

Summary
Recent studies have shown that factors involved in transcription-coupled mRNA processing are important for the maintenance of genome integrity. How these processes are linked and regulated in vivo remains largely unknown. In this study, we addressed in the mouse model the function of Omcg1, which has been shown to participate in co-transcriptional processes, including splicing and transcription-coupled repair. Using inducible mouse models, we found that Omcg1 is most critically required in intestinal progenitors. In absence of OMCG1, proliferating intestinal epithelial cells underwent abnormal mitosis followed by apoptotic cell death. As a consequence, the crypt proliferative compartment of the small intestine was quickly and totally abrogated leading to the rapid death of the mice. Lack of OMCG1 in embryonic stem cells led to a similar cellular phenotype, with multiple mitotic defects and rapid cell death. We showed that mutant intestinal progenitors and embryonic stem cells exhibited a reduced cell cycle arrest following irradiation, suggesting that mitotic defects may be consecutive to M phase entry with unrepaired DNA damages. These findings unravel a crucial role for pre-mRNA processing in the homeostasis of the small intestine and point to a major role of OMCG1 in the maintenance of genome integrity.
Key words: Cell cycle checkpoint, Embryonic stem cells, Gastrointestinal tract, Mitotic delay, Mitotic catastrophe, Pre-mRNA processing
Introduction
Cells are continuously exposed to DNA damaging agents, the nature and the level of which are highly variable from one cell to another, depending mainly on cellular physiology and microenvironment. Accordingly, the cellular response to DNA damage, which involves a tight coupling between cell cycle progression and repair, may also result in different outcomes depending on, for example, the cellular proliferation status or developmental potentialities. Hence, cells dividing at a high rate such as embryonic cells or adult blood and gut progenitor cells are particularly sensitive to irradiation (Heyer et al., 2000; Potten, 2004; Till and McCulloch, 2011). In contrast, adult stem cells and cancer initiating cells are thought to escape from DNA damage-induced elimination because they are quiescent (Fuchs, 2009; Moore and Lyle, 2011). Pluripotent cells of the early mammalian embryo and their ex vivo counterpart, the embryonic stem (ES) cells, differ in their capacity to repair damaged DNA compared to differentiated cells (Artus and Cohen-Tannoudji, 2008; Tichy and Stambrook, 2008). Inherent cell cycle properties of pluripotent cells participate in maintenance of genome integrity. Indeed, lack of G1 checkpoint may allow unrepaired DNA damage to become exacerbated during subsequent replication leading eventually to cell death. Recent data also suggest that stem cells and progenitors may respond differentially to DNA damage (Mohrin et al., 2010; Sotiropoulou et al., 2010). Upon irradiation, hematopoietic and keratinocyte stem cells expressed prosurvival factors and underwent DNA repair while downstream progenitors tended to be eliminated through apoptosis. Interestingly, radioprotection appeared independent of quiescence since resting and cycling hematopoietic stem cells were equally resistant. In absence of Ataxia Telangiectasia Mutated (ATM), a master kinase of the DNA Damage Response (DDR), difference between hematopoietic stem cell and progenitors was lost (Mohrin et al., 2010). In humans, mutations of several genes involved in DDR have been associated with premature aging syndromes (Jackson and Bartek, 2009). Similarly, mutant mice for genes involved in DNA repair allow to link DDR to stem cell depletion and aging (Ruzankina et al., 2007; Inomata et al., 2009). However, much remains to be learned about the genome maintenance mechanisms that counter DNA damage in vivo and their differential use in the various cell types that constitute developing and adult organisms.
Ovum mutant candidate gene 1 (Omcg1), also known as Ccdc16 and Zfp830, encodes for a nuclear zinc finger protein participating to the maintenance of genome integrity. Omcg1-deficiency in mouse embryonic fibroblasts induces a massive accumulation of double-strand breaks, checkpoint activation and replication forks collapse, as the consequence of R-loops formation (Houlard et al., 2011). These highly stable heteroduplexes, resulting from the hybridization of the nascent elongating RNA with the template DNA strand, form when pre-mRNA processing is defective (Li and Manley, 2005; Aguilera and Gómez-González, 2008; Tuduri et al., 2009). Although, the precise function of OMCG1 in mRNA processing is not fully understood yet, it has been shown that OMCG1 participates to splicing (Bessonov et al., 2008) and transcription-coupled repair (Kuraoka et al., 2008). Interestingly, OMCG1 is phosphorylated by ATR/ATM kinases in response to irradiation (Matsuoka et al., 2007; Houlard et al., 2011) opening the possibility that it may participate in the modulation of pre-mRNA processing events in response to DNA damage. In this study, we show that OMCG1 is critically required for survival of proliferating gut epithelial cells, using a conditional inactivation approach. Contrary to fibroblasts, Omcg1-deficiency did not dramatically affect replication but provoked severe mitotic defects in intestinal progenitor cells. Abnormal mitosis was also observed in Omcg1 mutant ES cells and, using time-lapse microscopy, we showed that mutant cells had reduced DNA damage checkpoints and delayed mitosis. Altogether, our data suggest that OMCG1 plays a critical role in the maintenance of genome integrity and document an efficient mode of elimination of damaged cells in rapidly dividing cell populations.
