

Summary
Focal adhesion kinase (FAK) is critically positioned to integrate signals from the extracellular matrix and cellular adhesion. It is essential for normal vascular development and has been implicated in a wide range of cellular functions including the regulation of cell proliferation, migration, differentiation, and survival. It is currently being actively targeted therapeutically using different approaches. We have used human endothelial cells as a model system to compare the effects of inhibiting FAK through several different approaches including dominant negatives, kinase inhibitors and shRNA. We find that manipulations of FAK signaling that result in inhibition of FAK 397 phosphorylation inhibit proliferation and migration. However, abolition of FAK expression using stable (shRNA) or transient (siRNA) approaches does not interfere with these cellular functions. The ability to regulate cell proliferation by FAK manipulation is correlated with the activation status of Rac, an essential signal for the regulation of cyclin-dependent kinase inhibitors. The knockdown of FAK, while not affecting cellular proliferation or migration, dramatically interferes with vascular morphogenesis and survival, mirroring in vivo findings. We propose a novel model of FAK signaling whereby one of the multifunctional roles of FAK as a signaling protein includes FAK as a phospho-regulated repressor of Rac activation, with important implications on interpretation of research experiments and therapeutic development.
Keywords: FAK, Rac, Endothelial, Proliferation, shRNA
Introduction
Focal Adhesion Kinase (FAK) is a prominent component of the cellular-substratum attachment point. It undergoes rapid phosphorylation on tyrosine residue 397 in response to adhesion, and numerous extracellular stimuli (Schaller, 2010). FAK has been widely investigated in many cell types and has been implicated in the regulation of numerous downstream cellular signals, including, activation of MAPK, activation of PI-3′-kinase, activation of Rac, as well as the phosphorylation of focal adhesion proteins such as paxillin (Zhao and Guan, 2009; Schaller, 2010).
Knocking out FAK in the mouse results in early embryonic lethality with disrupted angiogenesis (Ilić et al., 1995). Endothelial specific knockout mice recapitulate the phenotype and early embryonic lethality seen in the whole animal (Shen et al., 2005; Braren et al., 2006). Thus, the endothelial cell is a cell type where FAK has a critical biological role.
FAK has been implicated in regulating a wide array of endothelial cell functions including migration, proliferation, cell survival, as well as cellular permeability (Gilmore and Romer, 1996; Avraham et al., 2003; Ilic et al., 2003; Wu et al., 2003; Braren et al., 2006; Bryant et al., 2006; Holinstat et al., 2006). Many of these studies have utilized expression of a naturally occurring splice variant of FAK, FAK-Related Non-Kinase (FRNK). This protein lacks the N-terminal and kinase domain of FAK. The expression of FRNK inhibits the phosphorylation of endogenous FAK in a dominant negative fashion. Our previous studies demonstrated that expression of FRNK (or FAKY397F, a mutation of the autophosphorylation site), in primary endothelial cells, inhibited cellular proliferation by disrupting the normal regulation of cyclin-dependent kinase inhibitors (Bryant et al., 2006). Studies on the endothelial cells of knockout mice have revealed diverse mechanistic findings. Effects on the proliferation, migration, and survival of these cells have been reported in one study (Shen et al., 2005). In contrast, others found no effect of FAK knockout on proliferation or migration and suggested the formation and/or maintenance of vascular structures was the principal defect in FAK-knockout endothelial cells (Ilic et al., 2003; Braren et al., 2006). Experiments in adult mice using inducible Cre-lox technology have been similarly uncertain with respect to the role of FAK in post-natal angiogenesis (Weis et al., 2008; Lee et al., 2010; Tavora et al., 2010; Lechertier and Hodivala-Dilke, 2012). To understand better the role of FAK in regulating endothelial cell function we compared FAK knockdown to the expression of FRNK, FAKY397F, and a FAK kinase inhibitor (Slack-Davis et al., 2007). Surprisingly our data suggest a novel model for how FAK regulates cellular signal transduction with important implications for understanding the role of FAK in multiple cell types and how distinct therapeutic interventions may target distinct FAK signaling actions.
Results and Discussion
We used several different methods to interfere with FAK signaling. As shown in Fig. 1A, expression of either FRNK or FAKY397F resulted in a loss of FAK phosphorylation at 397. We next tested the effects of these manipulations on endothelial cell proliferation. As shown in Fig. 1A, and as we have reported previously, over-expression of FRNK (Bryant et al., 2006) or FAKY397F nearly completely inhibited the increase in DNA synthesis following stimulation with complete growth medium. This inhibition requires focal adhesion targeting via the FAT domain, as expressing FRNK with a C1034S mutation that disrupts focal adhesion binding has no effect on proliferation (Bryant et al., 2006). Similar results were obtained when we used the FAK inhibitor, PF573228 (Fig. 1B) or an additional FAK inhibitor, (FAK inhibitor 14, data not shown). Importantly the inhibition of both FAK 397 phosphorylation and proliferation were dose dependent with both inhibitors. We next used shRNA-expressing lentiviruses with several distinct sequences targeting FAK to stably knock down total FAK levels and phospho-FAK397 (Fig. 1C). Somewhat surprisingly, these cells were able to be passaged, with no apparent morphological change and had normal population doublings. Despite highly efficient knockdown of FAK and a near complete loss of detectable phospho-FAK397, endothelial cell proliferation was unaffected. We reasoned that perhaps this was an adaptive response to the loss of FAK, or perhaps due to the robust stimulus provided by complete growth medium. We, therefore, used two additional sequences to synthesize siRNA targeting FAK, permitting short-term transfection experiments. Electroporation of endothelial cells with FAK siRNA results in a robust knockdown of total FAK to less than 5% of starting levels (Fig. 2A). This is accompanied by a corresponding loss of phospho-FAK397. However, neither siRNA sequence that effectively knocked-down FAK, resulted in any significant inhibition of DNA synthesis, even when comparatively weaker single agents, such as FGF or VEGF were used as a stimulus (Fig. 2B). Thus, neither short-term (siRNA) nor long-term knock-down (shRNA) of FAK replicated the effects of FRNK, FAKY397F, or the FAK inhibitor.
