

Summary
Invasion and metastasis are aggressive cancer phenotypes that are highly related to the ability of cancer cells to degrade extracellular matrix (ECM). At the cellular level, specialized actin-rich structures called invadopodia mediate focal matrix degradation by serving as exocytic sites for ECM-degrading proteinases. Adhesion signaling is likely to be a critical regulatory input to invadopodia, but the mechanism and location of such adhesion signaling events are poorly understood. Here, we report that adhesion rings surround invadopodia shortly after formation and correlate strongly with invadopodium activity on a cell-by-cell basis. By contrast, there was little correlation of focal adhesion number or size with cellular invadopodium activity. Prevention of adhesion ring formation by inhibition of RGD-binding integrins or knockdown (KD) of integrin-linked kinase (ILK) reduced the number of ECM-degrading invadopodia and reduced recruitment of IQGAP to invadopodium actin puncta. Furthermore, live cell imaging revealed that the rate of extracellular MT1-MMP accumulation at invadopodia was greatly reduced in both integrin-inhibited and ILK-KD cells. Conversely, KD of MT1-MMP reduced invadopodium activity and dynamics but not the number of adhesion-ringed invadopodia. These results suggest a model in which adhesion rings are recruited to invadopodia shortly after formation and promote invadopodium maturation by enhancing proteinase secretion. Since adhesion rings are a defining characteristic of podosomes, similar structures formed by normal cells, our data also suggest further similarities between invadopodia and podosomes.
Keywords: Invadopodia, Adhesion rings, MT1-MMP, ILK, Integrin, Invasion
Introduction
An increasing number of studies have shown that deregulated ECM-integrin signaling is a critical driver of cancer aggressiveness, both in human tumors and in model systems (Lahlou and Muller, 2011; Lehmann et al., 2011; Parekh and Weaver, 2009; Provenzano et al., 2008a; Provenzano et al., 2008b; Stewart et al., 2004). One of the most feared and lethal consequences of deregulated adhesion signaling that can lead to metastatic spread is invasive tumor cell behavior; however, the underlying cellular mechanisms are complex and include cross-regulation of cell-cell adhesion, migration and extracellular matrix (ECM) degradation (Guo and Giancotti, 2004). ECM degradation is likely to be particularly critical for invasion of epithelial tumors surrounded by dense crosslinked basement membranes, since they cannot be traversed without proteolytic activity (Parekh and Weaver, 2009).
A number of studies have shown that actin-rich protrusions known as invadopodia are key cancer cell structures for ECM degradation due to the focused concentration of matrix-degrading proteases (Linder et al., 2011; Weaver, 2006). Similar structures have been identified in 3-dimensional systems and in mouse models, and are dependent for their activity on key invadopodium molecules (Gligorijevic et al., 2012; Quintavalle et al., 2010; Rottiers et al., 2009; Wolf et al., 2007). In addition, normal cells such as macrophages and osteoclasts that need to remodel matrix form similar structures called podosomes (Linder et al., 2011; Murphy and Courtneidge, 2011). Matrix metalloproteinases (MMPs) are particularly important for the ECM-degrading activity of invadopodia and recent papers have shown that vesicle trafficking is important for delivery of MMPs to initial invadopodium actin puncta. The transmembrane proteinase MT1-MMP in particular is trafficked from a late endocytic compartment to invadopodia, dependent on VAMP7, IQGAP, and the exocyst complex (Sakurai-Yageta et al., 2008; Steffen et al., 2008). MT1-MMP is essential in many systems for invadopodium activity, traversal of basement membranes and in vivo tissue invasion (Artym et al., 2006; Hotary et al., 2006; Sabeh et al., 2004).
We recently found that both the chemical composition and rigidity of ECM could regulate the activity of cancer cell invadopodia (Alexander et al., 2008; Liu et al., 2010; Parekh et al., 2011). Similarly, integrins and downstream adhesion adaptor proteins have also been found to regulate invadopodia (Chan et al., 2009; Lucas et al., 2010; Nakahara et al., 1996) and invadopodium-like structures formed in src-transformed cells (Badowski et al., 2008; Destaing et al., 2010). Although focal adhesions are the cancer cell structures most highly associated with ECM-integrin signaling, we and others have identified Myosin IIA- (Alexander et al., 2008), integrin α5 (Mueller et al., 1999), and RhoC- (Bravo-Cordero et al., 2011) positive structures surrounding invadopodia that resemble the adhesion rings of podosomes (Gimona et al., 2008; Linder and Aepfelbacher, 2003), suggesting direct on-site signaling. In addition, Spinardi et al. described a “podosome-like” structure formed in bladder carcinoma cells, with adhesion rings surrounding actin cores within hemidesmosome plaques (Spinardi et al., 2004). However, the presence of adhesion rings surrounding podosomes has been suggested to be a defining difference between podosomes and invadopodia (Gimona et al., 2008; Linder, 2009; Linder et al., 2011). Furthermore, unlike many podosome-forming cells, for which podosomes are the only adhesion structures, cancer cells usually contain focal adhesions (FA) which may compete with invadopodia for adhesion signaling proteins (Chan et al., 2009) or induce necessary signals from a distance (Wang et al., 2005). Therefore, whether cancer cell invadopodia constitute bona fide adhesion structures is unclear, as is the exact role of adhesion proteins in the invadopodium lifecycle.
In this study, we followed up on the intriguing possibility that invadopodia form adhesion rings and investigated how adhesion signaling regulates invadopodium activity. We find that virtually all invadopodia do form adhesion rings and that the adhesion rings are formed shortly after actin assembly. Inhibition of adhesion signaling by targeting RGD-binding integrins and integrin-linked kinase (ILK) led to a selective reduction in adhesion-ringed and ECM-degrading invadopodia, and reduced the rate of MT1-MMP accumulation at invadopodia. In addition, integrin- and ILK-inhibited cells exhibited a reduction in the recruitment of the vesicular adaptor and ILK-binding protein IQGAP to invadopodia. These data suggest a model in which integrin-ILK signaling at invadopodium precursor structures leads to adhesion ring formation, IQGAP recruitment and capture of MT1-MMP-containing vesicles to produce fully mature ECM-degrading invadopodia.
Results
Podosome-like adhesion rings form around invadopodia and correlate with invadopodium activity
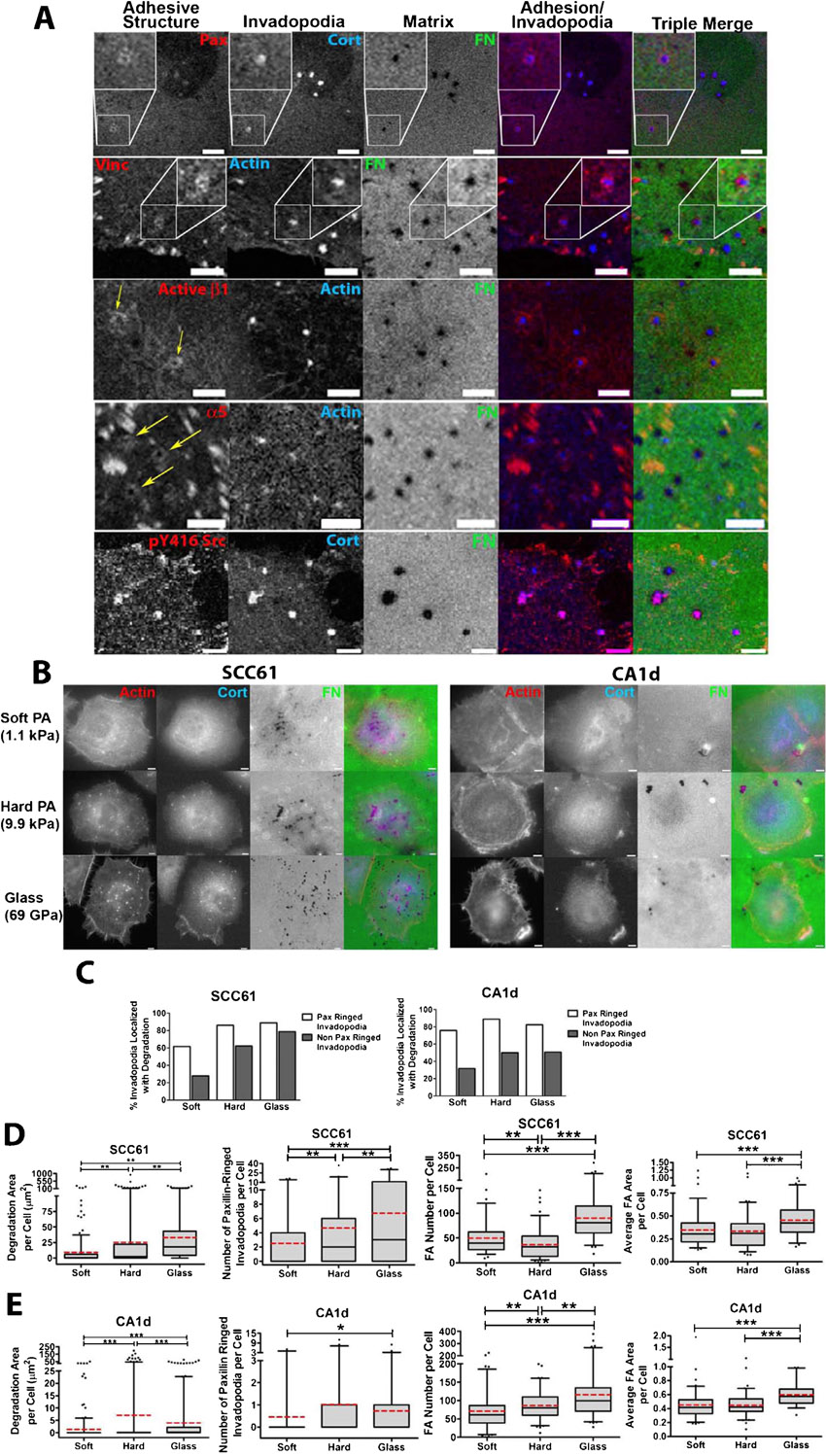
To understand how adhesion signaling associated with either FA or invadopodia regulates invadopodium activity, we began by immunostaining invadopodium-forming cancer cells for the FA markers paxillin or vinculin as well as the invadopodium markers cortactin or actin. CA1d breast cancer and SCC61 head and neck squamous cell carcinoma (HNSCC) cells have been previously described and were plated on invadopodium substrates coated with crosslinked 1% gelatin overlaid with FITC-fibronectin (FN) (Alexander et al., 2008; Clark et al., 2007). Interestingly, visualization by confocal microscopy revealed that paxillin and vinculin not only localized to FA but also to podosome-like adhesion rings surrounding actively degrading cortactin- or actin-containing invadopodia (Fig. 1A,B; supplementary material Fig. S1). Due to the high cytoplasmic levels of these adhesion proteins, these rings were not visible in wide-field epifluorescent images. Additional adhesion markers were also localized to either the rings (activated β1 integrin, α5 integrin), or invadopodium puncta (activated Src) (supplementary material Fig. S1).