Results
Acute Omcg1 inactivation leads to rapid disorganization of intestinal epithelium and death of adult mice
To address the function of Omcg1 in the adult, we performed acute ubiquitous Omcg1 inactivation using the Rosa26CreERT2 strain (Hameyer et al., 2007; Houlard et al., 2011). Omcg1flox/Δ;Rosa26CreERT2/+ (RT2OcKO) and Omcg1flox/+;Rosa26CreERT2/+ control mice received daily intra-peritoneal tamoxifen (TAM) injection and efficiency of allele deletion was monitored (Fig. 1A). Rapid and efficient conversion was achieved in all organs except the brain. Maximal levels of conversion were observed as soon as 24h after the first injection. Therefore, a single injection protocol was used for further experiments.
Fig. 1. Alterations of the intestine after acute ubiquitous Omcg1 deletion.

(A) PCR analysis of control mice 4 days after first TAM injection. (B) Hematoxylin and eosin staining of small intestine sections from control and RT2OcKO mice 3 days after first TAM injection.
Three days after the first injection, RT2OcKO mice appeared unwell with significant loss of body weight and rapidly declined afterwards. All RT2OcKO mice died on the fourth day after Cre induction whereas control mice were unaffected. RT2OcKO mice were euthanized before their death and autopsy revealed major alterations of their digestive tract including a distended and filled stomach and an intestine lacking spontaneous peristaltism. Histological analysis revealed a dramatic gut phenotype (Fig. 1B). The small intestine exhibited a thinner wall, less abundant and stunted villi and highly disorganized crypts. Large portion of the gut were almost devoid of normal epithelial structure. Although defects were also noticed in the intestinal wall as well as in other tissues such as heart and pancreas, the gut epithelium was by far, the most affected tissues in RT2OcKO mice. We therefore decided to focus our study on the gut epithelium.
To avoid indirect effects of whole body deletion on intestinal phenotype, we disrupted Omcg1 specifically in the gut epithelium using the Villin-CreERT2 transgenic line (El Marjou et al., 2004). A single injection in Omcg1flox/Δ;Villin-CreERT2tg/+ (VT2OcKO) and control (Omcg1flox/+;Villin-CreERT2tg/+) mice was sufficient to induce Omcg1flox deletion in 24h (Fig. 2A). Immunostaining of gut sections indicated that most intestinal epithelium nuclei were positive for OMCG1 (Fig. 2B). Apart from few scattered cells in the villi lamina propria and in the muscular layers, the other cells of the small intestine appeared to express lower levels of OMCG1. The protein could no longer be detected in the intestinal epithelium of TAM injected VT2OcKO mice from 24h onwards. Strikingly, VT2OcKO mice died 5 days after Cre induction and exhibited, one day before their death, macroscopic abnormalities of their digestive tracts similar to those found in RT2OcKO mice. Both small intestine and colon were affected and no regional phenotypic difference was observed along the length of the intestine. Altogether, these data indicate that Omcg1 is critically required in the intestine and that acute inactivation of Omcg1 in the gut epithelium is sufficient to cause the rapid death of adult mice.
Fig. 2. Inducible Omcg1 inactivation in the intestinal epithelium of adult mice.

(A) PCR analysis of control and VT2OcKO mice on tail (T) and intestinal epithelium (0h, 24h, 48h and 72h post TAM injection). (B) OMCG1 immunofluorescence staining of control and VT2OcKO intestine section 24h after TAM injection.
Omcg1 inactivation results in apoptosis and crypt disappearance
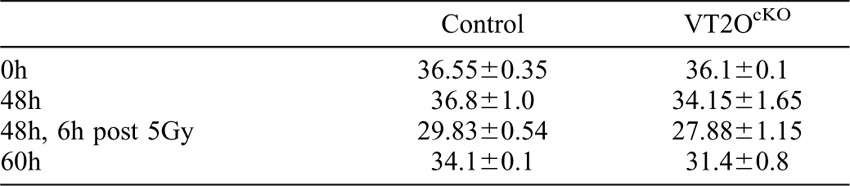
Examination of hematoxylin-eosin stained sections unravelled a dramatic change in the morphology of the crypts of VT2OcKO mice occurring between 48 and 72 hours after TAM injection (Fig. 3A). At 48 hours, VT2OcKO crypt architecture appeared slightly reduced compared to controls with a minimal reduction in the number of crypt cells (supplementary material Table S1). At 72 hours, many crypts were severely affected and contained cells displaying irregular cell shape, vacuolization or condensed chromatin. Cellular disintegration products reminiscent of Paneth cells cellular content were also frequently observed in the lumen. By contrast, villi appeared less affected exhibiting a normal distribution of goblet cells and the characteristic basal positioning of enterocyte nuclei. We however noted a moderate oedema of the villi with no sign of inflammation. At an intermediate time point (60 hours), a normal crypt organisation with Paneth cells residing at their bottom was observed. Noticeably at this stage, apoptotic bodies were observed in most mutant crypts suggesting that crypt cells are dying from massive apoptosis. To confirm this point, we performed cleaved caspase-3 immunostaining at 0, 48 and 60 hours after TAM injection (Fig. 3B,C). Only occasional apoptotic cells were found in uninjected control and VT2OcKO animals. In injected controls, we observed a slight increase in the number of caspase-3-positive crypt cells which is likely to reflect the known genotoxicity of Cre activity in mammalian cells (Naiche and Papaioannou, 2007; Higashi et al., 2009). Such adverse effects were very limited in our experimental design since a single TAM injection was used to temporarily activate the Cre recombinase. In injected VT2OcKO mice, the number of caspase-3-positive cells was significantly elevated at 48 hours and continued to increase at 60 hours. Apoptotic cells were observed along the entire height of the crypt as well as at the crypt-villus junction. Apart from a few caspase-3-positive cells at the tip of the villi, no sign of apoptosis was detected in VT2OcKO villi. These observations suggest that the sudden disorganization of the intestinal epithelium following loss of Omcg1 function results from the massive caspase-3-dependent apoptosis of intestinal crypt cells.