Fig. 1. Loss of FakY397 phosphorylation, but not FAK protein expression, is associated with inhibition of endothelial cell proliferation.

(A) HUVECs were infected with adenoviruses coding for GFP, GFP-FAKY397F, or GFP-FRNK. At 24 hours post infection CGM was added for 16 hours at which time cells were assessed for the level of phospho-FAK397 and ERK (loading control) (left panel), as well as BrdU incorporation (right panel). (B) Cells were pretreated with various doses of PF573228 for 30 minutes, followed by stimulation with complete growth medium for an additional 30 minutes. Cells were then assessed for the level of phospho-FAK397 and FAK (loading control) (left panel), as well as BrdU incorporation (right panel). (C) HUVECs were infected with lentiviruses harboring shRNAs targeting FAK (or control sequences) to knockdown FAK levels. At 72 hours post infection CGM was added for 16 hours at which time cells were assessed for the level of phospho-FAK397 and ERK (loading control) (left panel), as well as BrdU incorporation (right panel). All panels are representative of experiments performed at least three independent times. Data plotted as mean ± s.e.m.
Fig. 2. Transient knockdown of FAK does not inhibit human endothelial cell proliferation.

(A) Two independent sequences targeting FAK were transfected into HUVECs by electroporation. 48 hours post transfection cell lysates were analyzed for the expression of FAK by Western blotting. (B) Cells transfected with control siRNA, FAK-si#1, or FAK-si#2 were analyzed for incorporation of BrdU in response to VEGF (50 ng/ml), FGF (25 ng/ml), or Complete Growth Medium (CGM). Data plotted are averages of triplicate replicates ± s.e.m. Experiments are representative of similar experiments conducted at least three additional times.
Another function of endothelial cells that has been reported to require FAK activation is the regulation of migration (Avraham et al., 2003; Shen et al., 2005; Zhao and Guan, 2009). We also evaluated the effect on migration of inhibiting FAK using FRNK compared to the loss of FAK from the cell using siRNA-mediated knockdown. Similar to proliferation, we found that FRNK resulted in nearly complete inhibition of the stimulated transwell migration response. In contrast, siRNA-mediated knockdown had no appreciable effects on migration in this assay (Fig. 3A) Similar to proliferation, we failed to observe any inhibition of the migration response, even when weaker defined agonists were used (Fig. 3B).
Fig. 3. FRNK, but not FAK knockdown, inhibits endothelial cell migration.

(A) HUVECs were infected with adenoviruses coding for GFP or GFP-FRNK or transfected with siRNA targeting either GAPDH (control) or FAK. Cells were serum starved for 16 hours. Migration was then evaluated in response to CGM, using a transwell migration assay over 4 hours. Data are presented as number of cells migrated to the bottom in triplicate determinations on a single day ± s.e.m. Data are representative of experiments performed at least three times. (B) As in (A), cells were transfected with 2 different sequences directed against FAK and then evaluated for migration to VEGF or CGM. Data are presented as number of cells migrated to the bottom in triplicate determinations on a single day ± s.e.m. Data are representative of two independent experiments.
We considered that the effects of FRNK and FAKY397F might displace FAK from focal adhesions, and that mislocalized FAK might actually be the trigger for the cell cycle arrest we have observed. Mislocalization was previously implicated in fibroblast cell cycle arrest (Zhao et al., 1998). Therefore, we expressed FRNK in cells in which FAK had been knocked down by siRNA. Interestingly, we found that FRNK was equally effective at inhibiting cellular proliferation in the FAK knockdown cells (Fig. 4). Moreover overexpression of GFP-FAK can result in abundant FAK outside focal adhesions without any deleterious effects on cell proliferation, further arguing that FAK displacement is not responsible (Bryant et al., 2006).
Fig. 4. Endogenous FAK is not required for inhibition by FRNK.

(A) Cells were transfected with control siRNA or siRNA targeting FAK (FAKsi#2) and infected with either adenovirus expressing GFP or GFP-FRNK. Cells were analyzed at 48 hours by Western blot for total FAK, phospho-FAK397, as well as ERK2 as a loading control. The upper band detected with anti-FAK is total FAK, the lower band is expressed FRNK. Cells from (A) were simultaneously analyzed for cellular DNA synthesis using BrdU incorporation (B) or migration using a transwell migration assay (C) in the presence of complete growth medium.
Collectively, these data suggest that the physical presence of FAK or the signal transduction associated with FAK does not seem to be required for cellular proliferation in human endothelial cells. However, when FAT-domain containing regions of FAK are present in the cell, there is a requirement for FAKY397 to be phosphorylated for proliferation to proceed. The simple substitution of Y397 with a non-phosphorylated residue is sufficient to completely inhibit proliferation. The loss of the FAT domain binding by mutation at A.A. 1034, however, restores normal cell function as we have previously reported (Bryant et al., 2006). As the presence of FAK is not required for proliferation or migration (Figs 1–4), these data viewed collectively suggest that a non-activated FAK, bound through FAT-domain interaction may serve in a suppressive function. These data also suggest that different modes of FAK inhibition result in different functional outcomes, underscoring the notion that there are multiple independent mechanisms through which FAK regulates cellular functions (Zhao et al., 2010; Lechertier and Hodivala-Dilke, 2012).