Fig. 1. Adhesion ring localization at invadopodia correlates with invadopodium-associated extracellular matrix degradation.

(A) Confocal images of CA1d cells cultured on 1% crosslinked gelatin/FITC-FN (“Matrix”, green in triple merge) and immunostained for vinculin (Vinc) or paxillin (Pax) to mark adhesion structures (red in merges) and actin or cortactin (Cort) to mark invadopodia (blue in merges). Example focal adhesions (black arrows) and adhesion rings surrounding invadopodia (white arrows) are indicated. Dark areas in the matrix are where the FITC-FN/gelatin was degraded. Scale bars = 5 µm. (B) Zooms of boxed areas in A. Scale bars = 5 µm. (C) Percentage of adhesion ringed- and non-adhesion ringed-invadopodia colocalized with degradation of the underlying matrix in CA1d or SCC61 cells stained for paxillin and cortactin. Data are compiled from cells plated on soft PA, hard PA and glass conditions. Data separated by condition are shown in supplementary material Fig. S1. n≥180 cells per condition from 3 independent experiments. (D) Spearman correlation of the invadopodium ring or focal adhesion characteristics of an individual cell with the degradation area of FITC-FN underneath the same cell for CA1d or SCC61 cells plated on the noted underlying surfaces and stained for paxillin and cortactin. n≥60 cells per condition from 3 independent experiments. *P<0.05; **P<0.01; ***P<0.001 vs. no correlation.
In order to better understand the relationship between adhesion rings, FA, and invadopodium activity, we performed a correlative analysis of invadopodium-associated ECM degradation with the number of adhesion-ringed invadopodia or FA. To provide a richer dataset for analysis and because we previously found that cells modulate invadopodium activity according to substrate rigidity, we chose 3 rigidity conditions for the analysis and cultured CA1d or SCC61 cells on FITC-FN/1% gelatin overlaying polyacrylamide (PA) or glass invadopodium substrates of defined rigidity (Soft PA = 1 kPa, Hard PA = 10 kPa, glass = 1 GPa) as previously described (Alexander et al., 2008; Parekh et al., 2011). After 18 hours, the cultures were fixed and immunostained for cortactin and paxillin. Cortactin-positive invadopodium puncta have a characteristic morphology and size ≥1 µm (Clark et al., 2007), and were analyzed for association with ECM degradation and number per cell. FA number and size were analyzed from paxillin-positive structures at the periphery of the cell, and invadopodium-associated adhesion rings were identified manually (see Materials and Methods for more details, and Fig. 1 and supplementary material Fig. S1 for ringed invadopodium images). Overall, analysis across all conditions revealed that a higher percentage of adhesion-ringed invadopodium puncta were associated with degraded ECM than were non-ringed invadopodia (Fig. 1C; supplementary material Fig. S1C), suggesting that adhesion rings may promote invadopodium-associated degradation. We also correlated the number of FA and invadopodium ring structures per cell with the amount of degradation per cell. Interestingly, the number of paxillin-ringed invadopodia peaked on the same rigidity that elicited the highest invadopodium activity per cell, on the hard PA gel for the CA1d cells and on the glass surface for the SCC61 cells (supplementary material Fig. S1D,E). Using a Spearman's rank correlation to test the statistical dependence between two variables, we found that the number of adhesion-ringed invadopodia per cell significantly and positively correlated with ECM degradation area per cell on a cell-by-cell basis (Fig. 1D). The number of non-adhesion-ringed invadopodia also significantly correlated with ECM degradation per cell but to a lesser extent than adhesion-ringed invadopodia. By contrast, except for SCC61 cells plated on glass substrates, neither the number nor size of FA correlated with the degradation area per cell on a cell-by-cell basis (Fig. 1D; supplementary material Fig. S1D,E). We also found no relationship between FA dynamics and ECM degradation (live cell imaging, data not shown). Finally, we did not observe ECM degradation colocalized with FA or in streak-like FA-type distribution (Wang and McNiven, 2012). Altogether, these correlative data indicate that adhesion ringed-invadopodia are highly associated with ECM degradation.
Adhesion rings form shortly after invadopodium formation
Since SCC61 cells cultured on the glass surface degraded the most ECM and formed the most adhesion-ringed invadopodia (supplementary material Fig. S1D,E), this condition was used for subsequent mechanistic experiments to identify the role of adhesion rings in invadopodium function. Current models of invadopodium stages include early initiation of actin polymerization, followed shortly by recruitment of matrix metalloproteinases (MMPs) such as MT1-MMP and subsequent ECM degradation (Artym et al., 2006; Oser et al., 2009). Adhesion rings might be recruited early, before actin polymerization to trigger initiation of invadopodia, or alternatively, later, to promote persistence of invadopodia and/or recruitment of MMPs. To determine the real-time dynamics of adhesion rings forming at invadopodia, SCC61 cells stably expressing GFP-paxillin were transiently transfected with td-Tomato-F-Tractin (Tom-Tractin) to mark actin-positive invadopodia (Johnson and Schell, 2009; Laukaitis et al., 2001). These cells were then cultured overnight in medium without serum on MatTek dishes coated with 1% crosslinked gelatin/unlabeled FN (50 µg/ml). The next day, complete invadopodium medium (L-15+10% FBS, 5% Nu-serum, and 100 ng/ml EGF) was added to stimulate invadopodium formation and the basal cell surface was immediately imaged every 15 seconds for 1 hour by confocal microscopy (Fig. 2A; supplementary material Movie 1). In general, adhesion rings formed quickly after the formation of the Tom-Tractin-positive actin puncta, with respective mean and median times of 2.6 and 2 minutes after formation of the invadopodia (Fig. 2B). In addition, ∼90% of Tom-Tractin-positive invadopodium puncta recruited an adhesion ring over the lifetimes observed in the 1 hour movies (Fig. 2C). Finally, as has been previously noted (Bravo-Cordero et al., 2011), we frequently saw an oscillation of actin intensity at invadopodium puncta. These actin oscillations were paralled by ring oscillations and the recovery of GFP-paxillin intensity was generally slightly delayed in onset with respect to the Tom-Tractin intensity recovery (Fig. 2D kymograph). Occasionally, adhesion rings were lost from the invadopodia and did not recover (Fig. 2E kymograph) or arrived late (Fig. 2B), which we speculate may explain why many invadopodia in fixed analyses did not have surrounding rings. These data indicate that adhesion rings are typically recruited shortly after invadopodium initiation and suggest that they may contribute to the stability of invadopodia and/or recruitment of proteinases.
Fig. 2. Adhesion rings form following actin polymerization at invadopodia.

(A) Zoomed images of a single invadopodium over time from time-lapse confocal imaging of SCC61 cells expressing GFP-Paxillin (green in merge) and Tom-Tractin (Actin, red in merge). Scale bar = 3 µm. (B) Histogram showing the time between actin puncta formation and paxillin adhesion ring formation in live cell images from A; supplementary material Movie 1. (C) Quantification of the percentage of invadopodia with GFP-paxillin ring localization at any time during the movie. Data are mean and SEM. (D) Kymograph showing the lifetime of a typical invadopodium from time-lapse imaging in A. Oscillations are observed in both actin and paxillin signals. While both disappear concurrently, paxillin localization occurs after actin appearance at each oscillation. (E) Kymograph representing a subset of invadopodia from time-lapse imaging in A. For these invadopodia, the paxillin ring disappears prior to invadopodium disassembly. Time scale bars for D,E = 5 minutes.