Fig. 3. Omcg1 loss of function in intestinal epithelium leads to massive apoptosis of crypt cells.

(A) Histological analysis of control and VT2OcKO mice intestine at different time points following TAM injection. Asterisks: abnormal mitotic cells; arrowheads: round acidophilic cell with apoptotic nuclear bodies, arrows: dilated lymph vessels showing oedematous mucosa. (B) Cleaved caspase-3 immunostaining 0, 48 and 60 hours after injection. (C) Mean (± SD) percentage of caspase-3 positive cells per crypt (n-s: non-significant; ***: p<0.001, Mann-Whitney-Wilcoxon test).
Absence of double-strand breaks and mild reduction of proliferation in Omcg1-deficient crypts cells
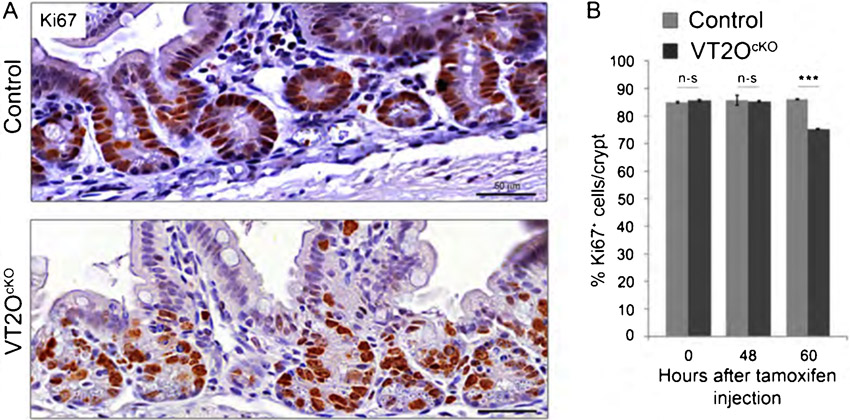
We previously reported that Omcg1 deficiency provokes massive accumulation of replication dependent double-strand breaks and early S phase blockade in embryonic fibroblasts (Houlard et al., 2011). We therefore asked whether similar defects could explain the phenotype observed in VT2OcKO mice. To investigate the capacity of cells to progress into S phase, we injected mice with 5-Bromo-2′-deoxyuridine (BrdU) 3 hours before sacrifice. We observed a moderate reduction in the number of BrdU-positive cells in VT2OcKO mice compared to control mice starting 48 hours after Cre activation (Fig. 4A,B) indicating that proliferation was not severely disrupted in mutant progenitor cells. This was confirmed using Ki67 immunostaining (supplementary material Fig. S1). We also search for the presence of double-strand breaks in Omcg1-deficient epithelial cells using γH2AX immunostaining (Fig. 4C). At 48 hours, γH2AX positive cells (Fig. 4C, asterisks) were observed frequently in mutant crypts and only rarely in control crypts. In all cases, γH2AX staining was intense and diffuse and generally concerned cells with abnormally condensed nuclei suggesting that H2AX phosphorylation is a secondary consequence of apoptotic DNA fragmentation (Rogakou et al., 2000). Double immunofluorescence staining for γH2AX and cleaved caspase-3 confirmed that these cells indeed correspond to cells already committed to apoptosis (supplementary material Fig. S2). Altogether, these observations indicate that, in contrast to the situation observed in fibroblasts, Omcg1-deficiency in gut epithelial cells is not associated with major chromosomal breaks and replication fork progression defects.
Fig. 4. Absence of proliferation arrest and double-strand breaks in Omcg1-deficient crypt cells.

(A) BrdU immunostaining of small intestine sections at 60h following TAM injection. (B) Mean (± SD) percentage of BrdU positive cells per crypt (n-s: non-significant; *: 0.01<p<0.05, Mann-Whitney-Wilcoxon test). (C) Anti-γH2AX immunostaining of control and VT2OCKO mice 48h following TAM injection. Wild-type mice 1h post irradiation (10 Gy) were used as positive controls.