Previous reports have suggested that in some circumstances PYK2 can become upregulated following the loss of FAK, and compensate for FAK functionally (Weis et al., 2008). As this has important implications for our data we evaluated this possibility. We found no significant changes in either PYK2 expression or phosphorylation of PYK2 at Y402, the site homologous to FAK397 following knockdown of FAK (Fig. 5A). These data are in agreement with the data originally published by Weis et al., where upregulation of PY2 was only seen after extended knockdown of PYK2 and was not evident at three days (Weis et al., 2008). In addition, we also performed experiments where we co-transfected endothelial cells with FAK siRNA and PYK2 siRNA. The addition of PYK2 siRNA had no effect on the proliferation response of endothelial cells to complete growth medium (Fig. 5B), further arguing that compensation is unlikely to account for the observed proliferation following FAK knockdown. Recent data from others have suggested PYK2 compensation may be more complex, as the use of different inducible CRE promoters to remove FAKfl/fl from adult mice, resulted in angiogenesis defects and no apparent compensation by PYK2 (Lee et al., 2010; Tavora et al., 2010).
Fig. 5. Proliferation response in FAK knockdown cells is not a consequence of PYK2 compensation.

(A) Cells transfected with siRNA targeting GAPDH or FAK were analyzed for the presence of phospho-PYK2 (Y402) or total PYK2 by western blotting 72 hours post transfection. (B) HUVECs were transfected with indicated siRNA or combination. At 24 hours cells were switched to serum-free medium for 16 hours followed by replacement with either serum-free medium (−) or complete growth medium (+). Cells were analyzed for BrdU incorporation and data are expressed as average percentage positive for incorporation from triplicate determinations. Error bars represent ± s.e.m. Identical results were seen in a separate experiment.
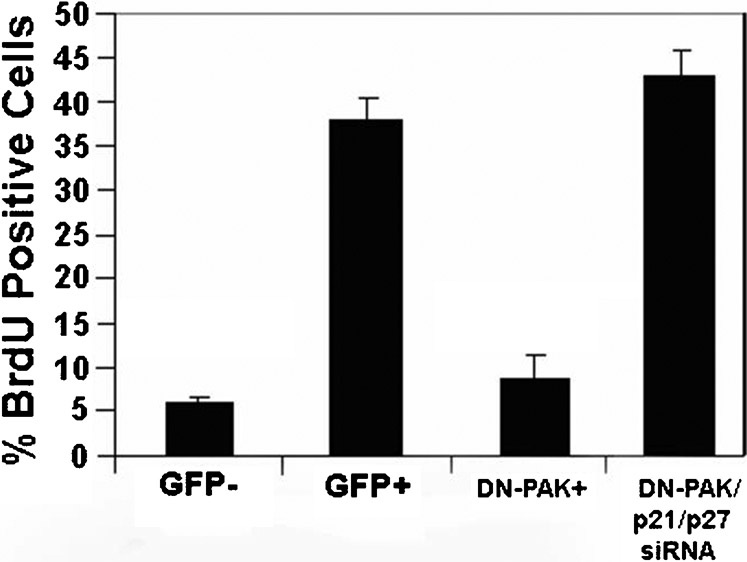
Previous studies have implicated FAK signaling as an important regulator of Rac activation in other cell types (Choma et al., 2007; Schaller, 2010) and Rac has been reported to regulate endothelial cell proliferation (Mettouchi et al., 2001), migration and capillary morphogenesis (Soga et al., 2001; Connolly et al., 2002). Given the established relationship between FAK and Rac and the pleitrophic role of Rac in endothelial cell biology, we sought to determine the requirements for FAK in Rac activation in endothelial cells. As shown in Fig. 6, expression of FRNK, FAKY397F, or pre-treatment with a FAK-kinase inhibitor, all result in an attenuation of Rac activation induced by complete growth medium. In contrast, the knockdown of FAK expression did not affect the mitogen-induced activation of Rac. These data parallel the results we observed on proliferation and suggest that deficient Rac activation could account for the inhibitory effects on proliferation and migration in response to expression of FRNK, FAKY397F and FAK inhibitors. To confirm a requirement of Rac signaling for endothelial cell proliferation under our conditions, we expressed dominant negative RacN17. As predicted, expression of RacN17 resulted in inhibition of cellular proliferation (Fig. 7A). Moreover, the nature of the cell cycle arrest is phenotypically identical to that we have previously published following expression of FRNK and FAKY397F (Bryant et al., 2006). This includes no changes in ERK phosphorylation and cyclin D accumulation but rather a diminished induction of SKP2 and corresponding stabilization of p27 and p21 levels (Fig. 7B). Similar results have also been seen in smooth muscle cells whereby both FAK and Rac alter levels of SKP and associated CDKI levels (Bond et al., 2004; Bond et al., 2008). As an alternative mechanism for inhibiting Rac, that would not interfere with GTPase cycling, we expressed dominant negative PAK by adenovirus. This also resulted in cell cycle arrest mediated by upregulation of CDKIs that was corrected by knockdown of p21 and p27 as we have previously published for FRNK (Bryant et al., 2006) (supplementary material Fig. S1). Taken together, these data argue that the inhibitory effects on cellular proliferation seen following inhibition of 397 phosphorylation by FRNK, FAKY397F, or PF573228, are the result of impaired signaling through Rac, despite the finding that FAK is not physically required for the activation of Rac. Given the documented role of Rac in cellular migration, it seems likely that this same mechanism also plays a part in the inhibition of migration observed following decreased FAK397 phosphorylation. While we have documented compromised Rac activation and an apparent requirement of Rac in proliferation response, we cannot discount other Rho-family GTPases, particularly CDC42. This GTPase can also be inhibited by the DN-PAK construct we employed and interestingly shares significant phenotypic overlap in the modulation of in vivo angiogenesis (Hoang et al., 2011a; Hoang et al., 2011b).
Fig. 6. Loss of FAK397 phosphorylation, but not FAK protein expression, is associated with impaired activation of RAC.