Integrin activity is critical for adhesion ring formation and invadopodium activity
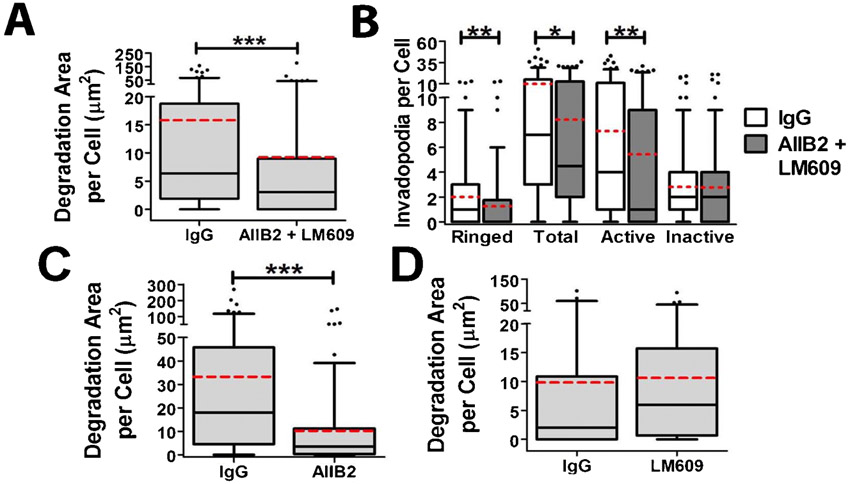
To directly test the role of adhesion proteins in invadopodium formation, adhesion ring formation, and invadopodium-associated ECM degradation, we blocked ligand binding of multiple integrin classes by incubating SCC61 cells with a peptide that blocks the binding of RGD-binding integrins to their ligands (gRGDsp) (Hayman et al., 1985) or with a combination of β1- and αvβ3-integrin blocking antibodies (AIIB2+LM609) (Cheresh, 1987; Hall et al., 1990) (Fig. 3; supplementary material Fig. S2). These integrin targets were chosen based on the ECM substrate used in our assay (FITC-FN) as well as their known contribution to podosome formation and activity (Destaing et al., 2010; Nakamura et al., 2007). We began with a standard immunofluorescence invadopodium assay and quantitated the ECM degradation area per cell. In addition, we quantitated the presence of various invadopodium features, including the total number of cortactin-positive characteristic invadopodium puncta, the presence of surrounding adhesion rings (“Ringed”), and whether or not cortactin puncta had associated ECM degradation (“Active” vs. “Inactive” invadopodia). Although it is possible that some of the “Inactive” invadopodia may represent other cortactin-positive structures, the size criteria of ≥1 µm diameter minimizes this possibility and use of this metric allows analysis of invadopodium maturation. Furthermore, we complement such analyses with live cell imaging (next paragraph). Compared to gRGEsp or IgG controls, integrin inhibition reduced the median area of ECM degradation per cell from 4 to 1.4 µm2 and 6.4 to 3.1 µm2, respectively (Fig. 3A,B; supplementary material Fig. S2). Integrin inhibition also significantly reduced the median number of adhesion-ringed invadopodia from 1 to 0 per cell, but had little effect on total invadopodium numbers (Fig. 3C; supplementary material Fig. S2). In addition, integrin inhibition with these reagents reduced the median number of active invadopodia from 3 to 1 per cell, while increasing or not affecting the number of inactive invadopodia (Fig. 3C; supplementary material Fig. S2). Of note, incubating cells with either β1 or αvβ3 blocking antibodies alone respectively inhibited or had no significant effect on the activity of invadopodia, suggesting that β1 may be the main integrin involved in our system (supplementary material Fig. S2C,D). Overall, these data indicate that integrins significantly contribute to adhesion ring formation and invadopodium-associated ECM degradation.
Fig. 3. Integrin activity is critical for adhesion ring formation and invadopodium activity.

(A–C) SCC61 cells were cultured on FITC-FN/1% gelatin (FN, green) and treated with RGD or RGE peptide control (250 µg/ml) for 16 hours then immunostained for paxillin (Pax, red) and cortactin (Cort, blue). (A) Confocal images. Zooms indicate typical invadopodia for condition. Scale bars = 5 µm. (B) Quantification of degradation area per cell. (C) Quantification of the number of paxillin-ringed cortactin-containing invadopodia (Ringed), total cortactin-positive invadopodia (Total), invadopodia localized with degradation of the FITC-FN matrix (Active), and invadopodia localized with intact FITC-FN (Inactive) n≥60 cells per condition from ≥3 independent experiments. (D–G) Live-cell imaging of SCC61 cells expressing Tom-Tractin, to mark invadopodia, and MT1-MMP-pHLuorin (MT1), to mark extracellular MT1-MMP was performed. (D) Quantification of invadopodium formation rate. (E) Representative kymographs of Tom-Tractin (Actin) and MT1 accumulation in invadopodia. Arrowheads indicate invadopodium formation time. Time scale bar = 5 minutes. (F) Invadopodium lifetime. (G) Quantification of MT1 accumulation at invadopodia over time, in which time zero is the appearance of the invadopodia and MT1 intensity at each time point is subtracted by the intensity at time zero. n≥11 cells, ≥300 invadopodia per condition from ≥3 independent experiments. Error bars on invadopodium formation graphs indicate SEM. For B,C,F, box and whiskers show respectively the 25–75th and 5–95th percentiles with the dotted red line indicating the mean and the black line indicating the median. *P<0.05; **P<0.01, ***P<0.001.
To directly determine if inhibition of integrins leads to a loss of proteinase recruitment, or conversely to alterations in invadopodium dynamics, we performed live cell imaging. To allow exclusive detection of extracellular MT1-MMP, SCC61 cells were engineered to stably express low levels of the critical invadopodium proteinase MT1-MMP tagged with superecliptic-pHLuorin-GFP (MT1-MMP-pHLuorin). The pHLuorin tag has greatly enhanced fluorescence at the extracellular pH of 7.4 and is quenched in the more acidic intracellular vesicles (Lizárraga et al., 2009; Miesenböck et al., 1998). After transient transfection with Tom-Tractin to visualize invadopodia (supplementary material Movie 2), dual color widefield images were captured on a Deltavision microscope at a frame rate of 1 per 15 seconds and computationally deconvolved to remove out of focus light. From these movies, the times of invadopodium formation and disassembly were determined as the IC50 points of the background-subtracted Tom-Tractin fluorescence intensity fit to a sigmoid curve at the initiation or disappearance of the invadopodia. Invadopodium lifetime was calculated as the time between formation and disassembly. Invadopodium formation rate was manually calculated as the number of invadopodia per cell per hour. MT1-MMP recruitment to invadopodia over time was measured as the background-subtracted fluorescence intensity of MT1-MMP-pHLuorin at each time point subtracted from the initial fluorescence intensity at the time of invadopodium formation. Using this approach, we found that inhibition of RGD-binding integrins with gRGDsp had no significant effect on the rate of invadopodium formation or on invadopodium lifetime compared to treatment with control gRGEsp peptide (Fig. 3D,F). However, there was a significant reduction in the accumulation of MT1-MMP at invadopodia. Thus, in control gRGEsp-treated cells, the MT1-MMP signal at Tom-Tractin-positive invadopodia increased steadily over the lifetime of individual invadopodia (Fig. 3E,G; supplementary material Movie 2). By contrast, in gRGDsp-treated cells, the overall MT1-MMP accumulation rate was significantly decreased, suggesting that the major effect of integrin inhibition was on invadopodium maturation (Fig. 3E,G; supplementary material Movie 3).
Integrin-linked kinase controls adhesion ring formation, invadopodium dynamics and activity
Our live and fixed imaging results with RGD blocking peptide indicated that integrins are involved in both adhesion ring formation and the accumulation of MT1-MMP at invadopodia. Since the majority of invadopodial MT1-MMP is derived from exocytosis of late endocytic vesicles (Steffen et al., 2008), we hypothesized that downstream signaling from integrins might facilitate this process. Although little is known about the relationship between integrin signaling and exocytosis (Balasubramanian et al., 2010; Gupton and Gertler, 2010; Wickström and Fässler, 2011; Wickström et al., 2010a), a potential downstream candidate is integrin-linked kinase (ILK), based on its reported function in linking caveolar vesicles to IQGAP at adhesions (Wickström et al., 2010a). Since cholesterol-rich caveolin-positive membranes and IQGAP have been separately shown to mediate proteinase trafficking to invadopodia (Caldieri et al., 2009; Sakurai-Yageta et al., 2008; Yamaguchi et al., 2009), ILK seemed a likely linker between integrins and MT1-MMP containing vesicles that might promote invadopodium degradative activity. To test that hypothesis, we knocked down ILK in SCC61 cells. Using two separate shRNA constructs in SCC61 cells, ILK expression was decreased by 58% and 82% in ILK-KD1 and -KD2 cells compared with a non-targeting control (Fig. 4B). When assessed by fixed cell immunofluorescence, ILK-KD cells exhibited a significant reduction in invadopodium-associated degradation, and the number of total, ringed, and active invadopodia per cell (Fig. 4A,C,D). Consistent with the hypothesized role in invadopodium maturation, there was no significant difference between control and ILK-KD cells in the number of inactive invadopodia per cell. Combining gRGDsp treatment with ILK-KD had no further effect on invadopodium-associated ECM degradation compared with either treatment alone, suggesting that they are likely to function in the same pathway (Fig. 4E).