Omcg1 is required for normal mitosis of intestinal epithelium progenitors
At 48 hours, mutant crypts exhibited an abnormally high number of mitotic cells showing disorganized mitoses with dispersed and unaligned chromosomes (Fig. 3A, asterisks). Such mitotic defects were not observed in mutant crypts at 24 hours and were less frequent at 60 hours. We performed histone H3 phosphorylated on serine 10 (HH3PS10) immunostaining on whole mount crypt-villi preparations. Strikingly, while control crypts usually contained 1 or 2 HH3PS10 positive cells, most mutant crypts contained a high number of such cells 48 hours following TAM injection (Fig. 5A). On average, a three-fold increase in the number of mitotic cells per crypts was found in VT2OcKO mice compared to control mice (Fig. 5B).
Fig. 5. Increased number of abnormal mitotic cells in Omcg1-deficient crypts.

(A) VT2OcKO and control villi with crypts attached at their bases were stained with Hoechst 33342 and anti-HH3PS10 antibody. (B) Number of mitotic cells per crypt was counted before and 48h after TAM injection (n-s: non-significant; *: 0.01 < p < 0.05, ***: p < 0.001, Mann-Whitney-Wilcoxon test). (C) Immunofluorescent staining on VT2OcKO and control intestine vibratome slices 48h after TAM injection. Hoechst 33342 (blue), anti-HH3PS28 antibody (red), anti-α-tubulin (green) and anti-γ-tubulin (white) were used for staining.
To address the nature of the mitotic defects of Omcg1-deficient gut epithelial cells, we carefully examined mitotic cells in term of chromosome distribution in relation to mitotic spindle and centrosomes. We performed staining on thick vibratome slices of three VT2OcKO and three control animals 48 hours after Cre induction and used optical section microscopy to visualize mitotic cells in their entire depth. In controls, all mitoses appeared normal and cells in metaphase exhibited aligned chromosomes and a pair of centrioles located at each poles of a regular shaped bipolar spindle (Fig. 5C). In sharp contrast, such figures were almost never observed in mutant crypts. The majority of Omcg1-deficient mitotic cells were unipolar with chromosomes spread along a V-shape spindle (Fig. 5C). The remaining cells exhibited either distorted bipolar like spindle or highly disorganized spindle (supplementary material Fig. S3). In some cells, isolated chromosomes were clearly visible. Surprisingly, a single centriole pair was observed even in cells with bipolar like spindle and some cells were totally lacking centrioles as judged by γ-tubulin staining. Altogether, these data suggest that Omcg1 is essential in the intestinal epithelium for proper mitotic spindle organisation and progression through M phase.
Mitotic defects consecutive to Omcg1 inactivation
The phenotype of Omcg1-deficient intestinal epithelium is highly reminiscent of the one we previously reported for early embryonic cells following constitutive Omcg1 inactivation. Indeed, mutant late blastocysts exhibited a high mitotic index associated with abnormal mitotic spindles and misaligned chromosomes (Artus et al., 2005). In the gut epithelium, the peak of abnormal mitoses slightly preceded the wave of massive apoptosis suggesting that mutant mitotic cells might progress to cell death. It is however not known whether apoptosis directly concerns mitotic cells or their progenies. To address this point, we performed HH3PS28 and cleaved caspase-3 co-immunostaining on three VT2OcKO and three control animals 48 hours after Cre induction. Examination of more than hundred crypts per animals failed to revealed doubly stained cell (not shown), strongly arguing against mutant cells elimination by an apoptotic process during mitosis.
To better characterize the mitotic phenotype of Omcg1-deficient cells, we generated Omcg1flox/null;Rosa26CreERT2/+ ES cells. Upon addition of 4-hydroxytamoxifen (4-OHT), Omcg1flox allele was rapidly and efficiently recombined and OMCG1 protein levels decreased progressively to reach very low levels by 48 hours (Fig. 6A,B). After 30 hours, treated cells stopped to proliferate and underwent massive cell death from 48 hours onwards (Fig. 6C). At 60 hours, a minority of viable cells was still present in the culture (supplementary material Fig. S4). During the period immediately preceding their death, ES cells exhibited an increased mitotic index as visualized by flow cytometry, Western blot and immunofluorescence analyses (Fig. 6A–D). Immunostaining revealed a high proportion of abnormal mitoses including disorganized, uni and multipolar spindles as well as isolated chromosomes (Fig. 6D).
Fig. 6. Abnormal mitotic progression in ES cells lacking OMCG1.

PCR (A) and Western blot (B) analyses of non-induced (NI) and induced Omcg1flox/null;Rosa26CreERT2/+ ES cells cultured for different time points after 4-OHT addition. Proliferation curves (C) and cell cycle profiles (D) of non-induced and induced ES cells. (E) Highly disorganized mitotic spindles (stars) and misaligned chromosomes (arrow) in mutant cells 48h after induction. (F) Immunofluorescence staining of ES cells either non-induced or 48 hours after induction for markers of DSB and DDR (green). Cells were counterstained with Hoechst 33342 (blue). ES cells 1 hour after 10 Gy of irradiation were included as control for Western blot and immunofluorescence analyses.
