experiments. Unpaired Student's t-test was used for statistical analysis and statistical significance is indicated as: **P<0.05.
Discussion
In our experiments adipogenic differentiation of LiSa-2 preadipocytes was induced by treating confluent cells with a cocktail containing insulin, cortisol and triiodthyronine. This model is well suited for monitoring adipocyte differentiation (Rogalla et al., 2010; van Beek et al., 2008; Wabitsch et al., 2000). Insulin is classically viewed as a promoter of adipogenesis as it increases the expression of CREB and PPAR-γ (Farmer, 2006). Cortisol enhances adipogenic differentiation by upregulating C/EBPs (Wu et al., 1996) and triiodthyronine increases lipid droplet formation (Mishra et al., 2010). The current accepted model of adipogenesis starts with the increased expression of C/EBPβ, which then induces the expression of PPAR-γ and C/EBPα forming a positive feed-back loop by activating each other's expression. This leads to the coordinated expression of hundreds of regulatory proteins and enzymes responsible for establishing the mature fat-cell phenotype (Fig. 6). CHOP is a major regulator of adipocyte differentiation. Its expression can be induced by curcumin (Scott and Loo, 2004) or by the curcumin-like compound piceatannol (as shown here) which leads to inhibition of adipocyte differentiation most likely by interfering with the adipogenic C/EBP isoforms (Batchvarova et al., 1995). This is associated with a significant reduction of the VEGF production and blocks lipid droplet formation by LiSa-2 cells. An increase of CHOP protein alone can retard adipogenesis, which is demonstrated in LiSa-2 cells permanently expressing Flag-CHOP. In Flag-CHOP-LiSa-2 cells PPAR-γ was reduced suggesting that overexpressed Flag-CHOP is in fact active as a negative regulator of transcription.
Fig. 6. Model of adipocyte differentiation and the impact of CHOP.
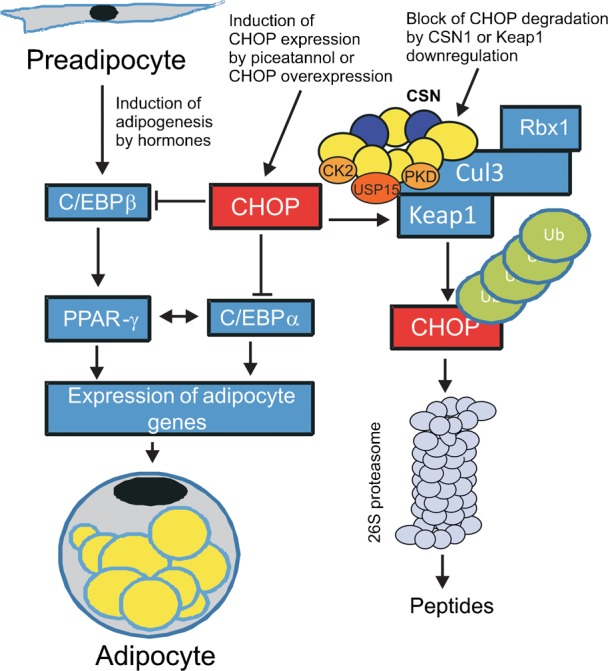
Adipocyte differentiation was induced by the hormones insulin, cortisol and triiodthyronine. This causes an increased expression of C/EBPβ, which then induces the expression of PPAR-γ and C/EBPα forming a positive feed-back loop by activating each other's expression. This is followed by the coordinated expression of proteins responsible for establishing the mature fat-cell phenotype. CHOP is a major negative regulator of adipocyte differentiation. It blocks the transactivation of pro-adipogenic C/EBPs. The expression of CHOP is induced by the curcumin-like compound piceatannol. In addition, CHOP can be stabilized by downregulation of the CSN most likely by exposing Keap1 to increased autocatalytic degradation via CRL3. Keap1 is presumably protected by USP15 associated to the CSN.