Fig. 4. Integrin-linked kinase controls adhesion ring formation, invadopodium dynamics and activity.

(A–E) SCC61 cells stably expressing shRNA against ILK1 (sh1, sh2) or non-targeting control (NTC) shRNA were cultured on FITC-FN/1% gelatin for 16 hours. (A) Confocal images. Scale bars = 5 µm. (B) Western blot for ILK in total cell lysates from SCC61 cells. Numbers indicate ILK band intensity of the indicated cell line as a ratio of the NTC ILK level, after normalization to the β-actin loading control. (C) Degradation area per cell. (D) Invadopodium characteristics. (E) NTC and ILK-KD cells were further treated with RGE or RGD peptides and analyzed for degradation. Note that there is only a significant difference within a cell line for NTC. n≥60 cells per condition from ≥3 independent experiments. (F–I) Live-cell imaging of NTC or ILK sh1 SCC61 cells expressing Tom-Tractin, to mark invadopodia, and MT1-MMP-pHLuorin (MT1), to mark extracellular MT1-MMP was performed and analyzed for (F) invadopodium formation rate, (G) Invadopodium lifetime, (H,I) MT1 accumulation over time at invadopodia. Time scale bars = 5 minutes. n≥11 cells, ≥300 invadopodia per condition from ≥3 independent experiments. Error bars on invadopodium formation graphs indicate SEM. For C,D,E,G, box and whiskers show respectively the 25–75th and 5–95th percentiles with the dotted red line indicating the mean and the black line indicating the median. *P<0.05; **P<0.01, ***P<0.001.
We also performed live cell imaging in GFP-paxillin, Tom-Tractin-expressing control and ILK-KD cells to analyze the effect of ILK-KD on MT1-MMP accumulation and invadopodium dynamics. Unlike the effect of RGD peptide (Fig. 3), ILK-KD led to a decrease in invadopodium formation, from a median of 28 to 12 invadopodia per cell per hour, and in invadopodium lifetime from a median of 35 to 21 minutes. (Fig. 4F,G). Similar to the effect of integrin inhibition with gRGDsp peptide, KD of ILK1 led to a large reduction in the rate of MT1-MMP recruitment to invadopodia. We note that unlike with RGD-inhibited cells, the decrease in extracellular MT1-MMP accumulation only occurred after the first 20 minutes (Fig. 4H,I; supplementary material Movies 4, 5). However, overall these data indicate that integrin and ILK-mediated adhesion signaling is critical for accumulation of extracellular MT1-MMP at invadopodia. Since the accumulation was quantitated at already-formed invadopodia, the decreases in protease accumulation for both integrin- and ILK-inhibited cells were independent of any effects on invadopodium formation or lifetime.
Integrins and ILK recruit IQGAP to invadopodia
Based on our finding that integrins and ILK affect invadopodium activity and MT1-MMP recruitment and the known role of the ILK-binding partner IQGAP in capturing caveolar vesicles (Sakurai-Yageta et al., 2008; Wickström et al., 2010a), we decided to test whether IQGAP localization at invadopodia was affected by integrin-ILK signaling. 3-D confocal imaging of IQGAP localization to actin- and vinculin-stained invadopodia demonstrated that IQGAP is localized in an overlapping distribution with both the actin puncta and the vinculin ring (Fig. 5A, horizontal line scans). Vertical line scans through the center of invadopodia also show that IQGAP is more concentrated at the top of the actin-positive area (Fig. 5A). To determine whether IQGAP is recruited to invadopodia dependent on integrins and ILK, RGD-inhibited or ILK-KD SCC61 cells were cultured overnight on invadopodium substrates and fixed and stained for actin and IQGAP. IQGAP and actin intensities at invadopodia were quantitated from confocal images taken under identical laser and camera conditions for all samples and the ratio of IQGAP to actin intensity at each invadopodium was plotted. Consistent with a model in which integrins and ILK facilitate IQGAP recruitment to invadopodia, inhibition of RGD-binding integrins or ILK-KD led to a decrease in the intensity of IQGAP at invadopodia (Fig. 5B–E). The median ratio of IQGAP:actin intensity decreased from 0.57 to 0.49 with RGD treatment and from 0.49 to 0.34 and 0.37 with ILK shRNA #1 and #2, respectively, compared to controls.
Fig. 5. Localization of IQGAP to invadopodia is dependent on integrins and ILK.

(A) Confocal images of SCC61 cells cultured on unlabeled-FN/1% gelatin for 16 hours and immunostained for actin (red), IQGAP (blue) and vinculin (Vinc, green). Confocal Z-stacks of boxed invadopodia are shown. Graphs indicate fluorescent intensity (in arbitrary units) of each marker over the indicated line scan in the X-Y or X-Z dimensions. (B–E) SCC61 cells treated with RGD or control RGE peptide (250 µg/ml) (B,D) or stably expressing shRNA against ILK1 (sh1, sh2) or non-targeting control (NTC) shRNA (C,E) were cultured on FITC-FN/1% gelatin (FN, green) for 16 hours then immunostained for actin (red) and IQGAP (blue). (B,C) Confocal images. Zooms indicate typical invadopodia for condition. (D,E) Ratio of IQGAP to actin intensity at invadopodia. Box and whiskers show respectively the 25–75th and 5–95th percentiles with the dotted red line indicating the mean and the black line indicating the median. ***P<0.001. Scale bars = 5 µm.
MT1-MMP regulates ECM degradation and invadopodium dynamics, but not adhesion ring formation
Recruitment of MT1-MMP and other proteinases is thought to be a late stage in invadopodium formation; however, several studies have reported reduced numbers of invadopodia per cell in proteinase-inhibited cells (Artym et al., 2006; Clark et al., 2007; Steffen et al., 2008), suggesting feedback from proteolytic activity. To test whether MT1-MMP acts upstream or downstream of adhesion ring formation, SCC61 cells transduced with shRNA targeting MT1-MMP were analyzed for invadopodia and adhesion ring formation and ECM degrading ability in fixed cell assays (Fig. 6). Consistent with previous reports (Artym et al., 2006; Steffen et al., 2008), ECM degradation area per cell was virtually abolished with MT1-MMP knockdown (Fig. 6C). Furthermore, there was a 4-fold decrease in the number of active invadopodia per cell and a corresponding increase in inactive invadopodium numbers (Fig. 6D). There was no significant change in either the number of adhesion-ringed invadopodia or the number of total invadopodia. Live cell imaging further revealed that the rate of invadopodium formation was reduced in MT1-MMP-KD cells (Fig. 6E). There was also a small but significant decrease in the median lifetime of invadopodia in MT1-MMP-KD cells from 45.5 to 35.5 minutes (Fig. 6F), suggesting that there is indeed a positive feedback loop that results from MT1-MMP activity at invadopodia (Artym et al., 2006; Clark et al., 2007; Steffen et al., 2008). These data are consistent with a model in which adhesion rings form upstream of MT1-MMP accumulation to promote ECM degradation. MT1-MMP activity itself also has significant effects on invadopodium dynamics, both in the formation and stability phases.
Fig. 6. MT1-MMP is required for invadopodium-associated ECM degradation, but not for adhesion ring localization.

(A–D) SCC61 cells stably expressing shRNA constructs against MT1-MMP (MT1 sh1 or sh2) or shLacZ control oligo were cultured on FITC-FN/gelatin substrates for 16 hours and then fixed and immunostained for vinculin (Vinc, red in merges) and actin (blue in merges). (A) Confocal images. Zoomed images show representative invadopodia for the condition. Scale bars = 5 µm. (B) Western blot of total cell lysates. Numbers indicate the ratio of MT1-MMP in each cell line normalized to the GAPDH loading control and then to shLacZ control. (C) Quantification of degradation area per cell. (D) Quantification of types of invadopodia per cell n≥60 cells per condition from ≥3 independent experiments. (E,F) Invadopodium formation (E) and lifetime (F) quantification of live-cell images of SCC61 cells stably expressing shLacZ or MT1 sh1 and transiently transfected with Tom-Tractin to mark invadopodia. n≥12 cells, ≥180 invadopodia per condition from ≥3 independent experiments. Error bars on invadopodium formation graphs indicate SEM. For C,D,F, box and whiskers show respectively the 25–75th and 5–95th percentiles with the dotted red line indicating the mean and the black line indicating the median. *P<0.05; **P<0.01, ***P<0.001.
