

Summary
Smad family proteins are essential intracellular mediators that regulate transforming growth factor-β (TGF-β) ligand signaling. In response to diverse stimuli, Smad7 is rapidly expressed and acts as a cytoplasmic inhibitor that selectively interferes with signals elicited from TGF-β family receptors. In addition, earlier works have indicated that retrovirally transduced Smad7 induces long-lasting cell proliferation arrest in a variety of mesenchymal cells through down-regulation of G1 cyclins. However, the molecular mechanisms underlying the cytostatic effects of Smad7 remain unknown. We show here that Smad7 can form a complex with endogenous histone deacetylase proteins HDAC-1 and HDAC-3 in NIH 3T3 mouse fibroblast cells. By contrast, forced expression of a dominant-negative variant of HDAC-1 efficiently protected cells against Smad7 proliferation inhibition, suggesting that Smad7 depends on the deacetylase activity of its associated HDAC-1 to arrest the cell cycle. Furthermore, Smad7 caused HDAC-1 bind to E2F-1 to form a ternary complex on chromosomal DNA containing an E2F-binding motif and leading to repression in the activity of the E2F target genes. Smad7 mutations that prevented its binding to either HDAC-1 or E2F-1 resulted in a significant decrease in Smad7-mediated inhibition of cell proliferation. The present results strongly suggest that nuclear Smad7 is a transcriptional corepressor for E2F, providing a molecular basis for the Smad7-induced arrest of the cell cycle.
Keywords: Cell cycle, Transcriptional corepressor, E2F, Histone deacetylase, Smad
Introduction
Smad family members have been identified as essential genes for the intracellular mediation of transforming growth factor-β (TGF-β) peptide signaling (Massague, 1998; Raftery and Sutherland, 1999). The signaling pathway of the TGF-β family, which includes bone morphogenetic protein (BMP) and activin, is involved in the regulation of critical cell processes such as development, growth, and differentiation (Sporn and Roberts, 1992; Hogan, 1996). Upon TGF-β ligand-induced assembly, which involves type-I and type-II receptor serine/threonine kinases, the type-I receptor becomes activated via phosphorylation by the type-II receptor and in turn phosphorylates the Smad proteins (Wrana et al., 1994; Yingling et al., 1996; Zhang et al., 1996). The Smad family has two conserved domains, MH1 and MH2, that are located in their N- and C-terminal regions, respectively (Heldin et al., 1997). While the MH1 domain is known to operate through direct and indirect interactions with DNA, the MH2 domain mediates associations between Smad proteins and with other factors such as coactivators and corepressors that influence gene expression (Massague and Wotton, 2000; Heldin et al., 2009; Wu and Hill, 2009). Based on their functions and similarities, Smads have been classified into three subgroups. The receptor-regulated Smads (R-Smads) are present in the cytoplasm and act as substrates for activated type-I receptor kinases, followed by migration to the nucleus (Moustakas et al., 2001; Inman et al., 2002). Common partner Smads (Co-Smads) form a complex with phosphorylated R-Smads that regulates the transcription of target genes (Chen et al., 1997; Massague and Wotton, 2000). The third group is composed of inhibitory Smads (I-Smads) that include Smad6 and Smad7, which are found in vertebrates and can interfere with TGF-β pathways (Hayashi et al., 1997; Imamura et al., 1997; Nakao et al., 1997; Bhushan et al., 1998; Hata et al., 1998; Nakayama et al., 1998). While R- and Co-Smad genes are ubiquitously expressed, induction of I-Smads occurs quickly in response to TGF-β and BMP (Nakao et al., 1997; Afrakhte et al., 1998; Takase et al., 1998). The induced I-Smads then bind to the cytoplasmic domain of the type-I receptor to prevent it from phosphorylating R-Smads, leading to the negative feedback seen in the TGF-β pathway (Heldin et al., 1997; Massague, 1998).
Smad7 in particular shows a correlation with disease: levels are low in normal tissues in vivo, but significantly higher in several disease states (Heldin et al., 2009). Smad7 principally resides in the nucleus but TGF-β leads to its partial cytoplasmic distribution (Itoh et al., 1998; Kavsak et al., 2000; Suzuki et al., 2002). Smad7 is involved in diverse biological processes, as illustrated by the variety of stimuli that can regulate its expression, such as γ-interferon, tumor necrosis factor-α, shear stress, and UV light (Topper et al., 1997; Ulloa et al., 1999; Bitzer et al., 2000; Quan et al., 2001). For instance, Smad7 participates in the control of myocyte and adipocyte differentiation and tumor cell metastasis independently of TGF-β signaling (Heikkinen et al., 2010; Miyake et al., 2010). Smad7 therefore appears to be more than a mere inhibitor in the TGF-β signaling pathway. Indeed, our previous study showed that Smad7 can arrest cell proliferation at G1 phase in mesenchymal cells following repressed levels of cyclins D1 and E (Kitamura et al., 2005).
The acetylation of the lysine side chains is a crucial posttranslational modification in numerous proteins and has crosstalk with phosphorylation, methylation and other covalent modifications, leading to dynamic signaling networks (Freiman and Tjian, 2003; Yang and Seto, 2008). Lysine acetylation was first identified in histones, the major protein component of chromatin that packs nuclear DNA into nucleosomes (Allfrey et al., 1964; Phillips, 1968). Histone hyperacetylation in the tail region leads to nucleosome remodeling as well as a more accessible chromatin structure, which correlates with gene transcription (Kouzarides, 1999; Johnstone, 2002; Yang and Seto, 2007). Besides histones, reversible acetylation has been seen in a number of transcription factors, other nuclear proteins, and various cytoplasmic regulators including p53, importin-α, α-tubulin and Hsp90. Such acetylation broadly modulates their cellular control by altering their localization, enzymatic activity, DNA-binding ability, protein-protein interaction, and gene transactivating function (Kouzarides, 2000; Spange et al., 2009). A balance between acetylation and ubiquintination has also been reported to affect the stability of p53 (Ito et al., 2002), Smad7 (Gronroos et al., 2002), c-Myc (Vervoorts et al., 2003), and Runx3 (Jin et al., 2004).
Histone deacetylases (HDACs) are enzymes that catalyze the removal of acetyl groups from acetyllysine residues within histones and other proteins (Johnstone, 2002; Yang and Seto, 2008) that have been modified by histone acetyltransferase (HAT) (Marmorstein and Roth, 2001; Lee and Workman, 2007). Nuclear HDACs have been identified as critical components in various transcription corepressor complexes including those that can incorporate nucleosome-remodeling subunits (Flaus and Owen-Hughes, 2001; Khochbin et al., 2001). In such molecular machinery, HDAC activity plays an important role in exerting repression via its interaction, either directly or indirectly, with DNA-binding transcription factors (de Ruijter et al., 2003; Yang and Seto, 2007). It has in fact been shown that recruited HDAC affects promoter activity for a variety of transcription factors including nuclear hormone receptors, nuclear factor κB, myocyte enhancer factor 2 and Sox2 (Nagy et al., 1997; Zhong et al., 2002; Tang and Goldman, 2006; Baltus et al., 2009). In addition, the HDAC complex is required for the repression of certain promoters responsive to the E2F family of transcription factors that control G1 cyclin genes in cell proliferation regulation (Sambucetti et al., 1999). Thus, by modulating transcription, the catalytic activity of HDACs directs cell fate through hormonal response, apoptosis, cell differentiation, multipotent cell status, as well as cell cycle progress (Nagy et al., 1997; Sambucetti et al., 1999; Zhong et al., 2002; Tang and Goldman, 2006; Baltus et al., 2009; Wilting et al., 2010; Yamaguchi et al., 2010). Accordingly, anomalous patterns of lysine acetylation associated with aberrant gene expressions have been closely linked to various disorders such as cancers, cardiovascular diseases and ageing (Johnstone, 2002; Yang and Seto, 2007). In particular, dysregulation of HDACs and their improper allocation to a number of oncogenes and tumor suppressor genes have been observed in multiple malignant cells (Cress and Seto, 2000; Timmermann et al., 2001; Bolden et al., 2006), suggesting that both abnormal HDAC activation and inactivation could result in unsuitable cell proliferation and eventual tumorigenesis, and that the coordinated function of HDACs is essential for accurate control of the cell cycle.
Our earlier data indicated that forced expression of Smad7 causes a long lasting arrest of the cell cycle in mesenchymal cells that are otherwise proliferating rapidly (Kitamura et al., 2005). Although the mechanisms underlying these phenomena remain obscure, the finding that Smad7 entirely localizes in the nucleus in the course of the cell cycle suggests that Smad7 may exert its inhibitory effect through a functional complex with nuclear factors responsible for cell cycle progression. In the present study, we show that Smad7 binds to HDAC-1 via its C-terminal region both in vitro and in vivo. We also show that when a dominant-negative form of an HDAC-1 mutant is ectopically coexpressed with Smad7, it frees cells from the Smad7-induced proliferation arrest. In addition, Smad7-truncated mutants lacking the ability to bind HDAC-1 lose their ability to inhibit proliferation, suggesting that Smad7 is dependent on the deacetylase activity of the associated HDAC-1 to arrest the cell cycle. Moreover, our data demonstrate that Smad7 can bring HDAC-1 to transcription factor E2F-1 and thereby causes the repression of several E2F-responsive genes, which is critical for cell cycle progress. These results reveal a potential molecular basis for the Smad7-induced cell cycle arrest, and further suggest a novel link between the TGF-β signal pathway and cell proliferation control.
Results
Association of Smad7 with HDAC-1 and HDAC-3 in NIH 3T3 cells
To examine whether Smad7 interacts physically in vivo with HDAC family members, we introduced an expression vector for N-terminally Flag-tagged mouse Smad7 (Flag-Smad7) into NIH 3T3 mouse fibroblast cells. Smad7-containing protein complexes were immunoprecipitated using an α-Flag antibody from nuclear extract prepared under non-denaturing conditions in order to prevent possible dissolution of nuclear complexes. Western blot analyses of the immune complexes showed that the α-Flag antibody coprecipitated HDAC-1 with Flag-Smad7 (Fig. 1A). Essentially identical results were obtained from the retroviral transduction of Myc-Smad7 (data not shown). These results indicate that endogenous HDAC-1 forms a complex with Smad7 expressed in NIH 3T3 cells and that their interaction is independent of the Smad7 expression method or Smad7 expression levels. In addition, endogenous HDAC-3 also formed a complex with Smad7 (Fig. 1A). These results are consistent with previous reports describing interactions between Smad7 and HDACs in 293T cells (Bai et al., 2000; Simonsson et al., 2005).
Fig. 1. Complex formation between Smad7 and specific HDAC proteins.