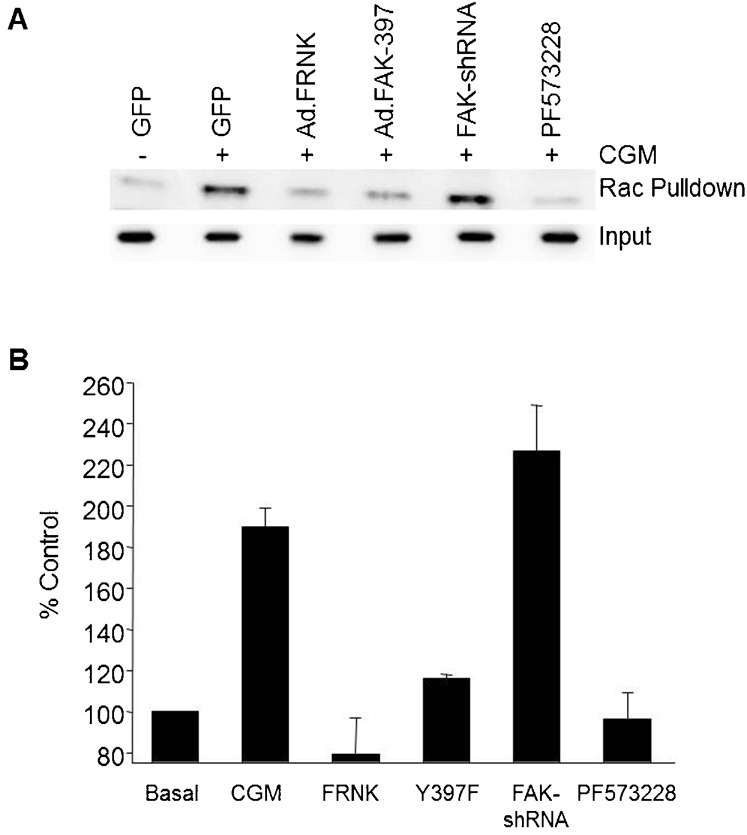
(A) Cells were manipulated by viral expression, chemical inhibition or shRNA mediated knockdown to manipulate FAK397 phosphorylation levels, as well as FAK expression similar to Fig. 1. These cells were then serum starved, followed by stimulation with CGM treatment for 30 minutes. Cells were probed for the presence of active Rac using a GST-PAK affinity assay. Bound Rac was detected by western blotting with anti-Rac. Equivalent input was monitored using ERK2. (B) Quantification of Rac activation following CCD-based densitometric analysis of western blots. Data are normalized to basal values for each experiment and represented as the mean ± range of two independent experiments.
Fig. 7. Dominant negative Rac interferes with HUVEC proliferation and Cyclin-Dependent Kinase Inhibitor regulation.

(A) HUVECS were infected with adenoviruses coding for GFP or RacN17 and then tested for the ability to induce DNA synthesis in response to CGM using BrdU incorporation. (B) HUVECs expressing RacN17 were analyzed for alterations in cell signaling and cell cycle control proteins by western analysis as indicated. These proteins show identical changes in response to expression of FRNK (Bryant et al., 2006).
That the knockdown of FAK in human cells did not show appreciable effects on migration or proliferation was quite surprising. As FAK is required for normal mouse vascular development, as well as pathological angiogenesis, (Ilic et al., 2003; Shen et al., 2005; Braren et al., 2006; Lee et al., 2010; Tavora et al., 2010) we were interested to determine if human endothelial cells required FAK for complex angiogenic functions such as morphogenesis. We utilized an in vitro, co-culture assay which closely mimics in vivo angiogenesis, including the formation of patent lumens, formation of tight junctions, and deposition of basement membrane proteins (Donovan et al., 2001). We found that knockdown of FAK markedly interfered with normal vascular morphogenesis in this assay, leading to a nearly complete loss of cells by day 14 (Fig. 8A). These effects were specific for FAK, as replacement with a non-targeted sequence for FAK allowed the formation of stable vascular structures. In addition, we also observed complete inhibition of the formation of endothelial cell vascular structures following treatment with the FAK inhibitor PF573,228 (Fig. 8B). These data imply that this complex phenotype requires both the physical presence of FAK and an active kinase, consistent with observations in vivo (Lim et al., 2010). Following these cultures over time, it appeared that the cells lacking FAK showed poorer branching and elongation as well as a progressive loss of cells (supplementary material Fig. S2). These data serve to confirm a critical signaling requirement for the presence of FAK in human endothelial cells. Thus, while not a requirement for in vitro proliferation and migration, the absence of FAK in human endothelial cells significantly impacts vascular morphogenesis and survival, largely phenocopying results from embryonic mouse explants (Ilic et al., 2003; Braren et al., 2006). In addition given the similarity of the observed phenotype to those reported in vivo for developmental angiogenesis (Ilic et al., 2003; Lim et al., 2010) (an inability to extend, elongate and stabilize early sprouts) (supplementary material Fig. S2) this may be an excellent model to use an “erase and replace” strategy to probe the structure-function requirements of FAK in developmental angiogenesis.
Fig. 8. FAK expression is required for vascular morphogenesis.
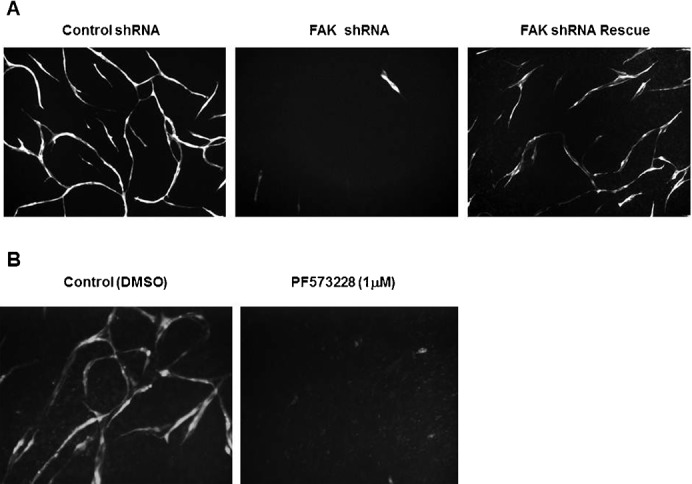
(A) HUVECs co-infected with lentiviruses that coded for: control shRNA and GFP; FAK-3′UTR shRNA and GFP; or FAK-3′UTRshRNA and FAK (Rescue) were co-cultured with primary human fibroblasts for 21 days. Endothelial vascular structures were visualized by monitoring RFP expression co-expressed with shRNA. Images were photographed at a final magnification of 100×. Data from the entire time course can be found in supplementary material Fig. S2. (B) HUVECs infected with GFP were plated as admixed co-cultures in the presence or absence of 1 µM PF573228. Cells were monitored and photographed at day 12 at a final magnification of 100×.