An important layer of CHOP regulation is the control of its protein stability. The fact that CHOP degradation is reduced by Keap1 knockdown in addition to our CSN-Cul3-Keap1-CHOP binding studies led us to conclude that CHOP is ubiquitinated by the CRL3Keap1 complex, which is regulated by the CSN. Interestingly, CHOP protein is stabilized in siCSN1-LiSa-2 cells leading to inhibition of adipogenesis. The complex relation between CHOP stability and CSN downregulation can be explained as follows. BTB proteins including Keap1 are ubiquitinated autocatalytically by CRL3 and subsequently degraded (Lo and Hannink, 2006; Wee et al., 2005). The CSN is associated with USP15 protecting BTB3 protein against destabilization as shown in S. pombe (Schmidt et al., 2009). We conclude that downregulation of the CSN is accompanied by a reduction of USP15-mediated protection of CRL3-bound Keap1 leading to an increase of CHOP. This causes inhibition of PPAR-γ expression and a blockade of adipogenesis.
As shown in our model (Fig. 6) upregulation of CHOP either by forced induction or by inhibition of degradation prevents adipocyte differentiation and consequently averts adipose tissue growth and obesity. Our results demonstrate that CHOP is a distinguished target for pharmacological intervention of obesity.
Materials and Methods
Cell culture, adipocyte differentiation and ORO staining
We used human liposarcoma cells (LiSa-2) to study adipocyte differentiation in vitro. Cells were cultured under standard conditions at 37°C and 5% CO2 and grown in Iscove/RPMI 4∶1 with 10% fetal calf serum (FCS; Biochrom, Berlin, Germany), 100 U/ml penicillin and 0.1 mg/ml streptomycin or serum-free, basal medium ((DMEM/F12 (1∶1)) supplemented with 10 mg/ml transferin, 15 mM NaHCO3, 15 mM HEPES, 33 mM biotin, 17 mM pantothenate, 100 U/ml penicillin and 0.1 mg/ml streptomycin. To induce differentiation of confluent LiSa-2 cells, cells were cultured in serum-free medium supplemented with 1 nM insulin, 20 pM triiodthyronine and 1 mM cortisol (adipogenic medium). LiSa-2 cells were plated on a 6-well-plate and cultured for 24 h with Iscove/RPMI medium. The culture medium was incubated with or without 50 µM piceatannol (Calbiochem, Darmstadt, Germany) in DMSO. Differentiation was monitored by a protocol described before (Wabitsch et al., 2000) and assessed by ORO staining using the Thermo Scientific HyClone complete AdvanceSTEMTM Adipogenic differentiation kit (Thermo Fisher Scientific, Schwerte, Germany). In this protocol provided by the manufacturer lipid droplets are stained with ORO and nuclei with hemotoxylin. Differentiated cells were harvested after 1, 8, 15 and 22 days of initiation of differentiation. During differentiation the medium was changed every other day.
siRNA knockdown cells, stable and transient transfections
LiSa-2 cell lines permanently expressing siRNA oligos against CSN1 or GFP as a control construct were generated using the pSUPER system (Peth et al., 2007). CHOP cDNA was obtained by PCR and cloned into pcDNA3.1 vector (Invitrogen, Freiburg, Germany) encoding an N-terminal Flag-tag. Flag-CHOP-pcDNA3.1 and Flag-pcDNA3.1 as a control construct were transfected into LiSa-2 cells. Single clones were used to obtain resistant cell lines with 1 µg/ml of puromycin for siCSN1 and siGFP cells or with 0.25 mg/ml of neomycin for Flag-CHOP-pcDNA3.1 and Flag-pcDNA3.1 cells. For transient transfection siKeap1 und siGFP (Cell Signaling, Boston, USA) were transfected in Flag-CHOP-LiSa-2 cells. 48 h after transfection the cells were lyzed. The supernatants were analyzed by SDS-PAGE and Western blotting.
Pulldowns, immunoprecipitation, glycerol gradients and Western blotting
Flag pulldowns were performed using Flag beads and anti-Flag antibody from Sigma-Aldrich (St. Louis, USA) (Huang et al., 2009). Xpress-His-CSN1 was pulled down by His beads as outlined before (Huang et al., 2009). Immunoprecipitations with the anti-CSN7 and the anti-CHOP antibodies were carried out as described (Uhle et al., 2003). CHX experiments were performed using a final concentration of CHX of 20 µg/ml (Hetfeld et al., 2005).
Glycerol gradient centrifugation analysis was performed as described (Huang et al., 2005).