(A) Association of Smad7 with endogenous HDAC-1 and HDAC-3, respectively, in NIH 3T3 mouse fibroblast cells. Nuclear extracts from NIH 3T3 cells transfected with a vector containing control or Flag-Smad7 were incubated with an anti-Flag antibody to immunoprecipitate a complex containing Flag-Smad7. Immune complexes (left) and total nuclear extracts (right) were examined by Western blotting using α-HDAC-1, α-HDAC-3 and α-Flag antibodies. (B,C) Requirement of the 381–408 C-terminal region for the association of Smad7 with HDAC-1. A series of truncated Smad7 variants with an N-terminal Myc-tag (B) were expressed together with HDAC-1-Flag in 293T cells and immunoprecipitated with α-Myc. Immune complexes were analyzed by Western blotting using α-Flag and α-Myc. Black boxes indicate amino acids 381-408, which are essential for the interaction with HDAC-1 (C). (D) NIH3T3 cells transduced with wild-type Smad7 or each deletion mutant by a retrovirus vector were seeded in a 96-well plate (3×103 cells/well) and incubated for 72 h. [3H]-thymidine was loaded for the last 4 h and the radioactivity incorporated into chromosomal DNA was measured by a microplate scintillation counter. Data are shown as means with S.E. bars from triplicate assays.
A previous coexpression study indicated that the MH2 domain (a.a. 261–426) was sufficient for Smad7 to interact with HDAC-1 (Simonsson et al., 2005). To determine the more precise Smad7 region required for HDAC-1 association and proliferation arrest, we cointroduced truncated versions of Smad7 with an N-terminal Myc-tag (Fig. 1B) and Flag-HDAC-1 into 293T cells. Following immunoprecipitation with a α-Myc antibody, Western blot analyses using an α-Flag antibody showed that a C-terminally truncated Smad7(1-408) accompanied a detectable amount of Flag-HDAC-1, whereas further truncated fragments (1–380 and 1–259) did not (Fig. 1C). These results suggest that the Smad7(381–408) region is essential for HDAC-1 interaction, and that additional residues within the Smad7(409–426) may enhance this interaction, similar to its role in association with the cytoplasmic domain of TGF-β receptor type-I (Hayashi et al., 1997).
To assess whether the Smad7 truncations can inhibit proliferation, NIH 3T3 cells were transduced with each mutant and subjected to DNA synthesis assays. Amounts of incorporated [3H]thymidine between 68 and 72 h post-infection showed that full-length Smad7(1–426) reduced DNA synthesis to 14% of normal levels, indicating the inhibition of cell proliferation (Fig. 1D). Similarly, two mutants, Smad7(1–408) and Smad7(260–426), showed inhibitory activity comparable to that of full-length Smad7, reducing thymidine incorporation to 19% and 15%, respectively. In sharp contrast, Smad7(1–380) and Smad7(1–259) mutants showed only small effects on DNA synthesis in parallel with their inability to bind HDAC-1. Together, these results demonstrate that a region of 28 residues (a.a. 381–408) in the C-terminal MH2 domain of Smad7 is essential for complex formation with HDAC-1 and the inhibition of cell proliferation.
Smad7 binds in vitro to a C-terminal region of HDAC-1
To clarify whether Smad7 interacts with HDAC proteins directly or indirectly, we performed in vitro binding assays using recombinant proteins purified from E. coli cells. The full length Smad7 was expressed as a GST fusion protein and collected on glutathione-coupled beads. Separately purified Flag-HDACs were obtained in solution from column-bound GST-Flag-HDACs by cleavage with a sequence-specific protease. The GST-Smad7 fusion and control GST bound to the beads were incubated with Flag-HDAC-1 and extensively washed. Western blot analyses revealed that GST-Smad7, but not GST only, bound to HDAC-1 (Fig. 2B). Similar results were obtained for HDAC-2 and HDAC-3 in vitro binding to GST-Smad7 (not shown).
Fig. 2. In vitro binding of HDAC-1 to Smad7.

The C-terminal region responsible for direct interaction with Smad7 was located outside the HDAC-1 deacetylase domain. The E. coli cell-derived Flag-HDAC-1 protein and indicated variants shown in (A) were incubated with control GST and GST-Smad7 bound to glutathione-coupled beads and then collected. Proteins bound to the beads were detected by Western blotting with α-Flag (B).
To map which HDAC-1 domains are recognized by Smad7 in the in vitro assays, we prepared a series of truncated HDAC-1 fragments with an N-terminal Flag-tag (Fig. 2A). Western blotting showed that HDAC-1 fragments that bound to GST-Smad7 commonly contained 155 residues (a.a. 328–482) from the C-terminal, which is outside the catalytic domain. These in vitro data indicate a direct binding of this C-terminal region to Smad7 and suggest that Smad7 can form a complex with HDAC-1 in vivo through similar interactions. A consistent interaction between Smad7 and a C-terminal fragment (a.a. 161–482) of HDAC-1 in cotransfected 293T cells was indeed previously reported (Simonsson et al., 2005).
A dominant negative form of HDAC-1 restores cell growth and proliferation from Smad7-induced arrest
HDAC-1 has been shown to play crucial roles in cell cycle progress by regulating gene expression. To assess the potential relationship between histone deacetylase activity and Smad7 effects, we prepared retroviral expression vectors for both the human wild-type HDAC-1 as well as a mutant, H141A HDAC-1, where the histidine 141 is substituted with an alanine residue. Previous reports showed in vitro that H141A HDAC-1 lacks deacetylase activity and can interfere with the function of endogenous HDAC-1 in myoblast cells (Hassig et al., 1998; Mal et al., 2001; Ito et al., 2002). In addition, a dominant-negative H141A HDAC-1 seemed to be helpful in clarifying the importance of HDAC-1 activity in Smad7-induced cell cycle arrest because both wild-type and H141A HDAC-1 can form similar protein complexes (Humphrey et al., 2008). By efficient infection and subsequent drug selection, NIH 3T3 cells were stably transduced with a vector expressing either the wild-type or the mutant H141A HDAC-1. Both Flag-tagged versions were detected by immunofluorescence microscopy at an equivalent level and in similar nuclear locations (Fig. 3A). After 72 h of infection, histone H3 was examined using α-Ac-K9/13 antibody specific for acetylated lysine residues at 9 and 13 in the N-terminal region. Interestingly, Western blotting revealed that acetylation of histone H3 was dramatically increased in H141A HDAC-1-expressing cells, thus indicating that the H141A HDAC-1 mutant was able to act as a dominant-negative variant against HDAC-1 in this system (Fig. 3B).
Fig. 3. Release of Smad7-induced cell cycle arrest by the H141A mutant of HDAC-1.

(A) Minimal effect on the level and localization of Smad7 when co-expressed with either wild-type or H141A HDAC-1. NIH 3T3 cells infected with combinations of retroviral vectors expressing the indicated protein: Smad7, wild-type HDAC-1, or an alanine substitution mutant for histidine 141 in HDAC-1 (H141A HDAC-1). Cells re-plated 48 h before fixation were single- or double-stained with rabbit α-Smad7 and mouse α-Flag antibody, followed by visualization with Alexa488-labeled α-rabbit Ig (green) and a Cy3-labeled α-mouse Ig (red) secondary antibody, respectively. Bars, 40 μm. (B) Effect of H141A HDAC-1 on the acetylation level of histone H3. NIH 3T3 cells were infected with control or retroviral vectors expressing Flag-tagged wild-type or H141A HDAC-1, and harvested 72 h later. Acetylation levels were examined by Western blotting using an antibody specific for acetylated histone H3 at both Lys9 and Lys13. Comparable protein levels were confirmed for total endogenous histone H3 in each sample and for exogenous wild-type and H141A HDAC-1 by using α-histone H3 and α-Flag, respectively. (C) Microscopic images of the cultures were taken to monitor the proliferation and morphology of NIH 3T3 cells expressing Smad7 and either wild-type or H141A HDAC-1. After 12 h of serial vector infection, cells were seeded (2×105/a well of 6-well plate) and periodically photographed until 96 h under a phase-contrast microscope. Bars, 200 μm. (D) Effects of H141A HDAC-1 on Smad7-induced proliferation arrest. After 12 h of infection, NIH 3T3 cells (5×103/a well of 96-well pate) were seeded to allow proliferation. The number of cells at 12, 24, 48, 72 and 96 h was measured as DNA content in each well. Transduced genes were as follows: •, two controls; ▴, H141A HDAC-1 and a control; △, wild-type HDAC-1 and a control; ◯, a control and Smad7; ▪, H141A HDAC-1 and Smad7; and □, wild-type HDAC-1 and Smad7. Data indicate means with S.D. from triplicate assays. (E) Effects of a dominant-negative HDAC-1 against Smad7 on the levels of various proteins. Cells infected with control, H141A HDAC-1, or Smad7 vectors were seeded (3×105 cells/a 60-mm dish) and harvested at 72 h of culture. Western blots show the levels of exogenous Flag-tagged H141A HDAC-1 and Smad7, and of endogenous HDAC-1, HDAC-2, Cyclin A, Cyclin D1 and Smad4 proteins.
To examine the phenotypes of forced expression of the wild-type and H141A HDAC-1 and their effects on Smad7-induced cell cycle arrest, NIH 3T3 cells were serially infected with different vector combinations. The primarily nuclear localization of Smad7 was minimally affected by the coexpression of either protein (Fig. 3A). After infection, cells were re-plated for dilution and monitored for proliferation by periodically recording microscopic observations as well as by DNA content analysis. Expression of exogenous wild-type or H141A HDAC-1 alone showed no apparent influence on cell proliferation under 10% FBS conditions (Fig. 3C) even though the levels of both wild-type and H141A HDAC-1 remained similar (Fig. 3B). When coexpressed with Smad7 however, H141A HDAC-1 effectively suppressed growth inhibition (Fig. 3C) with cells showing a proliferation curve that was very similar to that of control cells (Fig. 3D). As expected, 72 h after infection, the levels of cyclins D1 and A remarkably decreased in Smad7-expressing cells (Fig. 3E) (Kitamura et al., 2005). In sharp contrast, cells expressing both Smad7 and H141A HDAC-1 had almost the same levels of cyclin proteins, similarly to control cells infected with the empty vectors. The above results thus suggest that H141A HDAC-1 blocked the attenuated expression of cyclins D1 and A, resulting in cell cycle progression and cell proliferation. In the absence of ectopic Smad7, H141A HDAC-1 caused little change in the levels of these cyclins (Fig. 3E), which is consistent with the normal proliferation seen in H141A HDAC-1-expressing cells. It is therefore unlikely that a growth-promoting ability perhaps displayed by the inactivated form of HDAC-1 is responsible for rescuing cells from Smad7 arrest.
In addition, because the level of Smad7 was unchanged by H141A HDAC-1, it could be argued that the action of H141A HDAC-1 occurs downstream of Smad7. These results strongly suggest that the deacetylase activity of endogenous HDAC-1 mediates Smad7 effects on proliferation by physically associating with it. Exogenous expression of wild-type HDAC-1 produced little change on the effect of Smad7, suggesting that endogenous HDAC-1 is sufficient or that an unidentified limiting factor(s) contributes to this interaction.