FACS analysis of cell cycle profile recovery after nocadazole release suggested that progression through mitosis was delayed in mutant cells (supplementary material Fig. S5). Mitotic delay was confirmed using time-lapse microscopy on Omcg1flox/null;Rosa26CreERT2/+ ES cells stably expressing H2B-mRFP (Fig. 7A; supplementary material Movie 1). We observed that duration of cell division significantly increased with time. During the first half of the recorded period (26–36 h post Cre induction), mitosis progression was not significantly affected when compared to untreated cells. In contrast, cells entering M phase at later time points were delayed in their progression to anaphase. Formation of metaphase plate was severely perturbed with frequent unaligned or misoriented chromosomes. Of 27 cells entering mitosis in that period, 17 performed cytokinesis before the end of the movie with a 2-fold increase in the nuclear envelope breakdown (NEB) to onset of anaphase mean duration (Fig. 7B). For two of those, division generated three daughter cells. During this period, cell death occurring during mitosis was not observed.
Fig. 7. OMCG1 depletion leads to a reduced proliferation arrest in response to irradiation.

(A) Phase contrast and epifluorescence still images from movies of induced ES cells stably expressing H2B-mRFP (red) showing division from nuclear envelope breakdown (NEB) to end of cell division at different time points after 4-OHT addition. Times after NEB are indicated, arrows indicate dividing cells, and arrowheads point to misaligned chromosomes. (B) Mean (± SEM) duration of NEB to onset of anaphase and total cell division (n-s: non-significant; ***: p<0.001 ; t test). (C) Omcg1flox/null;Rosa26CreERT2/+ ES cells were seeded with OHT (+ OHT) or without OHT (− OHT), cultured for 33h, then irradiated with 5 Gy dose of γ-rays and immediately recorded by time-lapse microscopy. Graph shows the cumulative percentage of cells entering M phase over time after irradiation.
Attenuated DNA damage checkpoints in absence of Omcg1
We then search for DNA damage in Omcg1-deficient ES cells. We performed Western blot and immunofluorescence analyses using markers of DSBs (γH2AX and 53BP1) and DDR (phospho-ATM and p53) on ES cells treated with 4-OHT for 48 hours. As a positive control, we used ES cells recovered 1 hour after 10 Gy irradiation. Untreated ES cells exhibited high basal level of γH2AX staining that was significantly increased upon irradiation. A fraction of mutant ES cells showed increased γH2AX staining suggesting that chromosomal breaks were induced in absence of OMCG1 (Fig. 6F). This was further confirmed by the redistribution of 53BP1 staining into foci in some mutant cells (Fig. 6F). Dying cells characterized by condensed and fragmented DNA presented high levels of γH2AX (but not of 53BP1, not shown) that certainly contributed to the massive increased of γH2AX levels observed by Western blot (Fig. 6B). We also detected phospho-ATM staining in a small proportion of mutant cells (Fig. 6F) as well as elevated levels of p53 in the treated cell population (Fig. 6B,F). Altogether, these data indicate that Omcg1-deficiency in ES cells leads to chromosome breaks.
We reasoned that delay in sister chromatin dissociation could therefore reflect premature entry of Omcg1-deficient cells into mitosis with incompletely replicated or unrepaired DNA. Such possibility would imply a reduction of DNA damage checkpoints activities. To address this point, we examined by time-lapse microscopy the fate of ES cells exposed to 5 Gy irradiation a few hours before defects in mitotic progression were detected in deficient cells (Fig. 7C). Uninduced ES cells (n = 455) rapidly arrested to cycle and were able to progressively re-enter M phase 4 to 5 hours after irradiation. In contrast, mutant cells irradiated 33h after 4-OHT treatment (n = 346) were able to re-enter mitosis after a shorter period of time indicating that cell cycle arrest in response to DNA damage was reduced in Omcg1-deficient ES cells. We then monitored whether DNA damage checkpoint response was also affected in intestinal progenitors. 42 hours after Cre induction, VT2OcKO and control animals received 5 Gy dose of whole body irradiation. As expected, irradiation induced a potent apoptotic response of intestinal progenitors in both control and VT2OcKO mice. After 0.5 and 2.5 hours, mice were injected with BrdU and proliferation of intestinal progenitors was assessed by scoring mitotic cells 6 hours after irradiation (Fig. 8; supplementary material Fig. S6). In both control and VT2OcKO mice, a majority of mitotic cells stained positively for BrdU, and therefore most HH3PS28 positive cells indeed corresponded to cells that progressed through S phase and entered mitosis after irradiation. Interestingly, a higher number of mitotic cells per crypts was observed in VT2OcKO compared to control mice, suggesting that proliferation arrest in response to irradiation was reduced in mutant progenitors. Altogether, our data suggest that DNA damage checkpoints are not fully functional in Omcg1-deficient intestinal progenitors and ES cells.