Discussion
In this study, we examined the role of adhesion signaling in the regulation of invadopodium formation and activity. We find that podosome-like adhesion rings surround invadopodia shortly after their formation and their presence is highly correlated with invadopodium activity. Integrins and ILK are critical for adhesion ring formation, localization of the vesicular adaptor protein IQGAP and the transmembrane proteinase MT1-MMP to invadopodia, and ECM degradation. Overall these data indicate that adhesion signaling is critical for invadopodium maturation.
Adhesion ring formation is not a distinguishing feature of podosomes
Invadopodia and podosomes are both actin-rich structures that can degrade underlying matrix. Despite many similarities, including common molecular constituents and pathways of activation, invadopodia are currently classified as distinct structures that form in cancer cells and have separate properties from podosomes such as longer lifetimes, protrusive behavior, and morphological classifications (Gimona et al., 2008; Murphy and Courtneidge, 2011). One major distinction has been the presence of adhesion rings surrounding actin puncta in podosomes (Gimona et al., 2008; Linder, 2009; Linder et al., 2011). Here, we showed that two separate invadopodium-producing cancer cell lines exhibit adhesion ring formation around invadopodial protrusions. These rings consist of multiple adhesion components such as paxillin, vinculin, and activated β1 integrin and occur at the basal cell surface by confocal microscopy, suggesting that the ring structures represent bonafide adherent structures. However, they were less prominent with respect to staining intensity and morphology than typical podosome structures formed in cells such as macrophages, perhaps due to competition with focal adhesions for components (Chan et al., 2009) or due to greater distribution of adhesion components into a soluble cytoplasmic pool in cancer cells. Interestingly, in src-transformed fibroblasts, β1 integrin was shown to be important for not only the organization but also the formation of “invadosome” structures, which have similarities to both podosomes and invadopodia and are directly induced by mutated src kinase (Destaing et al., 2010). Although in our cancer cell invadopodium system we do find some role for ILK in invadopodium formation, the major role that we observed for both integrins and ILK was a very specific effect on invadopodium adhesion ring formation and maturation. Similarly, in osteoclasts, despite a small effect on actin podosome puncta diameter, knockout of integrin β1, β2, and/or αv or kindlin3 led to severe defects in podosome organization and bone resorption (Schmidt et al., 2011). In addition, Badowski et al. examined the role of paxillin in invadosome formation by src-transformed cells and podosome formation by osteoclasts. In those systems, they found that loss or mutation of paxillin primarily affected podosome organization and ECM degradation (Badowski et al., 2008). Overall, our data suggest that adhesion ring formation is a common feature of both podosomes and invadopodia and indicate that a major function of those rings is to promote recruitment of proteinases and ECM degradation.
A new model of invadopodium stages
Altogether, our data are consistent with a model in which adhesion rings are assembled shortly after invadopodium actin puncta assembly and promote recruitment of proteinases to allow ECM degradation (Fig. 7). RGD-binding integrins and ILK are crucial components of this process as inhibition or knock-down reduces adhesion ring formation, MT1-MMP recruitment to invadopodia and ECM degradation. Furthermore, inhibiting integrins in ILK-KD cells had no further effect on ECM degradation, suggesting that integrins and ILK reside in the same pathway with respect to invadopodium-associated ECM degradation. We place MT1-MMP at a later stage than adhesion formation since KD of MT1-MMP had no effect on the number of adhesion-ringed invadopodia but inhibition of adhesion signaling led to decreased recruitment of MT1-MMP. After exocytosis, MT1-MMP and other proteases might also interact with integrins via a direct docking mechanism (Gálvez et al., 2002; Mueller et al., 1999) that could provide additional positive feedback to enhance invadopodium lifetime and/or ECM degradation.
Fig. 7. Model of invadopodium maturation.

Initial actin puncta appearance is followed by adhesion ring structure formation including integrins (α,β), ILK, paxillin (Pax) and vinculin (Vinc). Ring formation leads to enhanced MT1-MMP recruitment and ECM degradation by invadopodia.
Whereas integrin inhibition with gRGDsp did not affect invadopodium formation or lifetime, ILK-KD did, indicating that ILK may not function solely downstream of integrins for those activities. Although ILK is thought to primarily function as a downstream effector of integrins in complex with PINCH and parvin (Sakai et al., 2003; Stanchi et al., 2009; Wickström et al., 2010b), ILK can also be regulated by growth factor signaling (Ho and Dagnino, 2012; Serrano et al., 2012) and PI3K/PTEN (Wu and Dedhar, 2001). Since invadopodium formation can be activated by both growth factor and PI3K signaling (Yamaguchi et al., 2005; Yamaguchi et al., 2011), one possibility is that the regulation of invadopodium dynamics by ILK may occur downstream of growth factor rather than integrin signaling. Another possibility is that the inhibition of RGD-binding integrins with gRGDsp was less effective at blocking the integrin-ILK-IQGAP-MT1-MMP pathway than ILK-KD. Since knockdown of MT1-MMP itself can affect invadopodium dynamics (Fig. 6), a more complete block of the pathway with ILK-KD might lead to the difference that we noted for invadopodium dynamics.
Regulation of exocytosis by integrins and ILK
Integrins and adhesion signaling have recently been shown to affect diverse exocytic processes. Assembly and localization of the exocyst complex were shown to be regulated by integrin adhesion and paxillin through activation of the RalA GTPase (Balasubramanian et al., 2010; Spiczka and Yeaman, 2008). β1 Integrin-ECM interaction was also recently shown to promote exocytosis of lipid rafts by a mechanism that links ILK, the scaffold protein IQGAP1 and the formin mDia1 to capture of microtubules and subsequent vesicle delivery to the plasma membrane (Wickström and Fässler, 2011; Wickström et al., 2010a) . Furthermore, integrin adhesion to collagen in 3D was shown to promote polarized trafficking of MT1-MMP (Bravo-Cordero et al., 2007). Of note, several microtubule motors have been shown to regulate exocytosis of MT1-MMP at podosomes (Cornfine et al., 2011; Wiesner et al., 2010). Furthermore, the exocyst complex localizes to invadopodia through IQGAP1 and is important for MT1-MMP localization to invadopodia (Sakurai-Yageta et al., 2008). In light of these data and our finding that IQGAP localization to invadopodia is reduced in integrin- and ILK-KD cells (Fig. 5), we speculate that adhesion rings surrounding invadopodia may promote targeted MT1-MMP recruitment to invadopodia through capture of vesicles via a similar ILK-IQGAP-mediated mechanism (Fig. 7).
Deregulation of β1 integrin and ILK signaling has been implicated in a wide range of aggressive tumor behaviors, including cancer invasion (Dai et al., 2003; Edwards et al., 2008; Guo and Giancotti, 2004; Lu et al., 2008; Poincloux et al., 2011; Sawai et al., 2006). With regard to ECM degradation and cancer invasion, our data suggest that at least one key mechanism may be via integrin-ILK-mediated regulation of protease secretion at invadopodia. Interestingly, ILK-regulated secretion has also been implicated in tumor angiogenesis (Edwards et al., 2008) and caveolar formation (Wickström et al., 2010a). Future studies should address how general is the secretory role of integrin-ILK signaling and to what extent invadopodia represent “hotspot” secretion sites for lipid raft-carried molecules.
Materials and Methods
Reagents
Antibodies: Cortactin, 4F11 (Upstate Biotechnology, Lake Placid, NY), Paxillin, Y113 (Abcam, Cambridge, MA), Vinculin, hVin-1 (Sigma, St. Louis, MO), IQGAP, H-109 (Santa Cruz, Santa Cruz, CA), Alexa Fluor 546 or 633 phalloidin (Invitrogen, Grand Island, NY). Peptides: Gly-Arg-Gly-Asp-Ser-Pro (GRGDSP, Sigma), and Gly-Arg-Gly-Glu-Ser-Pro (GRGESP, American Peptide Co., Sunnyvale, CA). tdTomato-F-tractin (Tom-Tract) was a gift of Dr. Robert Fischer at NHLBI and was created by cloning the 9-52 stretch of the F-actin binding protein ITPKA (Johnson and Schell, 2009) into pCMV-tdTomato (Clontech, Mountain View, CA).
Cell culture and stable cell lines
MCF10A-CA1d breast cancer cells were obtained from Dr. Fred Miller (Karmanos Institute, MI) and have been previously described (Santner et al., 2001). Cells were cultured in DMEM/F12 supplemented with 5% horse serum (HyClone, Thermo Scientific, Lafayette, CO), 0.1 µg/ml cholera toxin (Calbiochem, Merck KGaA, Darmstadt, Germany), 10 µg/ml insulin (Gibco, Invitrogen), 0.5 µg/ml hydrocortisone (Sigma), and 100 ng/ml EGF (Invitrogen) at 37°C with constant humidity. SCC61 HNSCC cells were previously described (Clark et al., 2007; Weichselbaum et al., 1986) and were cultured in DMEM supplemented with 20% fetal bovine serum (HyClone) and 0.4 mg/ml hydrocortisone at 37°C.