Smad7 brings HDAC-1 to E2F-1 transcription factor in a manner dependent on Lys359
The E2F family of transcription factors controls the cell cycle by regulating the expression of crucial genes, particularly during the G1-S transition. To investigate the potential role of E2F in the cell cycle arrest induced by Smad7 and HDAC-1, we first tested if Smad7 could interact with E2F-1, one of the most studied E2F members. Western blot analyses of Smad7-containing immune complexes prepared from the nuclear extracts of Flag-Smad7-expressing NIH 3T3 cells showed that complexes included endogenous E2F-1 (Fig. 4A), indicating that Smad7 associated in vivo with E2F-1 in the nucleus. A complex containing Myc-Smad7 and N-terminally HA-tagged mouse E2F-1 (HA-E2F-1) was also detected in cotransfected 293T cells (Fig. 4B). Coexpression of E2F-1 seemed to cause a decreased level of Smad7 possibly due to a transfection-related artifact in 293T cells (Fig. 4B), a phenomenon not observed in NIH 3T3 cells (Fig. 6A).
Fig. 4. Association of wild-type and K359A Smad7 with E2F-1.

(A) K359A Smad7 mutant lost growth inhibitory activity. NIH 3T3 cells were expressed with wild-type or K359A Smad7 and re-plated. The number of cells in each culture after 72 h of re-plating was measured (see Fig. 3D legend). Data represent means with S.D. from triplicate experiments. (B) Endogenous E2F-1 complexed with Smad7 but not with K359A mutant Smad7. NIH 3T3 cells were transfected with control, Flag-Smad7 or K359A mutant vectors, replated 24 h later and cultured a further 24 h. Following serum starvation for 8 h, the cells were incubated in DMEM containing 10% FBS to induce endogenous E2F-1. Nuclear extracts prepared 14 h later were incubated with α-Flag to immunoprecipitate complexes containing Flag-Smad7 or Flag-K359A Smad7. The immune complexes (left) and the nuclear extracts (right) were analyzed by Western blotting using α-E2F-1, and then α-Flag for comparable levels of Flag-Smad7 variants. (C) Defective effect of K359A on Smad7 association with E2F-1 in transfected 293T cells. Total cell extracts containing the indicated combinations of HA-tagged human E2F1, Myc-Smad7 and K359A Myc-Smad7 expressed in 293T cells were subjected to immunoprecipitation using α-Myc. The immune complexes (left panels) and the total lysates (right panels) were analyzed by Western blotting with α-HA to detect HA-E2F-1 (upper panels). Blots were re-probed with α-Myc for wild-type and K359A Myc-Smad7 (lower panels).
Fig. 6. Restoration of cell proliferation by E2F-1 from the Smad7-induced arrest in NIH 3T3 cells.

(A) Minimal effect of ectopic E2F-1 on the level of Smad7. NIH 3T3 cells were infected with retroviral vectors containing control, Smad7 or Flag-E2F-1, as indicated, and re-plated for further culture for another 72 h. The levels of Smad7 and Flag-E2F-1 were examined by Western blots of total cell extracts using α-E2F-1 and α-Smad7. (B) NIH 3T3 cells transduced in (A) were photographed periodically at the indicated time points to monitor their growth and morphology (see Fig. 3C legend). Bars, 200 μm. The number of cells in each culture after 72 h of re-plating was measured (see Fig. 3D legend). Data represent means with S.D. from triplicate experiments.
Because of the fact that HDAC-1 binds to Smad7, we monitored the interactions between HDAC-1 (wild-type and H141A mutant) and E2F-1 in the presence and absence of Smad7. For this purpose, lysates of cells expressing HA-E2F-1, HDAC-1-Flag and Myc-Smad7 in various combinations were subjected to immunoprecipitation with an α-Flag antibody, and the resultant immune complexes containing HDAC-1-Flag were analyzed by Western blotting. Interestingly, HA-E2F-1 was detected in HDAC1-Flag complexes only in the presence of Myc-Smad7. These results indicate that Smad7 can indeed mediate an interaction between HDAC-1 and E2F-1 through formation of a molecular complex (Fig. 5A, lanes 2, 4). Since H141A HDAC-1 also had the ability to form a complex containing E2F-1 in the presence of Smad7, we conclude that deacetylase activity is dispensable for HDAC-1 recruitment.
Fig. 5. Repression of E2F-regulated genes by Smad7-linked association between E2F-1 and HDAC-1.

(A) An in vivo complex of HDAC-1 and E2F-1 is mediated by Smad7 depending on Lys359. E2F1, Smad7, HDAC-1 and their variants, each with separate epitope tags, were expressed in 293T cells at the indicated combinations. Complexes containing HDAC-1-Flag or H141A HDAC-1-Flag were immunoprecipitated with α-Flag from total cell extracts. The immune complexes (left) and total lysates (right) were examined by Western blotting to detect E2F-1, Smad7 and HDAC-1 variants by using α-HA, α-Smad7 and α-Flag, respectively. (B) Smad7-induced repression of the Dhfr and Tk genes, both of which are regulated by E2F factors. (Left) Total RNA was prepared from NIH 3T3 cells infected with retro vectors for wild-type or K359A Smad7 and H141A HDAC-1, as shown, and cultured for 24 h. Complementary DNAs were synthesized from 1 µg RNA by reverse transcription with random primers. Equal aliquots from each reaction were used for PCR to amplify cDNA fragments of Dhfr (371 bp), Tk (411 bp), Cyclin A (459 bp) and Smad4 (358 bp), and subjected to agarose gel electrophoresis to detect the amplified products. (Right) Expression levels of wild-type/K359A Smad7 and H141A HDAC-1 were examined. Lysates from NIH3T3 cells infected as in (left) were analyzed by Western blotting using α-Smad7 or α-Flag antibody. (C) Association of Smad7 and HDAC-1 to the promoter region in the Dhfr gene that includes E2F-responsive sites. (Left) The promoter region of the mouse Dhfr gene is schematically represented. Hatched boxes denote E2F-responsive sites Fry et al., 1999). Lower arrows (P1 and P3; P2 and P6) indicate the positions of primer sets for PCR. The upper arrow (labeled as Dhfr) represents the major transcription start site Fry et al., 1999). A putative PCR fragment obtained from a control distal region (316 bp) and that from an E2F site-containing region (629 bp) proximal to the start site are shown as gray bars. (Right) Subregions in the promoter of the Dhfr gene were analyzed by chromatin immunoprecipitation assays by using control IgG and antibodies specific for Smad7, E2F-1 and HDAC-1, respectively. NIH 3T3 cells infected with control (−) or Smad7 vectors (+) were cultured in DMEM with 10% FBS for 14 h and harvested for assays. Genomic DNA fragments in the chromatin collected with each indicated antibody was used as a template for PCR. Products were subjected to electrophoresis to detect the fragments predicted in the left panel.
We have found that a substitution mutation of lysine 359 (K359A), although it did not affect the nuclear localization as observed in the wild-type (unpublished data), thoroughly removed the growth inhibition effect of Smad7 (Fig. 4A). To examine whether Lys359 is important for the association of Smad7 with E2F-1, a Flag-tagged K359A Smad7 (K359A Flag-Smad7) expressed in NIH 3T3 cells was immunoprecipitated from nuclear extracts with an α-Flag antibody. Western blots demonstrated that K359A Flag-Smad7 bound considerably less to endogenous E2F-1 than the wild-type Smad7 (Fig. 4B). A similar result was observed in cotransfected 293T cells expressing HA-E2F-1 and either wild-type or K359A mutant Myc-Smad7 (Fig. 4C).
We subsequently tested whether Lys359 was necessary for Smad7 linking HDAC-1 and E2F-1. For this purpose, combinations of Myc-Smad7 variants, HA-E2F-1 and HDAC-1-Flag expressed in 293T cells were analyzed through immunoprecipitation and Western blotting. K359A Myc-Smad7 bound to both HDAC-1-Flag and its H141A mutant with an efficiency similar to that of wild-type Smad7 (Fig. 5A, middle panel, lanes 4, 5, 7, 8), whereas the amount of K359A Myc-Smad7 binding to HA-E2F-1 was significantly lower when compared with that of wild-type Smad7 (Fig. 5A, lanes 4, 5). Consistently with this, HA-E2F-1 was not detected in immune complexes of HDAC-1 and K359A mutant Smad7 (Fig. 5A, upper panel, lanes 4, 5, 7, 8). Taken together, these data indicate that Lys359 of Smad7 is critical for association with E2F-1, but not with HDAC-1. In addition, we suggest that the proliferation-arresting activity of Smad7, which requires Lys359, acts as a bridge that links E2F-1 to the deacetylase activity of HDAC-1. Although the interaction of Smad7-HDAC-1 complex with E2F-1 appeared relatively weak in coimmunoprecipitation tests, it might be possible that Smad7-HDAC-1 interacts more strongly with complexes of E2F-1 and DP proteins, or E2F-1 and DNA, in vivo.
Smad7 induces repression of E2F-dependent transcription through HDAC activity
Upon observing that Smad7 can form a ternary complex with E2F-1 and HDAC-1, we next examined the effects of Smad7 on E2F-1 function by measuring the expression of genes regulated by E2F-1. Total RNA prepared from NIH 3T3 cells 24 h after infection by vectors holding either the wild-type Smad7 or its K359A mutant were subjected to semi-quantitative RT-PCR analyses for assessment of the transcript levels of various endogenous genes. As shown in Fig. 5B, Smad7 caused a significant transcript decrease in the dihydrofolate reductase (Dhfr) gene, which is known to be responsive to E2F-1 via an E2F-binding sequence (Blake and Azizkhan, 1989; DeGregori et al., 1997; Fry et al., 1997; Fry et al., 1999). In contrast, K359A Smad7, which is unable to bind to E2F-1, had little effect. We observed a similar transcript reduction in Smad7-expressing cells for the thymidine kinase (Tk) gene, but not for the Smad4 gene (Fig. 5B). Because the appearance and proliferation of cells, and the levels of cyclin A transcript were indistinguishable (Fig. 5B) (Kitamura et al., 2005), repression of the Dhfr and Tk genes seems to be a very early response to Smad7 that precedes the cell cycle arrest.
These results suggest that Smad7 caused specific genes to be silenced in a manner dependent on its binding to E2F-1. Moreover, this suggests that Smad7 acts as a bridge between E2F-1 and HDAC-1, the latter being able to remove acetyl groups to reconstitute the chromatin around its association sites. Indeed, H141A HDAC-1 alleviated the Smad7-mediated repression of Dhfr and Tk (Fig. 5B), further supporting the notion that Smad7 exerts its effects by bringing HDAC proteins to E2F-1 situated at the regulatory region of genes that are important for cell cycle progression.