Our data reveal several distinct types of FAK signal transduction taking place in vascular endothelial cells. One is an absolute requirement for FAK in the processes of vascular morphogenesis and ultimately cellular survival in complex microenvironments. In contrast, FAK was not required for endothelial cell proliferation or migration (at least in two-dimensional culture), though these processes were readily inhibited by interfering with FAKY397 phosphorylation, either through expression of non-phosphorylated mutants or treatment with kinase inhibitors. Furthermore, our findings suggest that there is a fundamental difference in activation of Rac when FAK is in the cell but Y397 cannot be phosphorylated compared to when FAKY397 levels are reduced by a loss of FAK expression. These data argue that FAK Y397, and indeed FAK itself, is not required for Rac activation, yet it does seem to play an important regulatory role. We found no evidence of PYK2 based compensation under the conditions we employed, consistent with a requirement for an evolved compensation response as originally reported as well as with recent reports from cells derived from knockout mice. These data also cannot be explained by a partial or insufficient knockdown of FAK for several reasons: 1) we find equivalent or lower levels of FAK 397 phosphorylation in knockdown cells compared to FRNK and FAKY397F; and 2) experiments with the FAK inhibitors clearly showed a dose dependent effect on proliferation, yet even near complete knockdown of FAK resulted in no effect on proliferation. Taking into account all of our observations, we propose that FAK activation serves not just as a scaffold for the assembly of downstream signals but also as a basal repressor of Rac activation, and possibly other signaling events as well, that is relieved following FAK activation and Y397 phosphorylation. We have schematically depicted this concept in the model represented by Fig. 9. In this model, non-phosphorylated FAK localized to focal adhesions by the FAT-domain (required) represses Rac activation (directly or indirectly) in the basal state. Upon stimulation and phosphorylation of Y397, changes in FAK binding partners and/or conformational shifts in the FAK protein itself, permit subsequent Rac activation, normal proliferation and migration (Fig. 9A). In cells lacking FAK, the FAK induced repression signal is relieved allowing normal Rac activation, proliferation, and migration to take place upon stimulation, despite the absence of FAK and the associated ability to scaffold downstream signaling molecules. However, reintroduction of FRNK under these circumstances (Fig. 4) inhibits these processes, suggesting that this is indeed a physical and repressive function of FAK mediated via FAT domain interactions (proliferation and migration are not inhibited by a mutant which cannot target to the Focal adhesion). This is in contrast to an active role of FAK in recruiting Rac regulators, though our data do not rule out that this possibility could also occur. There are clearly additional signaling functions of FAK that actively regulate vascular morphogenesis and survival, and these appear to have an absolute requirement for the presence of FAK (Fig. 9B). In cells where FAK 397 cannot be phosphorylated following cellular activation because either FAK lacks a kinase domain, lacks a Y397 residue, or is chemically inhibited (Fig. 9C), the repression of Rac cannot be relieved. In addition, the normal recruitment of Src, subsequent phosphorylation and generation of downstream signaling complexes is also compromised, leading to an inhibition of proliferation and migration, as well as morphogenesis and survival. This model is consistent with existing data on FAK in endothelial cells and many other cell types. However, it is likely that FAK represents a truly multi-functional signaling protein (Schaller, 2010), with this being just one aspect of its functionality. Recent experiments using knock-in mice suggest FAK has signaling roles that are both kinase-dependent and kinase independent (Lim et al., 2010; Zhao et al., 2010). While Rac repression may constitute a kinase-dependent role in primary endothelial cells, it also seems plausible that in cells with elevated Src activity or other situations that might provide for alternative mechanisms to phosphorylate FAK397, this repression could be relieved independently of the kinase activity of FAK itself. These types of events may underscore some of the variability observed with respect to efficacy of FAK inhibitors in the treatment of tumors in preclinical models (Lechertier and Hodivala-Dilke, 2012).
Fig. 9. A model of FAK as a phospho-regulated repressor of Rac activation and endothelial cell function.

(A) Under resting conditions FAK is bound to focal adhesion proteins through FAT-dependent interactions but remain unphosphorylated. Under these conditions Rac activation (orange ellipse) is not efficiently coupled, neither are other signals requiring SRC docking, subsequent phosphorylation and the scaffolding of other signaling proteins (e.g., CAS, Grb7, PI3′-Kinase, Grb2 etc.) (purple triangle). Activation of FAK, results in phosphorylation of tyrosine 397, with subsequent changes in conformation of FAK itself and/or binding partners that permits Rac activation, proliferation and migration. Other signals essential for vascular morphogenesis and survival are also coordinately activated. (B) When FAK is silenced, the physical loss of FAK removes the repression allowing full activation of Rac (upon appropriate stimulation) as well as normal proliferation and migration, even in the absence of FAK397 phosphorylation. However, coordination and activation of the additional signals required for vascular morphogenesis, which are FAK-dependent, is impaired. (C) When FAK is present but is inhibited (whether induced by chemical inhibitors or expression of mutant constructs), the FAT domain of FAK targets FAK but tyrosine 397 phosphorylation is disrupted. Under these conditions the repressive signal cannot be relieved and there is no efficient Rac activation. The altered 397 phosphorylation also disrupts the activation of the FAK-dependent additional signals required for vascular morphogenesis and survival. The loss of focal adhesion targeting via the FAT domain renders these mutants ineffective. Expression of these mutants in cells knocked down for FAK results in repression of Rac activation, proliferation and migration.