Tom-Tract was transiently transfected into SCC61 cells at a 1:1 µg DNA:µl Lipofectamine 2000 (Invitrogen) ratio for 6 hours. GFP-paxillin was cloned from pEGFP-N3-Paxillin, a gift of Dr. Donna Webb (Vanderbilt), into the pENTR-TOPO vector and recombined into the LZRS-GW-Neo retroviral vector, a gift from Dr. Al Reynolds (Vanderbilt), using the Gateway recombination system (Invitrogen). MT1-MMP-pHLuorin (MT1-pHLuor) was a gift from Dr. Philippe Chavrier (Institut Curie, Paris). MT1-pHLuor was PCR-cloned into the pLenti6 lentiviral vector (Invitrogen). Control (non-targeting shRNA (NTC), Addgene) and two shRNA constructs targeting ILK (sh1- 5′-CUGAACAAACACUCUGGCAUU-3′ (Thermo Scientific), sh2- 5′-GCAAUGACAUUGUCGUGAAGG-3′ (Sigma)) were obtained in the lentiviral vector pLKO.1-puro. MT1-MMP targeting shRNA (MT1 sh1- 5′-CAGCGATGAAGTCTTCACTTA -3′, sh2- 5′-CAGCCTCTCACTACTCTTTC -3′) or shLacZ as a control were subcloned into the lentiviral vector pLenti-BlockIt (Invitrogen). Retrovirus and lentivirus were produced using Phoenix (Dr. Garry Nolan, Stanford) or 293FT cells, respectively, and SCC61 cells were incubated with viral supernatant overnight and later selected with the appropriate markers.
Fixed cell microscopy and invadopodium analysis
The ECM degradation assay has been previously described (Bowden et al., 2001; Clark et al., 2007). Briefly, MatTek culture dishes (MatTek Corp., Ashland, MA) were coated with a thin layer of 1% gelatin that was crosslinked with 0.5% glutaraldehyde prior to addition of 50 µg/ml FITC-conjugated fibronectin (FITC-FN). Cells were cultured in a 1:1 ratio of DMEM:RPMI-1640 (or L-15 for live cell imaging) with 5% NuSerum (Gibco), 10% FBS, and 100 ng/ml EGF for 20 hours before fixation with 4% paraformaldehyde and immunostaining. Polyacrylamide substrate preparation was also previously described (Alexander et al., 2008; Parekh et al., 2011).
Widefield fluorescent images were captured on a Nikon Eclipse TE2000-E microscope with a 40× Plan Fluor oil immersion objective lens. Degradation area per cell was determined using MetaMorph software (Molecular Devices, Sunnyvale, CA) by tracing a region around the cellular actin footprint and measuring the thresholded area of dark spots in the FITC-FN in that region.
Fixed cell confocal images were taken using a Zeiss LSM 510 microscope with a Plan Apo 63× oil immersion objective lens with Argon-488 nm, HeNe-543 nm, and HeNe-633 nm lasers. Images were taken at a scan speed of 8 and 4 scans were averaged per acquisition. Degradation area per cell was determined as above. Invadopodia were identified as morphologically characteristic round actin or cortactin puncta ≥1 µm in diameter found at the bottom of the cell (Clark et al., 2007) and quantitated from confocal images. If ECM degradation was associated with an invadopodium, it was classified as “active”; otherwise it was classified as “inactive”. The presence of an adhesion ring was defined as paxillin or vinculin fluorescence above background surrounding the invadopodia. Invadopodium metrics (adhesion ringed, non-ringed, total, active and inactive invadopodia) were manually counted. For focal adhesion analyses on paxillin-immunostained cells, the central region of the cell with high background cytoplasmic fluorescence was excluded and the remaining periphery was thresholded. The number and area of peripheral focal adhesions per cell was then determined with the “Analyze Particles” function in ImageJ (NIH).
For IQGAP localization analyses, SCC61 cells were plated on 1% gelatin/FITC-FN and fixed and stained for actin and IQGAP. Images were obtained with the Zeiss LSM 510 confocal as described above. The pinhole was opened slightly (1.4 µm optical slice) to account for the slightly higher Z-axis localization of IQGAP at invadopodia. Equal laser power, digital gain and offset settings were used in each experiment and actin and IQGAP signals were within the dynamic range. Invadopodia were traced using the actin immunostaining and average intensity of the actin and IQGAP signals within the traced regions was obtained using ImageJ.
Live cell imaging
Adhesion ring imaging
For adhesion ring imaging, live cell confocal images were taken using a Zeiss LSM 710 microscope with a Plan Apo 63× oil immersion objective lens with Argon-488 nm and HeNe-561 nm lasers. Time-lapse images were obtained every 15 seconds at a scan speed of 8 with 4 images averaged per acquisition. Time of invadopodia and paxillin ring appearance was manually determined.
Imaging of Tom-Tractin alone or with MT1-pHLuorin
Imaging of Tom-Tractin alone or with MT1-pHLuorin was performed using an Applied Precision DeltaVision Core microscope with a Plan Apo 60× oil immersion objective lens. SCC61 cells expressing Tom-Tractin and/or MT1-pHLuorin were seeded in the ECM degradation assay on 1% gelatin/unlabeled-FN and placed in a heated microscope chamber at 37°C 2 hours prior to imaging. When used, RGE or RGD peptides were added at the time of cell seeding. Images were then obtained every 15 seconds and processed with 10 iterations of constrained iterative deconvolution using Softworx 5.0 (Applied Precision, Issaquah, WA). For live-cell invadopodium analyses, the actin-containing invadopodia were traced at their largest area and fluorescence intensity of Tom-Tract and MT1-pHLuorin was determined at each time point using Metamorph. The Tom-Tract and MT1-pHLuorin signal at each time point was background corrected at each invadopodium with an identical traced region placed in a similar area of the cell but lacking an invadopodium. With these data, invadopodium formation rate, lifetime, and MT1-pHLuorin accumulation at invadopodia were determined as described in the manuscript.
Statistics
Data sets were tested for normality using the Kolmogorov-Smirnov normality test in GraphPad InStat3 (GraphPad Software, La Jolla, CA). Groups of non-normal data were compared using the Mann-Whitney rank sum test, while parametric data were compared with an unpaired t-test. Non-normal data were plotted as box and whiskers, which show respectively the 25–75th and 5–95th percentiles with the dotted red line indicating the mean and the black line indicating the median. For the Spearman rank analysis performed in Fig. 1, comparisons between degradation area per cell and each other data set were performed in GraphPad InStat3. A perfect positive correlation = 1, no correlation = 0 and a perfect negative correlation = −1, and p values represent a correlation significantly different than no correlation.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health [R01GM075126, U01CA143069 to A.M.W., and CA68485, DK20593, DK58404, HD15052, DK59637 and EY08126 to the VUMC Cell Imaging Shared Resource, and the VICC support grant for additional Core facility support] and the Japan Society for the Promotion of Science [Travel Grant to D.H.].
Footnotes
Competing interests: The authors declare that there are no competing interests.