To analyze whether the mouse Dhfr gene is associated with E2F-1, Smad7, HDAC-1, or a combination thereof, its promoter region was subjected to chromatin immunoprecipitation assays. Cell lysates containing cross-linked chromatin were prepared from NIH 3T3 cells infected with either control or Smad7 vectors and then serum-stimulated for the induction of endogenous E2F-1. After sonication, immune complexes including fragmented genomic DNA were purified using an antibody specific to E2F-1, Smad7 or HDAC-1, and used as DNA templates for PCR. Primer sets were designed to amplify a DNA fragment harboring two plausible E2F-recognition sites (proximal region) or a negative control with no noticeable E2F site (distal region) (Blake and Azizkhan, 1989; Fry et al., 1997). The patterns of the amplified fragments indicated that Smad7 located at the proximal but not distal region (Fig. 5C), suggesting the existence of a target site for Smad7 in the former. Indeed, E2F-1 was found to bind Smad7 in this region (Fig. 5C, lane 7), presumably through its ability to recognize sequence-specific DNA, whereas Smad7 had no apparent influence on the association between E2F-1 and DNA (Fig. 5C, lane 8). Interestingly, the expression of Smad7 markedly increased the level of HDAC-1 in the proximal region (Fig. 5C, compare lanes 9, 10), implying a complex that included HDAC-1, Smad7 and E2F-1 at this site. Taken together with the above results that Smad7 linked HDAC-1 and E2F-1, and that Smad7 caused the diminished expression of Dhfr in an HDAC-1-dependent manner, we suggest that nuclear Smad7 may bring HDAC-1 to E2F-1, which is located at the promoter of Dhfr, to induce its repression.
Excess E2F-1 rescues growing cells from Smad7-induced arrest of proliferation
To evaluate the role of E2F in Smad7 proliferation arrest, we tested its effects on NIH 3T3 cells by transducing either or both exogenous E2F-1 and Smad7. At 72 h after serial introduction of the retroviral vectors, the levels of Flag-E2F-1 and Smad7 proteins were indeed substantial, but only slightly influenced by one another (Fig. 6A). While overexpression of E2F-1 is known to stimulate the activity of genes harboring E2F-responsive elements (Fry et al., 1999), the proliferation of cells expressing exogenous E2F-1 was comparable to that of control cells placed in 10% FBS for 72 h (Fig. 6B). In contrast to proliferation-inhibited cells expressing solely Smad7, cells expressing E2F-1 and Smad7 exhibited an almost normal proliferation with nontransformed phenotypes (Fig. 6B). These results indicate that excess amounts of E2F-1 can overcome the inhibitory effects of Smad7. Moreover, they strongly suggest that when endogenous E2F-1 is bound to Smad7, its activity is insufficient for stimulating the genes that are required for the cell cycle progression, and that this is a major mechanistic reason for the arrest of cell proliferation.
Endogenous Smad7 associates with HDAC-1 and E2F-1, and represses an E2F-responsive promoter
In proliferating NIH 3T3 fibroblast cells, endogenous levels of Smad7 that can cause cell cycle arrest are very low. To elucidate the physiological role of the linkage between Smad7, HDAC-1 and E2F-1 in untransfected cells, we used human lung A549 cells which have detectable levels of Smad7 protein expression. We initially examined Smad7 in immune complexes collected with α-HDAC-1 or α-E2F-1 from nuclear extracts of A549 cells. Western blot analyses with α-Smad7 showed that endogenous Smad7 associated with both endogenous HDAC-1 (Fig. 7A) and E2F-1 (Fig. 7B). Our data suggest their nuclear complex is present in untransfected cells in a manner similar to that in Smad7-transduced NIH 3T3 cells.
Fig. 7. Endogenous Smad7 regulating an E2F-responsive promoter.
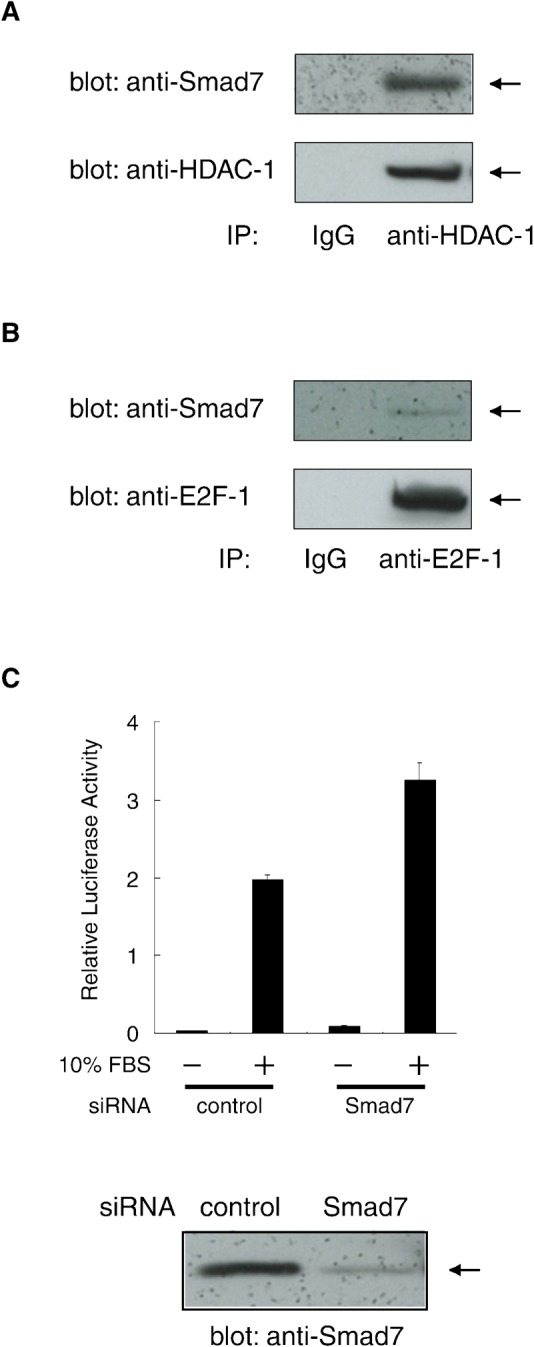
(A) Association of endogenous Smad7 and HDAC-1 proteins in A549 human lung cells. Nuclear extract from untransfected A549 cells were incubated with an α-HDAC-1 antibody or control IgG. Immune complexes were examined by Western blotting using α-Smad7 and α-HDAC-1 antibodies. (B) Association of endogenous Smad7 and E2F-1 proteins. Immune complexes prepared using α-E2F-1 or control IgG as in (A) were examined by Western blotting using α-Smad7 and α-E2F-1 antibodies. (C) Effect of Smad7 knock-down on the transcriptional activation of E2F-responsive promoter. (Upper panel) In combination with a vector for either Smad7-targeting or control siRNA, luciferase reporter (pGVB2-E2F×4) driven by an E2F-responsive promoter was transfected into A549 cells. Cells were serum-starved for 40 h, stimulated in medium containing 10% FBS for 18 h and then lysed for assays. Luciferase activity in the lysates is shown as means with S.D. from triplicate experiments. (Lower panel) Level of endogenous Smad7 in the lysates was examined by Western blotting using α-Smad7.
We next tested if endogenous Smad7 regulated the transcriptional activity of E2F factors. The E2F-responsive reporter plasmid pGVB2-E2F×4 was introduced into A549 cells in combination with either siRNA for Smad7 (siSmad7), or non-targeting control siRNA. Endogenous Smad7 was effectively and specifically knocked-down in siSmad7-transfected cells, as indicated by Western blotting (Fig. 7C, lower panel). Furthermore, upon FBS treatment for endogenous E2F, siSmad7 treatment induced a significant elevation (up to ∼160%) of E2F-responsive reporter activation as compared to control-treated cells (Fig. 7C, upper panel). These results suggest that Smad7 regulates E2F-1-mediated transcriptional activation through an endogenous repressive complex formed with HDAC-1 and E2F-1 in untransfected cells. Although enhanced transcriptional activity of E2F by siSmad7 was observed, its influence on proliferation of A549 cells was not evident (data not shown). This observation is presumably due to the cell-type-dependent effect of Smad7 on cell proliferation, which needs further investigation.
Discussion
The physiological roles of cells depend on multiple signaling pathways that control the cell fate through proliferation, differentiation, or growth arrest. The disruption of this control can lead to pathologies such as tumorigenesis. Maintaining cells in a quiescent state is therefore critical and it indeed occurs through a variety of mechanisms that regulate the activity of E2F family of transcription factors (Frolov and Dyson, 2004; Polager and Ginsberg, 2008). In this study, we found that Smad7 forms a nuclear complex with HDAC-1 and HDAC-3 and brings HDAC-1 to E2F-1, resulting in the repression of E2F-responsive genes. We also found that a Smad7 substitution mutant lacking the ability to bind to E2F-1 can neither act as a transcriptional corepressor nor induce proliferation arrest.
E2F family proteins are transcription factors that recognize specific sequences as a DNA-binding heterodimer with a DP protein and participate in cell cycle control by regulating gene expression (Lam and La Thangue, 1994; Trimarchi and Lees, 2002; Polager and Ginsberg, 2008). E2F-1, -2, and -3, which constitute an activator class of E2F family members, stimulate the gene expression required in S-phase and play important roles for the G1- to S-phase transition, whereas E2F-4, -5, and -6 normally act as dominant repressors to keep cells in G0-phase (Classon and Dyson, 2001; Trimarchi and Lees, 2002). The effects of E2F are controlled by pRb, which has the potential to arrest the cell cycle (Luo et al., 1998; Mulligan and Jacks, 1998; Classon and Dyson, 2001). pRb and its related members, p107 and p130, physically associate with E2F and repress E2F-responsive genes through mechanisms that involve HDAC recruitment (Brehm et al., 1998; Dyson, 1998; Magnaghi-Jaulin et al., 1998; Zhang et al., 2000). In the present study, we have shown that Smad7 causes HDAC-1 to link with E2F-1 at particular recognition sites to repress the activity of E2F target genes. Although the proposed action of Smad7 is seemingly similar to that of pRb i.e. acting as a bridge to form a ternary complex between E2F and HDAC, the role of Smad7 and pRb in cell cycle regulation appears to be quite different. The protein levels of pRb are fairly constant throughout the cell cycle and its function is primarily regulated by phosphorylation (Beijersbergen et al., 1995; Grana et al., 1998; Nevins, 1998). During the progression of G1-phase, pRb sequentially becomes phosphorylated due to the activation of cyclin-dependent kinases, including the cyclins D-Cdk4 and E-Cdk2, leading to the release of E2F which in turn allows the activation of the genes required for the cell cycle progression (Beijersbergen et al., 1995; Dyson, 1998; Grana et al., 1998; Nevins, 1998; Zhang et al., 2000). In contrast, the expression of Smad7 is largely regulated by various extracellular stimuli such as cytokines including TGF-β (Nakao et al., 1997), IFNγ (Ulloa et al., 1999) and TNFα (Bitzer et al., 2000). Therefore Smad7, as opposed to pRb (which maintains cell cycle order), may cause a transient arrest of the cell cycle in proliferating cells. In addition to the slowing down of proliferation due to the stimulation of Cdk inhibitors that occurs in response to the above cytokines (Polyak et al., 1994; Jeoung et al., 1995; Subramaniam et al., 1998; Seoane et al., 2001; Basile et al., 2003), the repressive effect of Smad7 on E2F responsive genes would also contribute to the cell cycle control at the level of transcription.