Fig. 8. Reduced proliferation arrest in response to irradiation in Omcg1-deficient intestinal progenitors.

(A) Experimental design. (B) Mean (± SD) percentage of BrdU-positive cells amongst HH3PS28-positive mitotic cells. (C) Mean (± SD) percentage of HH3PS28-positives cells per crypt (n-s: non-significant; ***: p<0.001 ; Mann-Whitney-Wilcoxon test).
Discussion
Recent studies have shown that factors involved in transcription-coupled mRNA processing are important for the maintenance of genome integrity (Aguilera and Gómez-González, 2008; Paulsen et al., 2009). How these processes are linked and regulated in vivo remains largely unknown. In this study, we addressed in vivo the function of Omcg1, which has been shown to participate in co-transcriptional processes, including splicing and transcription-coupled repair (Bessonov et al., 2008; Kuraoka et al., 2008; Houlard et al., 2011). We showed that Omcg1 is most critically required in intestinal progenitors. In absence of OMCG1, proliferating intestinal epithelial cells underwent abnormal mitosis followed by apoptotic cell death. As a consequence, the crypts proliferative compartment of the small intestine was quickly and totally abrogated leading to the rapid death of the mice. Together, these findings highlight a crucial role for pre-mRNA processing in the homeostasis of the small intestine.
Omcg1-deficiency in intestinal epithelial cells is first manifested by abnormal mitosis. This is also the case in early embryos and ES cells but in sharp contrast with embryonic fibroblasts, which show S-phase arrest and chromosomal breaks (Houlard et al., 2011). The reasons for such difference are currently unknown but a possibility would be that the primary function of OMCG1 differs between cell types. In fibroblast, OMCG1 would participate in the prevention of R-loop formation while in early embryonic cells it would regulate mitotic progression. Interestingly, genome-wide siRNA screens for mitotic defects performed in HeLa cells identified several known components of the splicing machinery (Kittler et al., 2004; Neumann et al., 2010). In yeast, mutations of splicing factors have also been associated with mitotic progression defects, although it was proposed to be a secondary consequence of pre-mRNA splicing defects (Biggins et al., 2001; Burns et al., 2002). Indeed, spindle defects and metaphase arrest observed in some of these mutants could be rescued by the introduction of an intronless version of TUB1, coding for α-tubulin. Recently, mitotic defects induced by depletion of the SR-related SON protein in cultured human cells has been shown to result from inefficient splicing of pre-mRNAs of genes associated with mitotic progression (Ahn et al., 2011). The mitotic delay observed after OMCG1 depletion could therefore be an indirect consequence of impaired processing of one or several pre-mRNA coding for proteins essential for cell division.
Another possibility would be that this difference reflects cellular differences in the response to the stress induced by Omcg1-deficiency. Indeed, premature entry into mitosis of cells with incomplete replication or unrepaired DNA is a well-described cause of delayed mitosis (Bartek et al., 2004; Rieder and Maiato, 2004). This phenomenon often referred as the poorly defined term “mitotic catastrophe”, implies the abrogation of checkpoints and was proposed to represent an alternative mode of elimination of damaged cells (Castedo et al., 2004; Vakifahmetoglu et al., 2008). Compared to fibroblasts, early embryonic cells and intestinal progenitors have a shortened cell cycle. Developmental constraints imposed on such highly proliferating cells are certainly not compatible with prolonged cell cycle arrest or delay. They may therefore be prone to a “remove rather than repair” response to damage. Importantly, 5 Gy-irradiated ES cells arrested to cycle for a few hours before re-entering in delayed mitosis. This transient arrest was shortened in absence of Omcg1 indicating that DNA damage checkpoints are attenuated in mutant ES cells. Similarly, we observed that Omcg1-deficient intestinal progenitors presented in vivo a reduced proliferation arrest response to irradiation. Whether this is indirectly due to the fact that mutant cells are in a sensitized situation with activated checkpoints prior to irradiation or reflects a more direct implication of OMCG1 in checkpoints activity remains to be determined. In respect to the latter point, it will be important to assess how ATR/ATM-mediated phosphorylation of OMCG1 impinges on its function (Matsuoka et al., 2007; Houlard et al., 2011).
In embryonic fibroblasts, primary defects induced by Omcg1-deficiency cause replication fork stalling, which in turns lead to double-strand breaks (Houlard et al., 2011). In intestinal mutant cells, no increase of γH2AX was detected, while DSBs were observed in a fraction of deficient ES cells. Such discrepancy may reflect differences in sensitivity of DSB detection in the two models. Anyhow, in neither cell types replication fork progression was as severely impaired as in fibroblasts. Because G1 phase duration is reduced in these rapidly dividing cells, S phase may occur before damage accrual raises a level incompatible with replication progression. ES cells and intestinal progenitors may also have a larger choice of DNA replication origins (Méchali, 2010) conferring increased flexibility to keep S phase progressing despite the presence of obstacles encountered by the replication machinery. Together with attenuated checkpoints, these two features may contribute to allow mutant cells to enter mitosis with defects that would induce delay in sister chromatid separation.