References
- Alexander N. R., Branch K. M., Parekh A., Clark E. S., Iwueke I. C., Guelcher S. A., Weaver A. M. (2008). Extracellular matrix rigidity promotes invadopodium activity. Curr. Biol. 18, 1295–1299 10.1016/j.cub.2008.07.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artym V. V., Zhang Y., Seillier-Moiseiwitsch F., Yamada K. M., Mueller S. C. (2006). Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodium formation and function. Cancer Res. 66, 3034–3043 10.1158/0008-5472.CAN-05-2177 [DOI] [PubMed] [Google Scholar]
- Badowski C., Pawlak G., Grichine A., Chabadel A., Oddou C., Jurdic P., Pfaff M., Albigès-Rizo C., Block M. R. (2008). Paxillin phosphorylation controls invadopodia/podosomes spatiotemporal organization. Mol. Biol. Cell 19, 633–645 10.1091/mbc.E06-01-0088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian N., Meier J. A., Scott D. W., Norambuena A., White M. A., Schwartz M. A. (2010). RalA-exocyst complex regulates integrin-dependent membrane raft exocytosis and growth signaling. Curr. Biol. 20, 75–79 10.1016/j.cub.2009.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden E. T., Coopman P. J., Mueller S. C. (2001). Invadopodia: unique methods for measurement of extracellular matrix degradation in vitro. Methods Cell Biol. 63, 613–627 10.1016/S0091-679X(01)63033-4 [DOI] [PubMed] [Google Scholar]
- Bravo-Cordero J. J., Marrero-Diaz R., Megías D., Genís L., García-Grande A., García M. A., Arroyo A. G., Montoya M. C. (2007). MT1-MMP proinvasive activity is regulated by a novel Rab8-dependent exocytic pathway. EMBO J. 26, 1499–1510 10.1038/sj.emboj.7601606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo-Cordero J. J., Oser M., Chen X., Eddy R., Hodgson L., Condeelis J. (2011). A novel spatiotemporal RhoC activation pathway locally regulates cofilin activity at invadopodia. Curr. Biol. 21, 635–644 10.1016/j.cub.2011.03.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldieri G., Giacchetti G., Beznoussenko G., Attanasio F., Ayala I., Buccione R. (2009). Invadopodia biogenesis is regulated by caveolin-mediated modulation of membrane cholesterol levels. J. Cell. Mol. Med. 13, 1728–1740 10.1111/j.1582-4934.2008.00568.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K. T., Cortesio C. L., Huttenlocher A. (2009). FAK alters invadopodia and focal adhesion composition and dynamics to regulate breast cancer invasion. J. Cell Biol. 185, 357–370 10.1083/jcb.200809110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheresh D. A. (1987). Human endothelial cells synthesize and express an Arg-Gly-Asp-directed adhesion receptor involved in attachment to fibrinogen and von Willebrand factor. Proc. Natl. Acad. Sci. USA 84, 6471–6475 10.1073/pnas.84.18.6471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark E. S., Whigham A. S., Yarbrough W. G., Weaver A. M. (2007). Cortactin is an essential regulator of matrix metalloproteinase secretion and extracellular matrix degradation in invadopodia. Cancer Res. 67, 4227–4235 10.1158/0008-5472.CAN-06-3928 [DOI] [PubMed] [Google Scholar]
- Cornfine S., Himmel M., Kopp P., El Azzouzi K., Wiesner C., Krüger M., Rudel T., Linder S. (2011). The kinesin KIF9 and reggie/flotillin proteins regulate matrix degradation by macrophage podosomes. Mol. Biol. Cell 22, 202–215 10.1091/mbc.E10-05-0394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai D. L., Makretsov N., Campos E. I., Huang C., Zhou Y., Huntsman D., Martinka M., Li G. (2003). Increased expression of integrin-linked kinase is correlated with melanoma progression and poor patient survival. Clin. Cancer Res. 9, 4409–4414. [PubMed] [Google Scholar]
- Destaing O., Planus E., Bouvard D., Oddou C., Badowski C., Bossy V., Raducanu A., Fourcade B., Albiges-Rizo C., Block M. R. (2010). β1A integrin is a master regulator of invadosome organization and function. Mol. Biol. Cell 21, 4108–4119 10.1091/mbc.E10-07-0580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards L. A., Woo J., Huxham L. A., Verreault M., Dragowska W. H., Chiu G., Rajput A., Kyle A. H., Kalra J., Yapp D. et al. (2008). Suppression of VEGF secretion and changes in glioblastoma multiforme microenvironment by inhibition of integrin-linked kinase (ILK). Mol. Cancer Ther. 7, 59–70 10.1158/1535-7163.MCT-07-0329 [DOI] [PubMed] [Google Scholar]
- Gálvez B. G., Matías-Román S., Yáñez-Mó M., Sánchez-Madrid F., Arroyo A. G. (2002). ECM regulates MT1-MMP localization with beta1 or alphavbeta3 integrins at distinct cell compartments modulating its internalization and activity on human endothelial cells. J. Cell Biol. 159, 509–521 10.1083/jcb.200205026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimona M., Buccione R., Courtneidge S. A., Linder S. (2008). Assembly and biological role of podosomes and invadopodia. Curr. Opin. Cell Biol. 20, 235–241 10.1016/j.ceb.2008.01.005 [DOI] [PubMed] [Google Scholar]
- Gligorijevic B., Wyckoff J., Yamaguchi H., Wang Y., Roussos E. T., Condeelis J. (2012). N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J. Cell Sci. 125, 724–734 10.1242/jcs.092726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Giancotti F. G. (2004). Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 5, 816–826 10.1038/nrm1490 [DOI] [PubMed] [Google Scholar]
- Gupton S. L., Gertler F. B. (2010). Integrin signaling switches the cytoskeletal and exocytic machinery that drives neuritogenesis. Dev. Cell 18, 725–736 10.1016/j.devcel.2010.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D. E., Reichardt L. F., Crowley E., Holley B., Moezzi H., Sonnenberg A., Damsky C. H. (1990). The alpha 1/beta 1 and alpha 6/beta 1 integrin heterodimers mediate cell attachment to distinct sites on laminin. J. Cell Biol. 110, 2175–2184 10.1083/jcb.110.6.2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayman E. G., Pierschbacher M. D., Ruoslahti E. (1985). Detachment of cells from culture substrate by soluble fibronectin peptides. J. Cell Biol. 100, 1948–1954 10.1083/jcb.100.6.1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho E., Dagnino L. (2012). Epidermal growth factor induction of front-rear polarity and migration in keratinocytes is mediated by integrin-linked kinase and ELMO2. Mol. Biol. Cell 23, 492–502 10.1091/mbc.E11-07-0596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotary K., Li X. Y., Allen E., Stevens S. L., Weiss S. J. (2006). A cancer cell metalloprotease triad regulates the basement membrane transmigration program. Genes Dev. 20, 2673–2686 10.1101/gad.1451806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson H. W., Schell M. J. (2009). Neuronal IP3 3-kinase is an F-actin-bundling protein: role in dendritic targeting and regulation of spine morphology. Mol. Biol. Cell 20, 5166–5180 10.1091/mbc.E09-01-0083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahlou H., Muller W. J. (2011). β1-integrins signaling and mammary tumor progression in transgenic mouse models: implications for human breast cancer. Breast Cancer Res. 13, 229 10.1186/bcr2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukaitis C. M., Webb D. J., Donais K., Horwitz A. F. (2001). Differential dynamics of alpha 5 integrin, paxillin, and alpha-actinin during formation and disassembly of adhesions in migrating cells. J. Cell Biol. 153, 1427–1440 10.1083/jcb.153.7.1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann B. D., Bauer J. A., Chen X., Sanders M. E., Chakravarthy A. B., Shyr Y., Pietenpol J. A. (2011). Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 121, 2750–2767 10.1172/JCI45014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder S. (2009). Invadosomes at a glance. J. Cell Sci. 122, 3009–3013 10.1242/jcs.032631 [DOI] [PubMed] [Google Scholar]
- Linder S., Aepfelbacher M. (2003). Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol. 13, 376–385 10.1016/S0962-8924(03)00128-4 [DOI] [PubMed] [Google Scholar]
- Linder S., Wiesner C., Himmel M. (2011). Degrading devices: invadosomes in proteolytic cell invasion. Annu. Rev. Cell Dev. Biol. 27, 185–211 10.1146/annurev-cellbio-092910-154216 [DOI] [PubMed] [Google Scholar]
- Liu S., Yamashita H., Weidow B., Weaver A. M., Quaranta V. (2010). Laminin-332-beta1 integrin interactions negatively regulate invadopodia. J. Cell. Physiol. 223, 134–142 10.1002/jcp.22018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizárraga F., Poincloux R., Romao M., Montagnac G., Le Dez G., Bonne I., Rigaill G., Raposo G., Chavrier P. (2009). Diaphanous-related formins are required for invadopodium formation and invasion of breast tumor cells. Cancer Res. 69, 2792–2800 10.1158/0008-5472.CAN-08-3709 [DOI] [PubMed] [Google Scholar]
- Lu X., Lu D., Scully M., Kakkar V. (2008). The role of integrins in cancer and the development of anti-integrin therapeutic agents for cancer therapy. Perspect. Medicin. Chem. 2, 57–73. [PMC free article] [PubMed] [Google Scholar]
- Lucas J. T., Jr, Salimath B. P., Slomiany M. G., Rosenzweig S. A. (2010). Regulation of invasive behavior by vascular endothelial growth factor is HEF1-dependent. Oncogene 29, 4449–4459 10.1038/onc.2010.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenböck G., De Angelis D. A., Rothman J. E. (1998). Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394, 192–195 10.1038/28190 [DOI] [PubMed] [Google Scholar]
- Mueller S. C., Ghersi G., Akiyama S. K., Sang Q. X., Howard L., Pineiro-Sanchez M., Nakahara H., Yeh Y., Chen W. T. (1999). A novel protease-docking function of integrin at invadopodia. J. Biol. Chem. 274, 24947–24952 10.1074/jbc.274.35.24947 [DOI] [PubMed] [Google Scholar]