In NIH 3T3 cells undergoing Smd7-induced proliferation arrest, endogenous HDAC-1 and HDAC-3 were found to form complexes with Smad7. These results are consistent with those of previous reports showing that both Smad6 and Smad7 can associate with various nuclear factors (Bai et al., 2000; Kavsak et al., 2000; Ichijo et al., 2005; Kollias et al., 2006). Comparative assays using a series of transiently overexpressed HDACs have also suggested a preferential interaction between Smad7 and HDAC-1 or HDAC-3 (Simonsson et al., 2005). In addition, the stable expression of a dominant-negative form of HDAC-1 (H141A HDAC-1), which has a point mutation at its catalytic center of deacetylase activity, can potently induce the reversion of Smad7-induced phenotypes that appeared in fibroblast cells. This is likely due to its competition with endogenous HDAC-1 for interacting factors such as Smad7 and other proteins. Along with the critical role of HDACs in a functional pRb complex (Brehm et al., 1998; Luo et al., 1998; Magnaghi-Jaulin et al., 1998), our findings strongly suggest that HDAC-1 activity is essential for Smad7's ability to arrest the cell cycle. The effects of the dominant-negative HDAC-1 on HDAC-3 action were not assayed in this study. Furthermore, we have not tested whether Smad7-associating HDAC-3 has a function similar to that of HDAC-1. Despite the unclear role of HDAC-3 on Smad7 activity, their interaction in vivo might be significant for adipogenesis because both Smad7 and a combination of HDAC-3 and pRb can influence adipocyte differentiation (Choy et al., 2000; Fajas et al., 2002).
The major function of R- and Co-Smad members is to regulate transcription by mediating upstream signals into the nucleus and by interacting with diverse transcription factors (Massague and Wotton, 2000; Moustakas et al., 2001) even though I-Smads have been shown to act as cytoplasmic antagonists in the Smad pathway (Hayashi et al., 1997; Hata et al., 1998). Our and others' findings showing that each I-Smad can bind to different nuclear factors such as DNA-binding proteins, including Hoxc-8 (Bai et al., 2000), glucocorticoid receptor (Ichijo et al., 2005), MyoD (Kollias et al., 2006) and E2F-1 (this study), therefore suggest that I-Smads also actively participate in transcriptional regulation in the nucleus. For example, Smad7 inactivates E2F-responsive genes in NIH 3T3 cells by complex formation with HDAC-1 and E2F-1, indicating that Smad7 can act as a transcriptional corepressor. Similarly, Smad6 is known to repress the osteopontin gene by binding to both Hoxc-8 and HDAC-1, thereby increasing their repression ability in Mv1Lu cells (Bai and Cao, 2002). Direct interaction of Smad7 with endogenous HDACs in NIH 3T3 cells and with recombinant ones in vitro suggest that the nuclear localization of Smad7 may be the result of its retention by partner molecules, such as nuclear HDAC complexes. In this regard, it would be interesting to investigate the similarity in the regulatory mechanism of target gene expression between nuclear Smad7 and R-/Co-Smads. This may provide an evolutionary understanding of the Smad family and help identify if regulation in the nucleus arose as an ancestral function prior to activity in the cytoplasm.
The MH2 domain plays critical and distinct roles in the Smad family through its interactions with various partner proteins (Heldin et al., 1997; Massague and Wotton, 2000). In the present study, we identified the Smad7 MH2 domain as an essential region of interaction with both HDAC-1 and E2F-1. Although these bindings simultaneously occurred in the MH2 domain, they are independent and not mutually exclusive. In particular, the docking site for E2F-1 strictly requires the Lys359 residue of Smad7 while that for HDAC-1 does not, suggesting separate binding locations. The MH2 domain can thus bind to HDAC-1 and E2F-1, linking them together. Although whether such an effect is specific to Smad7 is currently unknown, it was previously shown that Smad3 binds to p107 and either E2F-4 or E2F-5 by its MH2 domain, resulting in a functional link between these nuclear proteins (Chen et al., 2002). In view of the fact that the MH2 domain can associate with numerous kinds of molecules in the Smad pathway (Moustakas et al., 2001; Wu and Hill, 2009), this domain likely serves for intracellular signal crosstalk for bridging two or more factors, which can expand into interaction with other signaling networks.
A conserved pocket structure in the MH2 domain, the loop-strand pocket, has been shown to recognize the C-terminal end peptides of activated Smad1 and Smad2 for oligomerization, and has been suggested to act as a docking site for other proteins containing phospho-serine residues (Qin et al., 2001; Wu et al., 2001). Since the key residues composing the pocket structure are mostly conserved in Smad7, it may have such a site in its MH2 domain. Lys359, a residue crucial for Smad7 proliferation arrest activity, is one such key pocket residue in Smad1 and Smad2 (Qin et al., 2001; Wu et al., 2001) and is thus potentially involved in the interactions with other factors. Our K359A mutant showed little difference in its binding affinity to HDAC-1 compared to the wild-type Smad7. In contrast, binding to E2F-1 was significantly reduced, suggesting that the presumed pocket site is responsible for the interaction between Smad7 and E2F-1. Interestingly, E2F-1 has been shown to be phosphorylated at two Ser residues in G1-phase following serum stimulation on NIH 3T3 cells, hence inhibited its interaction with pRb (Fagan et al., 1994). Thus, Smad7 may recognize E2F-1, preferably in its phosphorylated form, which is free of pRb, and repress E2F-1 transcriptional activity by recruiting HDAC-1. It might also be possible that another protein in place of E2F-1 binds to the loop-strand pocket of Smad7, perhaps in a phosphorylation-dependent fashion. HDAC-1 interaction with Smad7 is nondisturbing and independent of the E2F-1 binding site and this may imply that Smad7 can associate in the pocket region with transcription factors other than E2F-1, the corresponding gene expression through HDAC activity.
The present results demonstrate a molecular basis for the nuclear action of Smad7 on cell cycle arrest. However, further studies are needed to elucidate the physiological relevance of this activity of Smad7. Based on this, we provide a model in which Smad7 brings HDAC-1 to E2F-1 targeted at the loop-strand pocket. This suggests that Smad7 suppresses the activity of diverse transcription factors that interact with this pocket in an HDAC-dependent manner. Smad7 may act as a transcriptional corepressor in a wide range of biological processes.
Materials and Methods
Cell culture
Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma D5796), supplemented with 10% heat-inactivated (56°C, 0.5 h) fetal bovine serum (FBS), and ampicillin and streptomycin at 0.1 mg/ml each. Cell proliferation was assessed by the quantity of DNA in cultures through measurement of the CyQUANT dye fluorescence (Molecular Probes). To monitor DNA replication, 3×103 cells per well were seeded in 96-well plates and labeled in medium containing [methyl-3H]-thymidine (0.5 mCi, Amersham) for the last 4 h of the 72 h culture. Insoluble genomic DNA prepared from cells lysed in 0.1% SDS was trapped onto 96-well filter plates (Packard Instrument) and applied to a scintillation counter for radioactivity counting of the incorporated thymidine.
DNA clones
Expression vectors for murine Smad7 and proviral vector pGψ+pur were prepared as described previously (Kitamura et al., 2000). K359A Smad7 mutant was prepared by replacing the wild-type Lys359 codon (AAA) with an Ala codon (GCA) using oligonucleotide-directed mutagenesis. For truncated variants (amino acid number: 1–408, 1–380, 1–259, 260–426, and 226–426), NheI-XbaI DNA fragments were generated by PCR and attached with a sequence that included the initiator Met and a Myc-tag at the N-terminal. All expression plasmids were constructed by inserting corresponding DNA fragments into an appropriate site of the expression vector pactEF (Okazaki and Sagata, 1995). Cloned cDNAs for C-terminally Flag-tagged human HDAC-1, and H141A mutants of HDAC-1 were kindly provided by Dr. N. Shindoh (Astellas Pharma, Tsukuba, Japan). Complementary DNAs for N-terminally HA-tagged E2F-1 were amplified by PCR from brain cDNA using primer sets designed in accordance with known sequences (INSD accession number: AF516106) and subcloned into the BamHI-XbaI sites of pcDNA3 (Invitrogen). Proviral plasmid DNA pGψ+bsd was constructed by inserting a 0.6-kb AatI-NgoMI fragment bearing the blasticidin resistance gene from pUB6/V5 (Invitrogen) in place of the pur gene in pGψ+pur. The XbaI-BamHI fragments of Myc-Smad7 and E2F-1 and the BamHI-XbaI fragments of HDAC-1-Flag and H141A HDAC-1-Flag were subcloned into both pGψ+pur and pGψ+bsd. For GST-fusion proteins, cDNAs encoding Smad7 and its variants were subcloned between the XbaI and BamHI sites in pGEX-6P-1X, which was made by inserting a linker with the internal XbaI and BamHI sites (complementary 5′-gatcgtctagaattgaattcccgggatccc-3′, 5′-tcgagggatcccgggaattcaattctagac-3′) into the BamHI and XhoI sites of pGEX-6P-1 (Pharmacia). For GST-Flag-HDAC proteins, the PCR-generated XbaI-BamHI DNA fragments encoding N-terminally Flag-tagged HDAC-1, its truncated variants (a.a. 1–191, 1–327, 192–482, and 328–482), HDAC-2 and HDAC-3 were inserted into pGEX-6P-1X in a similar manner. For construction of the E2F responsive luciferase reporter, pGVB2-E2Fx4, synthetic oligonucleotides containing four tandem E2F-binding sites from the E2A promoter (5′-aaacgcgtgaattctagttttcgcgcttaaattagttttcgcgcttaaattagttttcgcgcttaaattagttttcgcgcttaaatggatccagatctaa-3′, 5′-ttagatctggatccatttaagcgcgaaaactaatttaagcgcgaaaactaatttaagcgcgaaaactaatttaagcgcgaaaactagaattcacgcgttt-3′) (Helin, 1998) were ligated into the EcoRI-BglII sites of pGVB2 (Toyobo, Japan).
DNA transfection and retrovirus preparation
Embryonic kidney 293T cells were transfected with DNA constructs using a calcium-phosphate coprecipitation procedure, while the supernatants containing retroviruses were harvested as described previously (Kitamura et al., 2000). In some assays, cells in subconfluent cultures were transfected with the indicated vector DNA using FuGENE6 (Roche) (Aota et al., 2003). For double infection, NIH 3T3 cells were first incubated with HDAC-1, the H141A HDAC-1 mutant, E2F-1, or control vectors for 4 h and supplemented with fresh medium for 8 h. Thereafter the cells were infected with vectors expressing Smad7 or a control for 4 h and incubated for 8 h before re-plating.