A permissive role for FAK regulating proliferation and migration provides an essential adhesion checkpoint in cancer progression. Amplification of FAK signaling (and accompanying autoactivation) in cancer cells (Gabarra-Niecko et al., 2003; Mitra and Schlaepfer, 2006; Zhao and Guan, 2009), may provide an opportunity to bypass adhesion requirements and enhance metastatic potential, at least in part by relieving this repression. Similarly, our proposed model of FAK-mediated repression may provide an explanation for the paradoxical correlation linking low FAK expression levels with poor prognosis in several cancer types (Ayaki et al., 2001; Gabriel et al., 2006). The role of FAK in regulating tumor cell migration and metastasis, in addition to a critical role in angiogenesis, make FAK an emerging therapeutic target (Infusino and Jacobson, 2012; Ma, 2011). Our data suggest there may be important mechanistic differences between siRNA based approaches (Halder et al., 2005; Han et al., 2010) and chemical based inhibitors of FAK kinase activity (Parsons et al., 2008; Schultze and Fiedler, 2011). In addition, our data reveal a heretofore unappreciated repressive role of FAK, mediated through the FAT domain and regulated by Y397 phosphorylation. It remains to be seen if these differences can be exploited to clinical advantage.
Materials and Methods
Cell culture
HUVECs were purchased from several vendors and cultured as previously described (Bryant et al., 2006). Experiments were conducted in the presence of serum-free MCDB-131 and stimulation was performed with VEGF or complete growth medium (CGM).
Western blotting
We used the following anti-bodies: anti-FAK (Upstate, Lake Placid, NY), anti-FAK Y397, anti-ERK2 (Santa Cruz Biotechnology, Santa Cruz, CA). Western blotting conditions were performed as previously detailed (Meadows et al., 2001). Erk2 was routinely used to insure equal loading.
Measurement of DNA synthesis
HUVECs genetically manipulated as indicated were placed in serum-free conditions for 24 hours. After 24 hours, a mitogenic stimulus was added. Measurement of DNA synthesis by BrdU incorporation was performed as previously described (Meadows et al., 2004).
Recombinant adenovirus
The viruses described in this study were constructed using the AdEasy Adenoviral System (He et al., 1998). GFP-FRNK was generously provided by Dr. Allen Samarel (Loyola University Medical Center) (Heidkamp et al., 2002). Viruses were used at an MOI between 10–20.
Small-interfering RNA
Electroporation was used to transfect 125 ng of siRNA into 7.5×104 HUVECs resuspended in 75 µl of siRNA electroporation buffer (Ambion, Inc.). Cells were plated and recovered 24 hours in complete medium prior to any subsequent manipulation. An On-Target PLUS modified version of a FAK siRNA sequence selected from an unmasked Smartpool, was ordered from Dharmacon (cat NO. J-00316-05), FAK-si#1. We found modification of this siRNA (Jackson et al., 2006) is essential to avoid profound off-target effects. An additional FAK siRNA, was also synthesized with On-Target PLUS (GCGAUUAUAUGUUAGAGAUA), FAK-si#2 (Yano et al., 2004). A siRNA targeting GAPDH was used as a control (cat NO. D-001830-01-05).
Lentiviral expression
To stably knock down FAK, we used a sequence directed against the 3′-UTR (AGCATTGGGTCGGGAACTA) and converted to a hairpin for stable expression in the pFUGW lentiviral system (Lois et al., 2002) in which the GFP had been replaced with RFP. In some experiments, co-infections were performed with an additional virus generated from a FUGW-modified vector, whereby the GFP had been replaced with a FAK-IRES-GFP sequence. This sequence lacks the 3′-UTR and is not targeted by the 3′UTR directed shRNA. Cells were sterile sorted by FACS using RFP or RFP/GFP color selection to insure 100% infection of the desired populations, essentially as we have previously described (Bajaj et al., 2010). To confirm results, we also used two additional shRNA sequences directed against human FAK (D-1 and G-7) purchased from Open Biosystems in the GIPZ lentiviral vector.
Co-culture angiogenesis assay
HUVECs, modified with lentiviruses, were seeded with fibroblasts as previously described (Donovan et al., 2001; Bajaj et al., 2010). The medium was changed every third day and tube formation was monitored for at least 3 weeks. HUVECs were viewed with fluorescent microscopy to visualize infected cells and identical fields were photographed over time.
Supplementary Material
Acknowledgments
The authors thank C. Michael DiPersio for his critical reading of this manuscript. This study was supported by the National Institutes of Health Grant CA081419, the David E. Bryant Trust (to K.M.P.) and an individual American Heart Association Predoctoral Fellowship NO. 0515617T (to P.W.B.).
Footnotes
Competing interests: The authors declare that there are no competing interests.