A striking feature of Omcg1 loss of function in both intestinal progenitors and ES cells is the dramatic cell death that leads to the complete exhaustion of the cell populations in a few days. In addition to the high proliferation rate of these cells, concomitant induction of DNA damage with ineffective DNA damage checkpoint may contribute to such drastic phenotype. Delayed mitosis may also be directly involved in cell death. Indeed, a prolonged block at the metaphase-anaphase transition has been shown to induce an irreversible p53-dependent G1 arrest in normal human cells (Uetake and Sluder, 2010). Elevated p53 observed in mutant ES cells might reflect the activation of such checkpoint, if present in ES cells. Together with ATM phosphorylation, it may also report on the activation of the γH2AX-ATM-p53 pathway following the mitotic catastrophe observed when cells enter mitosis with damaged DNA (Imreh et al., 2011). Mitotic catastrophe was first described for proliferating epithelial cells of the small intestine following whole body irradiation of adult mice (Potten, 1990; Merritt et al., 1997). After radiation, cells stopped to cycle before undergoing a first wave of p53-dependent cell death involving PUMA but not the intrinsic pathway of apoptosis (Qiu et al., 2008; Kirsch et al., 2010). Later on, a second wave of p53-independent cell death was observed, characterized by progression through abnormal mitosis (Merritt et al., 1997). The mechanisms underlying this form of delayed mitosis-linked cell death in the gut epithelium is poorly understood. Inactivation of the key components of the DNA damage checkpoints, ATR and CHK1, results in the rapid death of crypt cells of the small intestine (Ruzankina et al., 2007; Qiu et al., 2008). However, no increase in the number of abnormal mitoses was reported, although Chk1-deficient intestinal progenitors were eliminated through p53-independent apoptosis. VT2OcKO mice and conditional ES cells may represent valuable models to study and better understand this process.
In conclusion, we have shown that Omcg1 is critically required in rapidly proliferating intestinal epithelium and ES cells. In absence of OMCG1, cells are rapidly eliminated after a prolonged and abnormal mitosis accompanied by defects in DNA damage checkpoints. Further works on OMCG1 will participate in a better understanding of mitosis associated cell death. Our observation that mutant cells display multiple types of chromosomal segregation defects and reduced DNA damage checkpoint activities suggests that OMCG1 may participate in genomic instabilities that are potentially deleterious for the organism. By analogy, it would be interesting to assess the potential implications of Omcg1 dysfunction in cancer, and in particular intestinal tumorigenesis.
Materials and Methods
Mice and ionizing radiation
Animals were housed in the Institut Pasteur animal facilities accredited by the French Ministry of Agriculture to perform experiments on live mice (accreditation B 75 15-06, issued on May 22, 2008) in appliance of the French and European regulations on care and protection of the Laboratory Animals (EC Directive 86/609, French Law 2001-486 issued on June 6, 2001). Protocols were approved by the veterinary staff of the Institut Pasteur animal facility and were performed in compliance with the NIH Animal Welfare Insurance NO. A5476-01 issued on July 2, 2007.
Four-six-week-old control and experimental littermates were injected intraperitoneally with TAM (75 mg/kg). Mice were exposed at a dose of 5 Gy at a rate of 1.07 Gy/min using an IBL 637 137Cesium irradiator. Non-irradiated mice received a single injection of BrdU (100 mg/kg) three hours before sacrifice, while irradiated mice were injected twice 5.5 and 2.5 hours before sacrifice.
Genotyping of mice and ES cells
Genotyping was performed by PCR after lysis of tissue and ES cell pellet in the following buffer: 50 mM Tris pH 8.5, 100 mM NaCl, 0.5% tween20, 100 mg/ml proteinase K at 56°C overnight (ON) followed by a 10 min incubation at 96°C. PCR amplification protocol consisted of an initial incubation at 94°C for 5 min, followed by 35 cycles at 94°C for 30 s, 60°C for 60 s, and 72°C for 60 s, and a final incubation at 72°C for 5 min. For Omcg1, PCR amplification with primers 1 (5′- GCCATAACAGGAAGTGACGCT), 2 (5′- GATGGGCGCATCGTAACCGTGC), 3 (5′- GGTGTTACACAGTGCGCAGCT) and 4 (5′- AGCGCCAACCCTTGGAGTTTA) allowed to discriminate Omcg1 alleles: wild type (238 bp, primers 1 and 3), βgeo/null (390 bp, primers 1 and 2), flox (326 bp, primers 1 and 3) and Δ (495 bp, primers 1 and 4).
Tissue preparation
For paraffin sections, the entire intestinal tract was dissected, flushed twice with ice cold PBS to remove any faecal content and perfused with ice cold PFA (4% in PBS). The small intestine was rolled up from the proximal to the distal end in concentric circles, fixed in 4% PFA at 4°C ON, dehydrated, embedded in paraffin wax, and sectioned using a microtome.