Immunoblotting and immunoprecipitation
Cells were lysed as described previously (Okazaki and Sagata, 1995). In immunoblotting experiments, a fixed amount (20 µg) of total protein was used per lane. The Smad7-specific antibody was affinity-purified from rabbit serum as described previously (Kitamura et al., 2000; Yoshikawa et al., 2000). For detection of human endogenous Smad7, rabbit anti-Smad7 antibody was obtained from Invitrogen. Rabbit antibodies against histone H3 (C-16), cyclin D1 (H-295), cyclin A (C-19), HDAC-1 (H-51), HDAC-2 (H-54), HDAC-3 (H-99), and E2F-1 (C-20) were obtained from Santa Cruz Biotechnology, and acetylated histone H3 (Lys9/14) from Upstate Biotechnology. Mouse monoclonal antibodies against the Myc epitope tag (9E10) and Smad4 (B-8) were provided by Santa Cruz Biotechnology; Flag (M2) by Sigma; and Rb (G3-245) and p27 (clone 57) by Pharmingen. A rat monoclonal antibody against HA-tag (3F10) was purchased from Roche. Horseradish peroxidase-conjugated IgGs against rabbit or rat Ig (Jackson ImmunoResearch) and against the mouse κ light chain of Ig (Southern Biotechnology) were used as secondary reagents.
For immunoprecipitation under nondenaturing conditions, cells were lysed as described above and extracted on ice for 0.5 h. The cell lysate was clarified by centrifugation at 16,000 g for 15 min at 4°C. Each supernatant was incubated on ice for 2 h with an appropriate antibody. The immune complexes were adsorbed onto protein A-Sepharose beads (Amersham Pharmacia) at constant rotation for 1 h at 4°C, followed by four washes with 1 ml of ice-cold lysis buffer, elution in SDS sample buffer and analysis as described above.
For preparation of the nuclear extract from Smad7 transfected cells, NIH 3T3 cells (2.5×106) were seeded in a 100-mm dish on the day before transfection with the indicated expression vectors. After 16 h, cells were rendered quiescent in medium containing reduced (0.2%) FBS for 24 h, followed by stimulation of the medium with 10% FBS for 14 h. Nuclei were isolated from cells (Smad7 transfected NIH 3T3 cells or non-transfected A549 cells) that were collected in 10 mM Hepes-KOH, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM PMSF and 0.2% NP-40, and then extracted in buffer containing 20 mM Hepes-KOH, pH 7.9, 0.4 M NaCl, 1 mM EGTA, 1 mM EDTA, 1 mM DTT and 1 mM PMFS on ice for 1 h. Each extract, including 300 µg of protein, was subjected to immunoprecipitation after three-fold dilution with buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl and 1 mM DTT.
In vitro binding assays with recombinant proteins
GST-Smad7 and GST-Flag-HDAC proteins were expressed in E. coli BL21(DE3) cells (Novagen) by induction with 0.5 mM IPTG at 15°C for 18 h. GST fusions in the cleared lysate from harvested cells were bound to glutathione-Sepharose 4B beads according to the manufacturer's instructions (Amersham Pharmacia). Flag-HDAC proteins were separated from the beads in solution by cleaving GST-Flag-HDACs with 80 µ/ml of a sequence-specific protease (PreScission; Amersham Pharmacia) at 4°C for 16 h.
Control GST and GST-Smad7 (each corresponding to 100 ng) bound to glutathione beads were incubated with Flag-HDAC protein (100 ng each) in 100 µl buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM DTT and 1% BSA) for 30 min at 4°C at constant rotation. The beads were extensively washed four times with 1 ml ice-cold buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA and 0.1% NP40. Protein complexes were eluted from the beads and denatured by boiling in SDS sample buffer for immunoblotting, as described above.
Reverse transcriptase-PCR analyses
Total RNA was extracted by ISOGEN solution (Wako) and used for the synthesis of cDNA using the SuperScript preamplification system (GIBCO) and then treated with RNaseH. PCR was performed with KOD Dash DNA polymerase (Toyobo, Osaka, Japan) as follows: denaturation at 96°C for 2 min followed by 25–30 cycles at 96°C for 30 s, 62°C for 2 s and 74°C for 30 s. Primer sets specific for murine Dhfr cDNA (upstream: 5′-tcaaagaaccaccacgaggag-3′, downstream: 5′-tgcatagctttaggaggggag-3′), tk cDNA (5′-agctacatcaatctgcccacc-3′, 5′-ggttcaagatgctgccgaaag-3′), cyclin A cDNA (5′-atgtcactgctggtccttcat-3′, 5′-tagttcacagccaaatgcagg-3′) and Smad4 cDNA (5′-ggtgctccattgcttac-3′, 5′-ctgccgcagatcaaagac-3′) were designed. PCR products were analyzed by agarose gel electrophoresis. Dilution series of cDNA were tested for quantitative evaluation of each RNA in order to ensure linear amplification of the targets.
Immunofluorescent staining
On the day following infection of the retrovirus vector, NIH 3T3 cells were re-plated on slides (eight-well Lab-Tek chamber, Nunc) and further cultured for 24 h. Cells were then fixed in Hepes-buffered saline containing 4% paraformaldehyde for 10 min and permeabilized with 0.2% Triton X-100 in TBS for 5 min. After blocking with 3% BSA in TBS for 1 h, cells were incubated with rabbit anti-Smad7 antibody or mouse anti-Flag antibody in 3% BSA for 2 h, followed by incubation with Alexa488-labeled α-rabbit Ig antibody or Cy3-labeled α-mouse Ig antibody, respectively, in 5% skimmed-milk in TBS for 1 h. Following each incubation, cells were extensively washed with TBST high salt buffer (20 mM Tris-HCl, pH 7.5, 1 M NaCl and 0.1% Tween-20) (Kitamura et al., 2000). Stained cells were examined by fluorescence microscopy and photographed (Axiophoto).
Chromatin immunoprecipitation (ChIP) assays
Cells were serum starved as described above, re-entered into the cell cycle by the addition of medium containing 10% FBS, and cultured for 14 h before being subjected to ChIP analyses (ChIP assay kit; Upstate Biotechnology). Antibodies used for immunoprecipitation are described above. DNA fragments brought down by immunoprecipitation were detected by PCR with KOD Dash DNA polymerase as follows: denaturation at 96°C for 2 min followed by 40 cycles at 96°C for 30 s, 62°C for 2 s and 74°C for 30 s. Primer sets were designed to have distal (P1: 5′-ctttcttctagtcagccaagc-3′, P3: 5′-ggagagcttattaatggtggg-3′) and proximal (P2: 5′-cccaccattaataagctctcc-3′, P6: 5′-gtttggcgcgaaatcgcagc-3′) regions in the promoter of the murine Dhfr gene (Fry et al., 1997).
siRNA transfection and reporter experiments
Small interfering RNA (siRNA) for Smad7 and non-targeting control siRNA were purchased from Santa Cruz Biotech (SC-36508 and SC-37007, respectively). Transfection of pGVB2-E2F×4 in combination with siRNA into A549 cells was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Briefly, 5×104 cells were plated in 24-well plates on the previous day of transfection. On the following day, the cells were transfected with pGVB2-E2F×4 (100 ng) in combination with siRNA (20 pmol) using Lipofectamine 2000. After 16 h transfection, the cells were washed with PBS and serum-starved in DMEM containing 0.2% FBS for 40 h, followed by stimulation with 10% FBS for 18 h. The cells were then lysed and subjected to luciferase assay using Luciferase Assay System (Promega).
Acknowledgments
We would like to thank Nobuaki Shindoh for the human cDNA encoding HDAC-1, H141A HDAC-1, HDAC-2 and HDAC-3. We also thank Ruriko Sakamoto for her kind help and Shin-ichi Aota for his valuable discussions. This research was supported in part by the New Energy and Industrial Technology Development Organization of Japan. This study was partly supported by the Ministry of Education, Culture, Sports, Science and Technology of Japan, Grant-in-Aid for Scientific Research (B) to K. O.
References
- Afrakhte M., Moren A., Jossan S., Itoh S., Sampath K., Westermark B., Heldin C. H., Heldin N. E., ten Dijke P. (1998). Induction of inhibitory Smad6 and Smad7 mRNA by TGF-beta family members. Biochem. Biophys. Res. Commun. 249, 505–511 10.1006/bbrc.1998.9170 [DOI] [PubMed] [Google Scholar]
- Allfrey V. G., Faulkner R., Mirsky A. E. (1964). Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 51, 786–794 10.1073/pnas.51.5.786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aota S., Nakajima N., Sakamoto R., Watanabe S., Ibaraki N., Okazaki K. (2003). Pax6 autoregulation mediated by direct interaction of Pax6 protein with the head surface ectoderm-specific enhancer of the mouse Pax6 gene. Dev. Biol. 257, 1–13 10.1016/S0012-1606(03)00058-7 [DOI] [PubMed] [Google Scholar]
- Bai S., Cao X. (2002). A nuclear antagonistic mechanism of inhibitory Smads in transforming growth factor-beta signaling. J. Biol. Chem. 277, 4176–4182 10.1074/jbc.M105105200 [DOI] [PubMed] [Google Scholar]
- Bai S., Shi X., Yang X., Cao X. (2000). Smad6 as a transcriptional corepressor. J. Biol. Chem. 275, 8267–8270 10.1074/jbc.275.12.8267 [DOI] [PubMed] [Google Scholar]
- Baltus G. A., Kowalski M. P., Zhai H., Tutter A. V., Quinn D., Wall D., Kadam S. (2009). Acetylation of sox2 induces its nuclear export in embryonic stem cells. Stem Cells 27, 2175–2184 10.1002/stem.168 [DOI] [PubMed] [Google Scholar]
- Basile J. R., Eichten A., Zacny V., Munger K. (2003). NF-kappaB-mediated induction of p21(Cip1/Waf1) by tumor necrosis factor alpha induces growth arrest and cytoprotection in normal human keratinocytes. Mol. Cancer Res. 1, 262–270 [PubMed] [Google Scholar]
- Beijersbergen R. L., Carlee L., Kerkhoven R. M., Bernards R. (1995). Regulation of the retinoblastoma protein-related p107 by G1 cyclin complexes. Genes Dev. 9, 1340–1353 10.1101/gad.9.11.1340 [DOI] [PubMed] [Google Scholar]
- Bhushan A., Chen Y., Vale W. (1998). Smad7 inhibits mesoderm formation and promotes neural cell fate in Xenopus embryos. Dev. Biol. 200, 260–268 10.1006/dbio.1998.8965 [DOI] [PubMed] [Google Scholar]
- Bitzer M., von Gersdorff G., Liang D., Dominguez-Rosales A., Beg A. A., Rojkind M., Bottinger E. P. (2000). A mechanism of suppression of TGF-beta/SMAD signaling by NF-kappa B/RelA. Genes Dev. 14, 187–197 10.1101/gad.14.2.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake M. C., Azizkhan J. C. (1989). Transcription factor E2F is required for efficient expression of the hamster dihydrofolate reductase gene in vitro and in vivo. Mol. Cell. Biol. 9, 4994–5002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolden J. E., Peart M. J., Johnstone R. W. (2006). Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 5, 769–784 10.1038/nrd2133 [DOI] [PubMed] [Google Scholar]