Proteins were separated and analyzed by SDS-PAGE and Western blotting using antibodies against Cul1 and Cul3 (Oncogene, Cambridge, USA), CSN1 and CSN8 (Enzo, Lausen, Switzerland), CSN3 (Uhle et al., 2003), CHOP, Keap1, PPAR-γ, Nrf2 and γ-tubulin (Santa Cruz, Santa Cruz, USA). Densitometry was carried out applying the ImageJ software.
VEGF production and statistics
For the quantification of VEGF protein levels in the supernatants of LiSa-2 cells human VEGF ELISA kit (RayBio, Norcross, USA) was employed. Statistics were calculated with Microsoft Excel and with the GraphPad InStat3 software. Error bars were calculated from standard deviations and unpaired Student's t-test was used for statistical analysis.
Acknowledgments
We thank Wolfgang Henke for critical reading of the manuscript. This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) DU 229/9-3 and DU 229/12-2 (to W.D.).
Footnotes
Competing interests: The authors declare that there are no competing interests.
References
- Aggarwal B. B. (2010). Targeting inflammation-induced obesity and metabolic diseases by curcumin and other nutraceuticals. Annu. Rev. Nutr. 30, 173–199 10.1146/annurev.nutr.012809.104755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alappat L., Awad A. B. (2010). Curcumin and obesity: evidence and mechanisms. Nutr. Rev. 68, 729–738 10.1111/j.1753-4887.2010.00341.x [DOI] [PubMed] [Google Scholar]
- Andersen C., Rayalam S., Della-Fera M. A., Baile C. A. (2010). Phytochemicals and adipogenesis. Biofactors 36, 415–422 10.1002/biof.115 [DOI] [PubMed] [Google Scholar]
- Baile C. A., Yang J. Y., Rayalam S., Hartzell D. L., Lai C. Y., Andersen C., Della-Fera M. A. (2011). Effect of resveratrol on fat mobilization. Ann. N. Y. Acad. Sci. 1215, 40–47 10.1111/j.1749-6632.2010.05845.x [DOI] [PubMed] [Google Scholar]
- Batchvarova N., Wang X. Z., Ron D. (1995). Inhibition of adipogenesis by the stress-induced protein CHOP (Gadd153). EMBO J. 14, 4654–4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braumann C., Tangermann J., Jacobi C. A., Müller J. M., Dubiel W. (2008). Novel anti-angiogenic compounds for application in tumor therapy - COP9 signalosome-associated kinases as possible targets. Mini Rev. Med. Chem. 8, 421–428 10.2174/138955708784223549 [DOI] [PubMed] [Google Scholar]
- Christiaens V., Lijnen H. R. (2010). Angiogenesis and development of adipose tissue. Mol. Cell. Endocrinol. 318, 2–9 10.1016/j.mce.2009.08.006 [DOI] [PubMed] [Google Scholar]
- Cope G. A., Suh G. S., Aravind L., Schwarz S. E., Zipursky S. L., Koonin E. V., Deshaies R. J. (2002). Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science 298, 608–611 10.1126/science.1075901 [DOI] [PubMed] [Google Scholar]
- Deshaies R. J., Joazeiro C. A. (2009). RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 10.1146/annurev.biochem.78.101807.093809 [DOI] [PubMed] [Google Scholar]
- Duda D. M., Borg L. A., Scott D. C., Hunt H. W., Hammel M., Schulman B. A. (2008). Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell 134, 995–1006 10.1016/j.cell.2008.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejaz A., Wu D., Kwan P., Meydani M. (2009). Curcumin inhibits adipogenesis in 3T3-L1 adipocytes and angiogenesis and obesity in C57/BL mice. J. Nutr. 139, 919–925 10.3945/jn.108.100966 [DOI] [PubMed] [Google Scholar]