References
- Avraham H. K., Lee T. H., Koh Y., Kim T. A., Jiang S., Sussman M., Samarel A. M., Avraham S. (2003). Vascular endothelial growth factor regulates focal adhesion assembly in human brain microvascular endothelial cells through activation of the focal adhesion kinase and related adhesion focal tyrosine kinase. J. Biol. Chem. 278, 36661–36668 10.1074/jbc.M301253200 [DOI] [PubMed] [Google Scholar]
- Ayaki M., Komatsu K., Mukai M., Murata K., Kameyama M., Ishiguro S., Miyoshi J., Tatsuta M., Nakamura H. (2001). Reduced expression of focal adhesion kinase in liver metastases compared with matched primary human colorectal adenocarcinomas. Clin. Cancer Res. 7, 3106–3112. [PubMed] [Google Scholar]
- Bajaj A., Zheng Q., Adam A., Vincent P., Pumiglia K. (2010). Activation of endothelial ras signaling bypasses senescence and causes abnormal vascular morphogenesis. Cancer Res. 70, 3803–3812 10.1158/0008-5472.CAN-09-2648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond M., Sala-Newby G. B., Newby A. C. (2004). Focal adhesion kinase (FAK)-dependent regulation of S-phase kinase-associated protein-2 (Skp-2) stability. A novel mechanism regulating smooth muscle cell proliferation. J. Biol. Chem. 279, 37304–37310 10.1074/jbc.M404307200 [DOI] [PubMed] [Google Scholar]
- Bond M., Wu Y. J., Sala-Newby G. B., Newby A. C. (2008). Rho GTPase, Rac1, regulates Skp2 levels, vascular smooth muscle cell proliferation, and intima formation in vitro and in vivo. Cardiovasc. Res. 80, 290–298 10.1093/cvr/cvn188 [DOI] [PubMed] [Google Scholar]
- Braren R., Hu H. Q., Kim Y. H., Beggs H. E., Reichardt L. F., Wang R. (2006). Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J. Cell Biol. 172, 151–162 10.1083/jcb.200506184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant P., Zheng Q., Pumiglia K. (2006). Focal adhesion kinase controls cellular levels of p27/Kip1 and p21/Cip1 through Skp2-dependent and -independent mechanisms. Mol. Cell. Biol. 26, 4201–4213 10.1128/MCB.01612-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choma D. P., Milano V., Pumiglia K. M., DiPersio C. M. (2007). Integrin alpha3beta1-dependent activation of FAK/Src regulates Rac1-mediated keratinocyte polarization on laminin-5. J. Invest. Dermatol. 127, 31–40 10.1038/sj.jid.5700505 [DOI] [PubMed] [Google Scholar]
- Connolly J. O., Simpson N., Hewlett L., Hall A. (2002). Rac regulates endothelial morphogenesis and capillary assembly. Mol. Biol. Cell 13, 2474–2485 10.1091/mbc.E02-01-0006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan D., Brown N. J., Bishop E. T., Lewis C. E. (2001). Comparison of three in vitro human ‘angiogenesis’ assays with capillaries formed in vivo. Angiogenesis 4, 113–121 10.1023/A:1012218401036 [DOI] [PubMed] [Google Scholar]
- Gabarra-Niecko V., Schaller M. D., Dunty J. M. (2003). FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev. 22, 359–374 10.1023/A:1023725029589 [DOI] [PubMed] [Google Scholar]
- Gabriel B., zur Hausen A., Stickeler E., Dietz C., Gitsch G., Fischer D. C., Bouda J., Tempfer C., Hasenburg A. (2006). Weak expression of focal adhesion kinase (pp125FAK) in patients with cervical cancer is associated with poor disease outcome. Clin. Cancer Res. 12, 2476–2483 10.1158/1078-0432.CCR-05-1867 [DOI] [PubMed] [Google Scholar]
- Gilmore A. P., Romer L. H. (1996). Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Mol. Biol. Cell 7, 1209–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder J., Landen C. N., Jr, Lutgendorf S. K., Li Y., Jennings N. B., Fan D., Nelkin G. M., Schmandt R., Schaller M. D., Sood A. K. (2005). Focal adhesion kinase silencing augments docetaxel-mediated apoptosis in ovarian cancer cells. Clin. Cancer Res. 11, 8829–8836 10.1158/1078-0432.CCR-05-1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H. D., Mangala L. S., Lee J. W., Shahzad M. M. K., Kim H. S., Shen D. Y., Nam E. J., Mora E. M., Stone R. L., Lu C. H. et al. (2010). Targeted gene silencing using RGD-labeled chitosan nanoparticles. Clin. Cancer Res. 16, 3910–3922 10.1158/1078-0432.CCR-10-0005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He T. C., Zhou S., da Costa L. T., Yu J., Kinzler K. W., Vogelstein B. (1998). A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA 95, 2509–2514 10.1073/pnas.95.5.2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidkamp M. C., Bayer A. L., Kalina J. A., Eble D. M., Samarel A. M. (2002). GFP-FRNK disrupts focal adhesions and induces anoikis in neonatal rat ventricular myocytes. Circ. Res. 90, 1282–1289 10.1161/01.RES.0000023201.41774.EA [DOI] [PubMed] [Google Scholar]
- Hoang M. V., Nagy J. A., Senger D. R. (2011a). Active Rac1 improves pathologic VEGF neovessel architecture and reduces vascular leak: mechanistic similarities with angiopoietin-1. Blood 117, 1751–1760 10.1182/blood-2010-05-286831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang M. V., Nagy J. A., Senger D. R. (2011b). Cdc42-mediated inhibition of GSK-3β improves angio-architecture and lumen formation during VEGF-driven pathological angiogenesis. Microvasc. Res. 81, 34–43 10.1016/j.mvr.2010.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holinstat M., Knezevic N., Broman M., Samarel A. M., Malik A. B., Mehta D. (2006). Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. J. Biol. Chem. 281, 2296–2305 10.1074/jbc.M511248200 [DOI] [PubMed] [Google Scholar]
- Ilić D., Furuta Y., Kanazawa S., Takeda N., Sobue K., Nakatsuji N., Nomura S., Fujimoto J., Okada M., Yamamoto T. et al. (1995). Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 377, 539–544 10.1038/377539a0 [DOI] [PubMed] [Google Scholar]
- Ilic D., Kovacic B., McDonagh S., Jin F., Baumbusch C., Gardner D. G., Damsky C. H. (2003). Focal adhesion kinase is required for blood vessel morphogenesis. Circ. Res. 92, 300–307 10.1161/01.RES.0000055016.36679.23 [DOI] [PubMed] [Google Scholar]