- Brehm A., Miska E. A., McCance D. J., Reid J. L., Bannister A. J., Kouzarides T. (1998). Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391, 597–601 10.1038/35404 [DOI] [PubMed] [Google Scholar]
- Chen C. R., Kang Y., Siegel P. M., Massague J. (2002). E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell 110, 19–32 10.1016/S0092-8674(02)00801-2 [DOI] [PubMed] [Google Scholar]
- Chen X., Weisberg E., Fridmacher V., Watanabe M., Naco G., Whitman M. (1997). Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature 389, 85–89 10.1038/38008 [DOI] [PubMed] [Google Scholar]
- Choy L., Skillington J., Derynck R. (2000). Roles of autocrine TGF-beta receptor and Smad signaling in adipocyte differentiation. J. Cell Biol. 149, 667–682 10.1083/jcb.149.3.667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Classon M., Dyson N. (2001). p107 and p130: versatile proteins with interesting pockets. Exp. Cell Res. 264, 135–147 10.1006/excr.2000.5135 [DOI] [PubMed] [Google Scholar]
- Cress W. D., Seto E. (2000). Histone deacetylases, transcriptional control, and cancer. J. Cell. Physiol. 184, 1–16 [DOI] [PubMed] [Google Scholar]
- de Ruijter A. J., van Gennip A. H., Caron H. N., Kemp S., van Kuilenburg A. B. (2003). Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 370, 737–749 10.1042/BJ20021321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregori J., Leone G., Miron A., Jakoi L., Nevins J. R. (1997). Distinct roles for E2F proteins in cell growth control and apoptosis. Proc. Natl. Acad. Sci. USA 94, 7245–7250 10.1073/pnas.94.14.7245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson N. (1998). The regulation of E2F by pRB-family proteins. Genes Dev. 12, 2245–2262 10.1101/gad.12.15.2245 [DOI] [PubMed] [Google Scholar]
- Fagan R., Flint K. J., Jones N. (1994). Phosphorylation of E2F-1 modulates its interaction with the retinoblastoma gene product and the adenoviral E4 19 kDa protein. Cell 78, 799–811 10.1016/S0092-8674(94)90522-3 [DOI] [PubMed] [Google Scholar]
- Fajas L., Egler V., Reiter R., Hansen J., Kristiansen K., Debril M. B., Miard S., Auwerx J. (2002). The retinoblastoma-histone deacetylase 3 complex inhibits PPARgamma and adipocyte differentiation. Dev. Cell 3, 903–910 10.1016/S1534-5807(02)00360-X [DOI] [PubMed] [Google Scholar]
- Flaus A., Owen-Hughes T. (2001). Mechanisms for ATP-dependent chromatin remodelling. Curr. Opin. Genet. Dev. 11, 148–154 10.1016/S0959-437X(00)00172-6 [DOI] [PubMed] [Google Scholar]
- Freiman R. N., Tjian R. (2003). Regulating the regulators: lysine modifications make their mark. Cell 112, 11–17 10.1016/S0092-8674(02)01278-3 [DOI] [PubMed] [Google Scholar]
- Frolov M. V., Dyson N. J. (2004). Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J. Cell Sci. 117, 2173–2181 10.1242/jcs.01227 [DOI] [PubMed] [Google Scholar]
- Fry C. J., Slansky J. E., Farnham P. J. (1997). Position-dependent transcriptional regulation of the murine dihydrofolate reductase promoter by the E2F transactivation domain. Mol. Cell. Biol. 17, 1966–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry C. J., Pearson A., Malinowski E., Bartley S. M., Greenblatt J., Farnham P. J. (1999). Activation of the murine dihydrofolate reductase promoter by E2F1. A requirement for CBP recruitment. J. Biol. Chem. 274, 15883–15891 [DOI] [PubMed] [Google Scholar]
- Grana X., Garriga J., Mayol X. (1998). Role of the retinoblastoma protein family, pRB, p107 and p130 in the negative control of cell growth. Oncogene 17, 3365–3383 10.1038/sj.onc.1202575 [DOI] [PubMed] [Google Scholar]
- Gronroos E., Hellman U., Heldin C. H., Ericsson J. (2002). Control of Smad7 stability by competition between acetylation and ubiquitination. Mol. Cell 10, 483–493 10.1016/S1097-2765(02)00639-1 [DOI] [PubMed] [Google Scholar]
- Hassig C. A., Tong J. K., Fleischer T. C., Owa T., Grable P. G., Ayer D. E., Schreiber S. L. (1998). A role for histone deacetylase activity in HDAC1-mediated transcriptional repression. Proc. Natl. Acad. Sci. USA 95, 3519–3524 10.1073/pnas.95.7.3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata A., Lagna G., Massague J., Hemmati-Brivanlou A. (1998). Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev. 12, 186–197 10.1101/gad.12.2.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H., Abdollah S., Qiu Y., Cai J., Xu Y. Y., Grinnell B. W., Richardson M. A., Topper J. N., Gimbrone M. A., Jr, Wrana J. L. et al. (1997). The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell 89, 1165–1173 10.1016/S0092-8674(00)80303-7 [DOI] [PubMed] [Google Scholar]
- Heikkinen P. T., Nummela M., Jokilehto T., Grenman R., Kahari V. M., Jaakkola P. M. (2010). Hypoxic conversion of SMAD7 function from an inhibitor into a promoter of cell invasion. Cancer Res. 70, 5984–5993 10.1158/0008-5472.CAN-09-3777 [DOI] [PubMed] [Google Scholar]
- Heldin C. H., Miyazono K., ten Dijke P. (1997). TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 390, 465–471 10.1038/37284 [DOI] [PubMed] [Google Scholar]
- Heldin C. H., Landstrom M., Moustakas A. (2009). Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 21, 166–176 10.1016/j.ceb.2009.01.021 [DOI] [PubMed] [Google Scholar]
- Helin K. (1998). Regulation of cell proliferation by the E2F transcription factors. Curr. Opin. Genet. Dev. 8, 28–35 10.1016/S0959-437X(98)80058-0 [DOI] [PubMed] [Google Scholar]
- Hogan B. L. (1996). Bone morphogenetic proteins in development. Curr. Opin. Genet. Dev. 6, 432–438 10.1016/S0959-437X(96)80064-5 [DOI] [PubMed] [Google Scholar]
- Humphrey G. W., Wang Y. H., Hirai T., Padmanabhan R., Panchision D. M., Newell L. F., McKay R. D., Howard B. H. (2008). Complementary roles for histone deacetylases 1, 2, and 3 in differentiation of pluripotent stem cells. Differentiation 76, 348–356 10.1111/j.1432-0436.2007.00232.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichijo T., Voutetakis A., Cotrim A. P., Bhattachryya N., Fujii M., Chrousos G. P., Kino T. (2005). The Smad6-histone deacetylase 3 complex silences the transcriptional activity of the glucocorticoid receptor: potential clinical implications. J. Biol. Chem. 280, 42,067–42,077 10.1074/jbc.M509338200 [DOI] [PubMed] [Google Scholar]
- Imamura T., Takase M., Nishihara A., Oeda E., Hanai J., Kawabata M., Miyazono K. (1997). Smad6 inhibits signalling by the TGF-beta superfamily. Nature 389, 622–626 10.1038/39355 [DOI] [PubMed] [Google Scholar]
- Inman G. J., Nicolas F. J., Hill C. S. (2002). Nucleocytoplasmic shuttling of Smads 2, 3, and 4 permits sensing of TGF-beta receptor activity. Mol. Cell 10, 283–294 10.1016/S1097-2765(02)00585-3 [DOI] [PubMed] [Google Scholar]
- Ito A., Kawaguchi Y., Lai C. H., Kovacs J. J., Higashimoto Y., Appella E., Yao T. P. (2002). MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 21, 6236–6245 10.1093/emboj/cdf616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh S., Landstrom M., Hermansson A., Itoh F., Heldin C. H., Heldin N. E., ten Dijke P. (1998). Transforming growth factor beta1 induces nuclear export of inhibitory Smad7. J. Biol. Chem. 273, 29,195–29,201 [DOI] [PubMed] [Google Scholar]
- Jeoung D. I., Tang B., Sonenberg M. (1995). Effects of tumor necrosis factor-alpha on antimitogenicity and cell cycle-related proteins in MCF-7 cells. J. Biol. Chem. 270, 18367–18373 [DOI] [PubMed] [Google Scholar]
- Jin Y. H., Jeon E. J., Li Q. L., Lee Y. H., Choi J. K., Kim W. J., Lee K. Y., Bae S. C. (2004). Transforming growth factor-beta stimulates p300-dependent RUNX3 acetylation, which inhibits ubiquitination-mediated degradation. J. Biol. Chem. 279, 29409–29417 10.1074/jbc.M313120200 [DOI] [PubMed] [Google Scholar]
- Johnstone R. W. (2002). Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 1, 287–299 10.1038/nrd772 [DOI] [PubMed] [Google Scholar]
- Kavsak P., Rasmussen R. K., Causing C. G., Bonni S., Zhu H., Thomsen G. H., Wrana J. L. (2000). Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol. Cell 6, 1365–1375 10.1016/S1097-2765(00)00134-9 [DOI] [PubMed] [Google Scholar]
- Khochbin S., Verdel A., Lemercier C., Seigneurin-Berny D. (2001). Functional significance of histone deacetylase diversity. Curr. Opin. Genet. Dev. 11, 162–166 10.1016/S0959-437X(00)00174-X [DOI] [PubMed] [Google Scholar]
- Kitamura K., Aota S., Sakamoto R., Yoshikawa S. I., Okazaki K. (2000). Smad7 selectively interferes with different pathways of activin signaling and inhibits erythroid leukemia cell differentiation. Blood 95, 3371–3379 [PubMed] [Google Scholar]
- Kitamura K., Aota S., Sakamoto R., Emori T., Okazaki K. (2005). Smad7 induces G0/G1 cell cycle arrest in mesenchymal cells by inhibiting the expression of G1 cyclins. Dev. Growth Differ. 47, 537–552 10.1111/j.1440-169X.2005.00829.x [DOI] [PubMed] [Google Scholar]
- Kollias H. D., Perry R. L., Miyake T., Aziz A., McDermott J. C. (2006). Smad7 promotes and enhances skeletal muscle differentiation. Mol. Cell. Biol. 26, 6248–6260 10.1128/MCB.00384-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. (1999). Histone acetylases and deacetylases in cell proliferation. Curr. Opin. Genet. Dev. 9, 40–48 10.1016/S0959-437X(99)80006-9 [DOI] [PubMed] [Google Scholar]
- Kouzarides T. (2000). Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 19, 1176–1179 10.1093/emboj/19.6.1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam E. W., La Thangue N. B. (1994). DP and E2F proteins: coordinating transcription with cell cycle progression. Curr. Opin. Cell Biol. 6, 859–866 10.1016/0955-0674(94)90057-4 [DOI] [PubMed] [Google Scholar]
- Lee K. K., Workman J. L. (2007). Histone acetyltransferase complexes: one size doesn't fit all. Nat. Rev. Mol. Cell Biol. 8, 284–295 10.1038/nrm2145 [DOI] [PubMed] [Google Scholar]
- Luo R. X., Postigo A. A., Dean D. C. (1998). Rb interacts with histone deacetylase to repress transcription. Cell 92, 463–473 10.1016/S0092-8674(00)80940-X [DOI] [PubMed] [Google Scholar]