- Farmer S. R. (2006). Transcriptional control of adipocyte formation. Cell Metab. 4, 263–273 10.1016/j.cmet.2006.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Füllbeck M., Huang X., Dumdey R., Frommel C., Dubiel W., Preissner R. (2005). Novel curcumin- and emodin-related compounds identified by in silico 2D/3D conformer screening induce apoptosis in tumor cells. BMC Cancer 5, 97 10.1186/1471-2407-5-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannß R., Dubiel W. (2011). COP9 signalosome function in the DDR. FEBS Lett. 585, 2845–2852 10.1016/j.febslet.2011.04.027 [DOI] [PubMed] [Google Scholar]
- Hetfeld B. K., Helfrich A., Kapelari B., Scheel H., Hofmann K., Guterman A., Glickman M., Schade R., Kloetzel P. M., Dubiel W. (2005). The zinc finger of the CSN-associated deubiquitinating enzyme USP15 is essential to rescue the E3 ligase Rbx1. Curr. Biol. 15, 1217–1221 10.1016/j.cub.2005.05.059 [DOI] [PubMed] [Google Scholar]
- Huang X., Hetfeld B. K. J., Seifert U., Kähne T., Kloetzel P. M., Naumann M., Bech-Otschir D., Dubiel W. (2005). Consequences of COP9 signalosome and 26S proteasome interaction. FEBS J. 272, 3909–3917 10.1111/j.1742-4658.2005.04807.x [DOI] [PubMed] [Google Scholar]
- Huang X., Langelotz C., Hetfeld-Pechoc B. K., Schwenk W., Dubiel W. (2009). The COP9 signalosome mediates beta-catenin degradation by deneddylation and blocks adenomatous polyposis coli destruction via USP15. J. Mol. Biol. 391, 691–702 10.1016/j.jmb.2009.06.066 [DOI] [PubMed] [Google Scholar]
- Kato J. Y., Yoneda-Kato N. (2009). Mammalian COP9 signalosome. Genes Cells 14, 1209–1225 10.1111/j.1365-2443.2009.01349.x [DOI] [PubMed] [Google Scholar]
- Koppen A., Kalkhoven E. (2010). Brown vs white adipocytes: the PPARgamma coregulator story. FEBS Lett. 584, 3250–3259 10.1016/j.febslet.2010.06.035 [DOI] [PubMed] [Google Scholar]
- Leppert U., Henke W., Huang X., Müller J. M., Dubiel W. (2011). Post-transcriptional fine-tuning of COP9 signalosome subunit biosynthesis is regulated by the c-Myc/Lin28B/let-7 pathway. J. Mol. Biol. 409, 710–721 10.1016/j.jmb.2011.04.041 [DOI] [PubMed] [Google Scholar]
- Li X., Huang H. Y., Chen J. G., Jiang L., Liu H. L., Liu D. G., Song T. J., He Q., Ma C. G., Ma D. et al. (2006). Lactacystin inhibits 3T3-L1 adipocyte differentiation through induction of CHOP-10 expression. Biochem. Biophys. Res. Commun. 350, 1–6 10.1016/j.bbrc.2006.08.188 [DOI] [PubMed] [Google Scholar]
- Lo S. C., Hannink M. (2006). CAND1-mediated substrate adaptor recycling is required for efficient repression of Nrf2 by Keap1. Mol. Cell. Biol. 26, 1235–1244 10.1128/MCB.26.4.1235-1244.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A., Zhu X. G., Ge K., Cheng S. Y. (2010). Adipogenesis is differentially impaired by thyroid hormone receptor mutant isoforms. J. Mol. Endocrinol. 44, 247–255 10.1677/JME-09-0137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peth A., Boettcher J. P., Dubiel W. (2007). Ubiquitin-dependent proteolysis of the microtubule end-binding protein 1, EB1, is controlled by the COP9 signalosome: possible consequences for microtubule filament stability. J. Mol. Biol. 368, 550–563 10.1016/j.jmb.2007.02.052 [DOI] [PubMed] [Google Scholar]
- Pollmann C., Huang X., Mall J., Bech-Otschir D., Naumann M., Dubiel W. (2001). The constitutive photomorphogenesis 9 signalosome directs vascular endothelial growth factor production in tumor cells. Cancer Res. 61, 8416–8421. [PubMed] [Google Scholar]
- Rogalla S., Geier C. F., Braumann C., Ordemann J., Müller J. M., Dubiel W. (2010). The COP9 signalosome has a role in adipogenesis. Proceedings Of The 45th Congress Of The European Society For Surgical Research, ESSR: Geneva (Switzerland), June 9-12, 2010, (ed. Cikirikcioglu M.), Vol. 45, pp. 17–23 Geneva, Switzerland: Medimond International Proceedings. [Google Scholar]