- Infusino G. A., Jacobson J. R. (2012). Endothelial FAK as a therapeutic target in disease. Microvasc. Res. 83, 89–96 10.1016/j.mvr.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson A. L., Burchard J., Leake D., Reynolds A., Schelter J., Guo J., Johnson J. M., Lim L., Karpilow J., Nichols K. et al. (2006). Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA 12, 1197–1205 10.1261/rna.30706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechertier T., Hodivala-Dilke K. (2012). Focal adhesion kinase and tumour angiogenesis. J. Pathol. 226, 404–412 10.1002/path.3018 [DOI] [PubMed] [Google Scholar]
- Lee J., Borboa A. K., Chun H. B., Baird A., Eliceiri B. P. (2010). Conditional deletion of the focal adhesion kinase FAK alters remodeling of the blood-brain barrier in glioma. Cancer Res. 70, 10131–10140 10.1158/0008-5472.CAN-10-2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S. T., Chen X. L., Tomar A., Miller N. L. G., Yoo J., Schlaepfer D. D. (2010). Knock-in mutation reveals an essential role for focal adhesion kinase activity in blood vessel morphogenesis and cell motility-polarity but not cell proliferation. J. Biol. Chem. 285, 21526–21536 10.1074/jbc.M110.129999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C., Hong E. J., Pease S., Brown E. J., Baltimore D. (2002). Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295, 868–872 10.1126/science.1067081 [DOI] [PubMed] [Google Scholar]
- Ma W. W. (2011). Development of focal adhesion kinase inhibitors in cancer therapy. Anticancer Agents Med. Chem. 11, 638–642. [DOI] [PubMed] [Google Scholar]
- Meadows K. N., Bryant P., Pumiglia K. (2001). Vascular endothelial growth factor induction of the angiogenic phenotype requires Ras activation. J. Biol. Chem. 276, 49289–49298 10.1074/jbc.M108069200 [DOI] [PubMed] [Google Scholar]
- Meadows K. N., Bryant P., Vincent P. A., Pumiglia K. M. (2004). Activated Ras induces a proangiogenic phenotype in primary endothelial cells. Oncogene 23, 192–200 10.1038/sj.onc.1206921 [DOI] [PubMed] [Google Scholar]
- Mettouchi A., Klein S., Guo W., Lopez-Lago M., Lemichez E., Westwick J. K., Giancotti F. G. (2001). Integrin-specific activation of Rac controls progression through the G(1) phase of the cell cycle. Mol. Cell 8, 115–127 10.1016/S1097-2765(01)00285-4 [DOI] [PubMed] [Google Scholar]
- Mitra S. K., Schlaepfer D. D. (2006). Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 18, 516–523 10.1016/j.ceb.2006.08.011 [DOI] [PubMed] [Google Scholar]
- Parsons J. T., Slack-Davis J., Tilghman R., Roberts W. G. (2008). Focal adhesion kinase: targeting adhesion signaling pathways for therapeutic intervention. Clin. Cancer Res. 14, 627–632 10.1158/1078-0432.CCR-07-2220 [DOI] [PubMed] [Google Scholar]
- Schaller M. D. (2010). Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J. Cell Sci. 123, 1007–1013 10.1242/jcs.045112 [DOI] [PubMed] [Google Scholar]
- Schultze A., Fiedler W. (2011). Clinical importance and potential use of small molecule inhibitors of focal adhesion kinase. Anticancer Agents Med. Chem. 11, 593–599. [DOI] [PubMed] [Google Scholar]
- Shen T. L., Park A. Y. J., Alcaraz A., Peng X., Jang I., Koni P., Flavell R. A., Gu H., Guan J. L. (2005). Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J. Cell Biol. 169, 941–952 10.1083/jcb.200411155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack-Davis J. K., Martin K. H., Tilghman R. W., Iwanicki M., Ung E. J., Autry C., Luzzio M. J., Cooper B., Kath J. C., Roberts W. G. et al. (2007). Cellular characterization of a novel focal adhesion kinase inhibitor. J. Biol. Chem. 282, 14845–14852 10.1074/jbc.M606695200 [DOI] [PubMed] [Google Scholar]
- Soga N., Namba N., McAllister S., Cornelius L., Teitelbaum S. L., Dowdy S. F., Kawamura J., Hruska K. A. (2001). Rho family GTPases regulate VEGF-stimulated endothelial cell motility. Exp. Cell Res. 269, 73–87 10.1006/excr.2001.5295 [DOI] [PubMed] [Google Scholar]
- Tavora B., Batista S., Reynolds L. E., Jadeja S., Robinson S., Kostourou V., Hart I., Fruttiger M., Parsons M., Hodivala-Dilke K. M. (2010). Endothelial FAK is required for tumour angiogenesis. EMBO Mol. Med. 2, 516–528 10.1002/emmm.201000106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis S. M., Lim S. T., Lutu-Fuga K. M., Barnes L. A., Chen X. L., Göthert J. R., Shen T. L., Guan J. L., Schlaepfer D. D., Cheresh D. A. (2008). Compensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell FAK. J. Cell Biol. 181, 43–50 10.1083/jcb.200710038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M. H., Guo M., Yuan S. Y., Granger H. J. (2003). Focal adhesion kinase mediates porcine venular hyperpermeability elicited by vascular endothelial growth factor. J. Physiol. 552, 691–699 10.1113/jphysiol.2003.048405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano H., Mazaki Y., Kurokawa K., Hanks S. K., Matsuda M., Sabe H. (2004). Roles played by a subset of integrin signaling molecules in cadherin-based cell-cell adhesion. J. Cell Biol. 166, 283–295 10.1083/jcb.200312013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Guan J. L. (2009). Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 28, 35–49 10.1007/s10555-008-9165-4 [DOI] [PubMed] [Google Scholar]
- Zhao J. H., Reiske H., Guan J. L. (1998). Regulation of the cell cycle by focal adhesion kinase. J. Cell Biol. 143, 1997–2008 10.1083/jcb.143.7.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Peng X., Sun S., Park A. Y., Guan J. L. (2010). Role of kinase-independent and -dependent functions of FAK in endothelial cell survival and barrier function during embryonic development. J. Cell Biol. 189, 955–965 10.1083/jcb.200912094 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}