- Magnaghi-Jaulin L., Groisman R., Naguibneva I., Robin P., Lorain S., Le Villain J. P., Troalen F., Trouche D., Harel-Bellan A. (1998). Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature 391, 601–605 10.1038/35410 [DOI] [PubMed] [Google Scholar]
- Mal A., Sturniolo M., Schiltz R. L., Ghosh M. K., Harter M. L. (2001). A role for histone deacetylase HDAC1 in modulating the transcriptional activity of MyoD: inhibition of the myogenic program. EMBO J. 20, 1739–1753 10.1093/emboj/20.7.1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein R., Roth S. Y. (2001). Histone acetyltransferases: function, structure, and catalysis. Curr. Opin. Genet. Dev. 11, 155–161 10.1016/S0959-437X(00)00173-8 [DOI] [PubMed] [Google Scholar]
- Massague J. (1998). TGF-beta signal transduction. Annu. Rev. Biochem. 67, 753–791 10.1146/annurev.biochem.67.1.753 [DOI] [PubMed] [Google Scholar]
- Massague J., Wotton D. (2000). Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 19, 1745–1754 10.1093/emboj/19.8.1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake T., Alli N. S., McDermott J. C. (2010). Nuclear function of Smad7 promotes myogenesis. Mol. Cell. Biol. 30, 722–735 10.1128/MCB.01005-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustakas A., Souchelnytskyi S., Heldin C. H. (2001). Smad regulation in TGF-beta signal transduction. J. Cell Sci. 114, 4359–4369 [DOI] [PubMed] [Google Scholar]
- Mulligan G., Jacks T. (1998). The retinoblastoma gene family: cousins with overlapping interests. Trends Genet. 14, 223–229 10.1016/S0168-9525(98)01470-X [DOI] [PubMed] [Google Scholar]
- Nagy L., Kao H. Y., Chakravarti D., Lin R. J., Hassig C. A., Ayer D. E., Schreiber S. L., Evans R. M. (1997). Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell 89, 373–380 10.1016/S0092-8674(00)80218-4 [DOI] [PubMed] [Google Scholar]
- Nakao A., Afrakhte M., Moren A., Nakayama T., Christian J. L., Heuchel R., Itoh S., Kawabata M., Heldin N. E., Heldin C. H. et al. (1997). Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 389, 631–635 10.1038/39369 [DOI] [PubMed] [Google Scholar]
- Nakayama T., Gardner H., Berg L. K., Christian J. L. (1998). Smad6 functions as an intracellular antagonist of some TGF-beta family members during Xenopus embryogenesis. Genes Cells 3, 387–394 10.1046/j.1365-2443.1998.00196.x [DOI] [PubMed] [Google Scholar]
- Nevins J. R. (1998). Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ. 9, 585–593 [PubMed] [Google Scholar]
- Okazaki K., Sagata N. (1995). The Mos/MAP kinase pathway stabilizes c-Fos by phosphorylation and augments its transforming activity in NIH 3T3 cells. EMBO J. 14, 5048–5059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips D. M. (1968). N-Terminal acetyl-peptides from two calf thymus histones. Biochem. J. 107, 135–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polager S., Ginsberg D. (2008). E2F - at the crossroads of life and death. Trends Cell Biol. 18, 528–535 10.1016/j.tcb.2008.08.003 [DOI] [PubMed] [Google Scholar]
- Polyak K., Kato J. Y., Solomon M. J., Sherr C. J., Massague J., Roberts J. M., Koff A. (1994). p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 8, 9–22 10.1101/gad.8.1.9 [DOI] [PubMed] [Google Scholar]
- Qin B. Y., Chacko B. M., Lam S. S., de Caestecker M. P., Correia J. J., Lin K. (2001). Structural basis of Smad1 activation by receptor kinase phosphorylation. Mol. Cell 8, 1303–1312 10.1016/S1097-2765(01)00417-8 [DOI] [PubMed] [Google Scholar]
- Quan T., He T., Voorhees J. J., Fisher G. J. (2001). Ultraviolet irradiation blocks cellular responses to transforming growth factor-beta by down-regulating its type-II receptor and inducing Smad7. J. Biol. Chem. 276, 26349–26356 10.1074/jbc.M010835200 [DOI] [PubMed] [Google Scholar]
- Raftery L. A., Sutherland D. J. (1999). TGF-beta family signal transduction in Drosophila development: from Mad to Smads. Dev. Biol. 210, 251–268 10.1006/dbio.1999.9282 [DOI] [PubMed] [Google Scholar]
- Sambucetti L. C., Fischer D. D., Zabludoff S., Kwon P. O., Chamberlin H., Trogani N., Xu H., Cohen D. (1999). Histone deacetylase inhibition selectively alters the activity and expression of cell cycle proteins leading to specific chromatin acetylation and antiproliferative effects. J. Biol. Chem. 274, 34940–34947 10.1074/jbc.274.49.34940 [DOI] [PubMed] [Google Scholar]
- Seoane J., Pouponnot C., Staller P., Schader M., Eilers M., Massague J. (2001). TGFbeta influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat. Cell Biol. 3, 400–408 10.1038/35070086 [DOI] [PubMed] [Google Scholar]
- Simonsson M., Heldin C. H., Ericsson J., Gronroos E. (2005). The balance between acetylation and deacetylation controls Smad7 stability. J. Biol. Chem. 280, 21,797–21,803 10.1074/jbc.M503134200 [DOI] [PubMed] [Google Scholar]
- Spange S., Wagner T., Heinzel T., Kramer O. H. (2009). Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 41, 185–198 10.1016/j.biocel.2008.08.027 [DOI] [PubMed] [Google Scholar]
- Sporn M. B., Roberts A. B. (1992). Transforming growth factor-beta: recent progress and new challenges. J. Cell Biol. 119, 1017–1021 10.1083/jcb.119.5.1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam P. S., Cruz P. E., Hobeika A. C., Johnson H. M. (1998). Type I interferon induction of the Cdk-inhibitor p21WAF1 is accompanied by ordered G1 arrest, differentiation and apoptosis of the Daudi B-cell line. Oncogene 16, 1885–1890 10.1038/sj.onc.1201712 [DOI] [PubMed] [Google Scholar]
- Suzuki C., Murakami G., Fukuchi M., Shimanuki T., Shikauchi Y., Imamura T., Miyazono K. (2002). Smurf1 regulates the inhibitory activity of Smad7 by targeting Smad7 to the plasma membrane. J. Biol. Chem. 277, 39919–39925 10.1074/jbc.M201901200 [DOI] [PubMed] [Google Scholar]
- Takase M., Imamura T., Sampath T. K., Takeda K., Ichijo H., Miyazono K., Kawabata M. (1998). Induction of Smad6 mRNA by bone morphogenetic proteins. Biochem. Biophys. Res. Commun. 244, 26–29 10.1006/bbrc.1998.8200 [DOI] [PubMed] [Google Scholar]
- Tang H., Goldman D. (2006). Activity-dependent gene regulation in skeletal muscle is mediated by a histone deacetylase (HDAC)-Dach2-myogenin signal transduction cascade. Proc. Natl. Acad. Sci. USA 103, 16977–16982 10.1073/pnas.0601565103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermann S., Lehrmann H., Polesskaya A., Harel-Bellan A. (2001). Histone acetylation and disease. Cell. Mol. Life Sci. 58, 728–736 10.1007/PL00000896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topper J. N., Cai J., Qiu Y., Anderson K. R., Xu Y. Y., Deeds J. D., Feeley R., Gimeno C. J., Woolf E. A., Tayber O. et al. (1997). Vascular MADs: two novel MAD-related genes selectively inducible by flow in human vascular endothelium. Proc. Natl. Acad. Sci. USA 94, 9314–9319 10.1073/pnas.94.17.9314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimarchi J. M., Lees J. A. (2002). Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 3, 11–20 10.1038/nrm714 [DOI] [PubMed] [Google Scholar]
- Ulloa L., Doody J., Massague J. (1999). Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature 397, 710–713 10.1038/17826 [DOI] [PubMed] [Google Scholar]
- Vervoorts J., Luscher-Firzlaff J. M., Rottmann S., Lilischkis R., Walsemann G., Dohmann K., Austen M., Luscher B. (2003). Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep. 4, 484–490 10.1038/sj.embor.embor821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilting R. H., Yanover E., Heideman M. R., Jacobs H., Horner J., van der Torre J., DePinho R. A., Dannenberg J. H. (2010). Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J. 29, 2586–2597 10.1038/emboj.2010.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrana J. L., Attisano L., Wieser R., Ventura F., Massague J. (1994). Mechanism of activation of the TGF-beta receptor. Nature 370, 341–347 10.1038/370341a0 [DOI] [PubMed] [Google Scholar]
- Wu J. W., Hu M., Chai J., Seoane J., Huse M., Li C., Rigotti D. J., Kyin S., Muir T. W., Fairman R. et al. (2001). Crystal structure of a phosphorylated Smad2. Recognition of phosphoserine by the MH2 domain and insights on Smad function in TGF-beta signaling. Mol. Cell 8, 1277–1289 10.1016/S1097-2765(01)00421-X [DOI] [PubMed] [Google Scholar]
- Wu M. Y., Hill C. S. (2009). Tgf-beta superfamily signaling in embryonic development and homeostasis. Dev. Cell 16, 329–343 10.1016/j.devcel.2009.02.012 [DOI] [PubMed] [Google Scholar]
- Yamaguchi T., Cubizolles F., Zhang Y., Reichert N., Kohler H., Seiser C., Matthias P. (2010). Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 24, 455–469 10.1101/gad.552310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. J., Seto E. (2007). HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26, 5310–5318 10.1038/sj.onc.1210599 [DOI] [PubMed] [Google Scholar]
- Yang X. J., Seto E. (2008). Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol. Cell 31, 449–461 10.1016/j.molcel.2008.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yingling J. M., Das P., Savage C., Zhang M., Padgett R. W., Wang X. F. (1996). Mammalian dwarfins are phosphorylated in response to transforming growth factor beta and are implicated in control of cell growth. Proc. Natl. Acad. Sci. USA 93, 8940–8944 10.1073/pnas.93.17.8940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa S. I., Aota S., Shirayoshi Y., Okazaki K. (2000). The ActR-I activin receptor protein is expressed in notochord, lens placode and pituitary primordium cells in the mouse embryo. Mech. Dev. 91, 439–444 10.1016/S0925-4773(99)00320-2 [DOI] [PubMed] [Google Scholar]
- Zhang H. S., Gavin M., Dahiya A., Postigo A. A., Ma D., Luo R. X., Harbour J. W., Dean D. C. (2000). Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell 101, 79–89 10.1016/S0092-8674(00)80625-X [DOI] [PubMed] [Google Scholar]
- Zhang Y., Feng X., We R., Derynck R. (1996). Receptor-associated Mad homologues synergize as effectors of the TGF-beta response. Nature 383, 168–172 10.1038/383168a0 [DOI] [PubMed] [Google Scholar]
- Zhong H., May M. J., Jimi E., Ghosh S. (2002). The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol. Cell 9, 625–636 10.1016/S1097-2765(02)00477-X [DOI] [PubMed] [Google Scholar]