- Saha A., Deshaies R. J. (2008). Multimodal activation of the ubiquitin ligase SCF by Nedd8 conjugation. Mol. Cell 32, 21–31 10.1016/j.molcel.2008.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmaler T., Dubiel W. (2010). Control of Deneddylation by the COP9 Signalosome. Subcell. Biochem. 54, 57–68 10.1007/978-1-4419-6676-6_5 [DOI] [PubMed] [Google Scholar]
- Schmidt M. W., McQuary P. R., Wee S., Hofmann K., Wolf D. A. (2009). F-box-directed CRL complex assembly and regulation by the CSN and CAND1. Mol. Cell 35, 586–597 10.1016/j.molcel.2009.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer K., Bozko P. M., Dubiel W., Naumann M. (2007). CSN controls NF-kappaB by deubiquitinylation of IkappaBalpha. EMBO J. 26, 1532–1541 10.1038/sj.emboj.7601600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott D. W., Loo G. (2004). Curcumin-induced GADD153 gene up-regulation in human colon cancer cells. Carcinogenesis 25, 2155–2164 10.1093/carcin/bgh239 [DOI] [PubMed] [Google Scholar]
- Taguchi K., Motohashi H., Yamamoto M. (2011). Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells 16, 123–140 10.1111/j.1365-2443.2010.01473.x [DOI] [PubMed] [Google Scholar]
- Uhle S., Medalia O., Waldron R., Dumdey R., Henklein P., Bech-Otschir D., Huang X., Berse M., Sperling J., Schade R. et al. (2003). Protein kinase CK2 and protein kinase D are associated with the COP9 signalosome. EMBO J. 22, 1302–1312 10.1093/emboj/cdg127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beek E. A., Bakker A. H., Kruyt P. M., Vink C., Saris W. H., Franssen-van Hal N. L., Keijer J. (2008). Comparative expression analysis of isolated human adipocytes and the human adipose cell lines LiSa-2 and PAZ6. Int J Obes (Lond) 32, 912–921 10.1038/ijo.2008.10 [DOI] [PubMed] [Google Scholar]
- Vona-Davis L., Rose D. P. (2009). Angiogenesis, adipokines and breast cancer. Cytokine Growth Factor Rev. 20, 193–201 10.1016/j.cytogfr.2009.05.007 [DOI] [PubMed] [Google Scholar]
- Wabitsch M., Brüderlein S., Melzner I., Braun M., Mechtersheimer G., Möller P. (2000). LiSa-2, a novel human liposarcoma cell line with a high capacity for terminal adipose differentiation. Int. J. Cancer 88, 889–894 [DOI] [PubMed] [Google Scholar]
- Wee S., Geyer R. K., Toda T., Wolf D. A. (2005). CSN facilitates Cullin-RING ubiquitin ligase function by counteracting autocatalytic adapter instability. Nat. Cell Biol. 7, 387–391 10.1038/ncb1241 [DOI] [PubMed] [Google Scholar]
- Wei N., Serino G., Deng X. W. (2008). The COP9 signalosome: more than a protease. Trends Biochem. Sci. 33, 592–600 10.1016/j.tibs.2008.09.004 [DOI] [PubMed] [Google Scholar]
- White U. A., Stephens J. M. (2010). Transcriptional factors that promote formation of white adipose tissue. Mol. Cell. Endocrinol. 318, 10–14 10.1016/j.mce.2009.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., Bucher N. L., Farmer S. R. (1996). Induction of peroxisome proliferator-activated receptor gamma during the conversion of 3T3 fibroblasts into adipocytes is mediated by C/EBPbeta, C/EBPdelta, and glucocorticoids. Mol. Cell. Biol. 16, 4128–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C., Wee S., Rhee E., Naumann M., Dubiel W., Wolf D. A. (2003). Fission yeast COP9/signalosome suppresses cullin activity through recruitment of the deubiquitylating enzyme Ubp12p. Mol. Cell 11, 927–938 10.1016/S1097-2765(03)00136-9 [DOI] [PubMed] [Google Scholar]