- Murphy D. A., Courtneidge S. A. (2011). The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 12, 413–426 10.1038/nrm3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara H., Nomizu M., Akiyama S. K., Yamada Y., Yeh Y., Chen W.-T. (1996). A mechanism for regulation of melanoma invasion. Ligation of alpha6beta1 integrin by laminin G peptides. J. Biol. Chem. 271, 27221–27224 10.1074/jbc.271.44.27221 [DOI] [PubMed] [Google Scholar]
- Nakamura I., Duong, le T., Rodan S. B., Rodan G. A. (2007). Involvement of alpha(v)beta3 integrins in osteoclast function. J. Bone Miner. Metab. 25, 337–344 10.1007/s00774-007-0773-9 [DOI] [PubMed] [Google Scholar]
- Oser M., Yamaguchi H., Mader C. C., Bravo-Cordero J. J., Arias M., Chen X., Desmarais V., van Rheenen J., Koleske A. J., Condeelis J. (2009). Cortactin regulates cofilin and N-WASp activities to control the stages of invadopodium assembly and maturation. J. Cell Biol. 186, 571–587 10.1083/jcb.200812176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh A., Weaver A. M. (2009). Regulation of cancer invasiveness by the physical extracellular matrix environment. Cell Adhes. Migr. 3, 288–292 10.4161/cam.3.3.8888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh A., Ruppender N. S., Branch K. M., Sewell-Loftin M. K., Lin J., Boyer P. D., Candiello J. E., Merryman W. D., Guelcher S. A., Weaver A. M. (2011). Sensing and modulation of invadopodia across a wide range of rigidities. Biophys. J. 100, 573–582 10.1016/j.bpj.2010.12.3733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poincloux R., Collin O., Lizárraga F., Romao M., Debray M., Piel M., Chavrier P. (2011). Contractility of the cell rear drives invasion of breast tumor cells in 3D Matrigel. Proc. Natl. Acad. Sci. USA 108, 1943–1948 10.1073/pnas.1010396108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano P. P., Inman D. R., Eliceiri K. W., Beggs H. E., Keely P. J. (2008a). Mammary epithelial-specific disruption of focal adhesion kinase retards tumor formation and metastasis in a transgenic mouse model of human breast cancer. Am. J. Pathol. 173, 1551–1565 10.2353/ajpath.2008.080308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano P. P., Inman D. R., Eliceiri K. W., Knittel J. G., Yan L., Rueden C. T., White J. G., Keely P. J. (2008b). Collagen density promotes mammary tumor initiation and progression. BMC Med. 6, 11 10.1186/1741-7015-6-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintavalle M., Elia L., Condorelli G., Courtneidge S. A. (2010). MicroRNA control of podosome formation in vascular smooth muscle cells in vivo and in vitro. J. Cell Biol. 189, 13–22 10.1083/jcb.200912096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottiers P., Saltel F., Daubon T., Chaigne-Delalande B., Tridon V., Billottet C., Reuzeau E., Génot E. (2009). TGFbeta-induced endothelial podosomes mediate basement membrane collagen degradation in arterial vessels. J. Cell Sci. 122, 4311–4318 10.1242/jcs.057448 [DOI] [PubMed] [Google Scholar]
- Sabeh F., Ota I., Holmbeck K., Birkedal-Hansen H., Soloway P., Balbin M., Lopez-Otin C., Shapiro S., Inada M., Krane S. et al. (2004). Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J. Cell Biol. 167, 769–781 10.1083/jcb.200408028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai T., Li S., Docheva D., Grashoff C., Sakai K., Kostka G., Braun A., Pfeifer A., Yurchenco P. D., Fässler R. (2003). Integrin-linked kinase (ILK) is required for polarizing the epiblast, cell adhesion, and controlling actin accumulation. Genes Dev. 17, 926–940 10.1101/gad.255603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai-Yageta M., Recchi C., Le Dez G., Sibarita J. B., Daviet L., Camonis J., D'Souza-Schorey C., Chavrier P. (2008). The interaction of IQGAP1 with the exocyst complex is required for tumor cell invasion downstream of Cdc42 and RhoA. J. Cell Biol. 181, 985–998 10.1083/jcb.200709076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santner S. J., Dawson P. J., Tait L., Soule H. D., Eliason J., Mohamed A. N., Wolman S. R., Heppner G. H., Miller F. R. (2001). Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast Cancer Res. Treat. 65, 101–110 10.1023/A:1006461422273 [DOI] [PubMed] [Google Scholar]
- Sawai H., Okada Y., Funahashi H., Matsuo Y., Takahashi H., Takeyama H., Manabe T. (2006). Integrin-linked kinase activity is associated with interleukin-1 alpha-induced progressive behavior of pancreatic cancer and poor patient survival. Oncogene 25, 3237–3246 10.1038/sj.onc.1209356 [DOI] [PubMed] [Google Scholar]
- Schmidt S., Nakchbandi I., Ruppert R., Kawelke N., Hess M. W., Pfaller K., Jurdic P., Fässler R., Moser M. (2011). Kindlin-3-mediated signaling from multiple integrin classes is required for osteoclast-mediated bone resorption. J. Cell Biol. 192, 883–897 10.1083/jcb.201007141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano I., McDonald P. C., Lock F. E., Dedhar S. (2012). Role of the integrin-linked kinase (ILK)/Rictor complex in TGFβ-1-induced epithelial-mesenchymal transition (EMT). Oncogene [Epub ahead of print] 10.1038/onc.2012.30 [DOI] [PubMed] [Google Scholar]
- Spiczka K. S., Yeaman C. (2008). Ral-regulated interaction between Sec5 and paxillin targets Exocyst to focal complexes during cell migration. J. Cell Sci. 121, 2880–2891 10.1242/jcs.031641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinardi L., Rietdorf J., Nitsch L., Bono M., Tacchetti C., Way M., Marchisio P. C. (2004). A dynamic podosome-like structure of epithelial cells. Exp. Cell Res. 295, 360–374 10.1016/j.yexcr.2004.01.007 [DOI] [PubMed] [Google Scholar]
- Stanchi F., Grashoff C., Nguemeni Yonga C. F., Grall D., Fässler R., Van Obberghen-Schilling E. (2009). Molecular dissection of the ILK-PINCH-parvin triad reveals a fundamental role for the ILK kinase domain in the late stages of focal-adhesion maturation. J. Cell Sci. 122, 1800–1811 10.1242/jcs.044602 [DOI] [PubMed] [Google Scholar]
- Steffen A., Le Dez G., Poincloux R., Recchi C., Nassoy P., Rottner K., Galli T., Chavrier P. (2008). MT1-MMP-dependent invasion is regulated by TI-VAMP/VAMP7. Curr. Biol. 18, 926–931 10.1016/j.cub.2008.05.044 [DOI] [PubMed] [Google Scholar]
- Stewart D. A., Cooper C. R., Sikes R. A. (2004). Changes in extracellular matrix (ECM) and ECM-associated proteins in the metastatic progression of prostate cancer. Reprod. Biol. Endocrinol. 2, 2 10.1186/1477-7827-2-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Botvinick E. L., Zhao Y., Berns M. W., Usami S., Tsien R. Y., Chien S. (2005). Visualizing the mechanical activation of Src. Nature 434, 1040–1045 10.1038/nature03469 [DOI] [PubMed] [Google Scholar]
- Wang Y., McNiven M. A. (2012). Invasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK-p130Cas complex. J. Cell Biol. 196, 375–385 10.1083/jcb.201105153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver A. M. (2006). Invadopodia: specialized cell structures for cancer invasion. Clin. Exp. Metastasis 23, 97–105 10.1007/s10585-006-9014-1 [DOI] [PubMed] [Google Scholar]
- Weichselbaum R. R., Dahlberg W., Beckett M., Karrison T., Miller D., Clark J., Ervin T. J. (1986). Radiation-resistant and repair-proficient human tumor cells may be associated with radiotherapy failure in head- and neck-cancer patients. Proc. Natl. Acad. Sci. USA 83, 2684–2688 10.1073/pnas.83.8.2684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickström S. A., Fässler R. (2011). Regulation of membrane traffic by integrin signaling. Trends Cell Biol. 21, 266–273 10.1016/j.tcb.2011.02.003 [DOI] [PubMed] [Google Scholar]
- Wickström S. A., Lange A., Hess M. W., Polleux J., Spatz J. P., Krüger M., Pfaller K., Lambacher A., Bloch W., Mann M. et al. (2010a). Integrin-linked kinase controls microtubule dynamics required for plasma membrane targeting of caveolae. Dev. Cell 19, 574–588 10.1016/j.devcel.2010.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickström S. A., Lange A., Montanez E., Fässler R. (2010b). The ILK/PINCH/parvin complex: the kinase is dead, long live the pseudokinase! EMBO J. 29, 281–291 10.1038/emboj.2009.376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner C., Faix J., Himmel M., Bentzien F., Linder S. (2010). KIF5B and KIF3A/KIF3B kinesins drive MT1-MMP surface exposure, CD44 shedding, and extracellular matrix degradation in primary macrophages. Blood 116, 1559–1569 10.1182/blood-2009-12-257089 [DOI] [PubMed] [Google Scholar]
- Wolf K., Wu Y. I., Liu Y., Geiger J., Tam E., Overall C., Stack M. S., Friedl P. (2007). Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat. Cell Biol. 9, 893–904 10.1038/ncb1616 [DOI] [PubMed] [Google Scholar]
- Wu C., Dedhar S. (2001). Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J. Cell Biol. 155, 505–510 10.1083/jcb.200108077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H., Lorenz M., Kempiak S., Sarmiento C., Coniglio S., Symons M., Segall J., Eddy R., Miki H., Takenawa T. et al. (2005). Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J. Cell Biol. 168, 441–452 10.1083/jcb.200407076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H., Takeo Y., Yoshida S., Kouchi Z., Nakamura Y., Fukami K. (2009). Lipid rafts and caveolin-1 are required for invadopodium formation and extracellular matrix degradation by human breast cancer cells. Cancer Res. 69, 8594–8602 10.1158/0008-5472.CAN-09-2305 [DOI] [PubMed] [Google Scholar]
- Yamaguchi H., Yoshida S., Muroi E., Yoshida N., Kawamura M., Kouchi Z., Nakamura Y., Sakai R., Fukami K. (2011). Phosphoinositide 3-kinase signaling pathway mediated by p110α regulates invadopodium formation. J. Cell Biol. 193, 1275–1288 10.1083/jcb.201009126 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}