For agarose sections, 1 cm jejunum was cut opened longitudinally, rinsed in PBS, fixed in 4% PFA at room temperature (RT) for 2 hours, rinsed in PBS, included in low melting agarose and sectioned using a vibratome.
For isolation of villi with attached crypts, we adapted the protocol described in (Guo et al., 2009). Briefly, 7 cm of jejunum was cut opened longitudinally, rinsed in ice cold PBS and put on ice into a 15 ml falcon containing 10 ml ice cold BSS buffer with protease and phosphatase inhibitors (1.5 mM KCl, 96 mM NaCl, 27 mM sodium citrate, 8 mM KH2PO4, 5.6 mM Na2HPO4, 15 mM EDTA and 1 mM dithiothreitol). After a 10 min preincubation on ice, jejunum was vortexed at medium speed at 4°C for 5 min three to five times. After each round of mechanical dissociation, pieces of tissues were decanted and supernatant was replaced with fresh BSS solution. Each supernatant was then examined under light microscopy to select the fraction containing the higher proportion of villi with crypts still attached. This fraction was then passed through a 70 µm cell strainer (BD Falcon) to remove debris and isolated cells, and rinsed in ice cold PBS with inhibitors. Pellet was obtained by gravity flow and fixed for whole mount immunofluorescent staining in 4% PFA at RT for 30 min. Typically, eight to twelve crypts per villi were obtained for jejunum of 4-week-old animals.
Histology and immunostaining
Hematoxylin and eosin staining was carried out on 4 µm paraffin sections. For immunochemistry, antigens were retrieved by boiling slides for 20 min in specific buffers (supplementary material Table S2). Sections were then incubated in blocking solution (2.5% normal horse serum, 0.25% Triton X-100 in PBS) for 45 min at RT. Incubation with primary antibodies was performed in the blocking solution at 4°C ON. Sections were rinsed several times (0.05% tween in PBS) and endogenous peroxydase activity was quenched with 3% H2O2 in PBS for 20 min. Specific binding was detected using ImmPRESS-HRP (vectorlabs). Sections were counterstained with hematoxylin, dehydrated and mounted in Eukitt. For BrdU immunodetection, staining was performed as above, except for an additional step in HCl 2N for 30 min after antigen retrieval. Automated whole slide imaging was performed using a MiraxScan device (Carl Zeiss microImaging) equipped with a 20× objective lens. The system was set to run in automated batch mode with automated focus and tissue finding. For immunofluorescence staining on paraffin sections, 10% goat serum, 0.25% Triton X-100 in PBS was used as blocking buffer. 100 µm vibratome sections and crypts-villi preparations were processed for immunofluorescence in solution. Intestinal tissues were carefully removed from agarose and post fixed in 4% PFA supplemented with 15 µM Taxol at RT for 3 min. For crypts-villi preparations, blocking and rinsing solutions were supplemented with 0.6% bovine serum albumin in order to limit tissue aggregations. Fluorochome-conjugated secondary antibodies were incubated at RT for 45 min and nuclei were counterstained with 10 mM Hoechst 33342 for 3 min. Sections and tissues were mounted in vectashield and images were acquired with an inverted microscope Zeiss Axiovert 200M with a Zeiss apotome system controlled by the Zeiss axiovision 4.4 software. The CCD camera used was a Roper Scientific Coolsnap HQ. Contrast adjustment and cropping was performed in Photoshop (Adobe). Primary and secondary antibodies used in this study are listed in supplementary material Table S2.
Western blot
ES cell pellets were resuspended in lysis buffer (Tris HCl 50mM pH 7.4, NaCl 150mM, NP40 1%, sodium deoxycholate 1%, SDS 1%, glycerol 0.1%) supplemented with Complete and PhosStop (Roche), and then digested for 30 min at 4°C with benzonase (Sigma, 2.5 units/μl). Proteins were denatured in 1× Laemli buffer at 95°C for 10 min and an equivalent of 2×105 cells per well comb were loaded on a polyacrylamide gel. After migration, proteins were transferred on nitrocellulose membrane (Biorad) and incubated ON at 4°C with primary antibodies. Membranes were incubated with peroxidase labelled secondary antibodies at RT for 45 min, rinsed in PBS and signals were visualized using ECl (Pierce).
Assaying crypt cells number, proliferation and apoptosis
For each analysis, at least 25 full longitudinal crypt sections containing paneth cells were scored from at least 3 mice of each genotype. Number of cells by crypt was determined from hematoxylin and eosin stained sections. Number of HH3PS28, Ki67 and BrdU positive cells were determined from whole slide images of immunostained sections. Total number of mitotic cells per crypt was also determined from microscopic examination of whole mount HH3PS10 immunostaining of crypt-villi preparations. Histone H3 phosphorylation at serine 10 and serine 28 coincides with the initiation of mitotic chromosomes condensation and antibodies recognizing these modifications are classically used to mark mitotic cells. Because HH3PS28 antibody was raised in rat, it was preferentially used in co-immunostaining analyses. Statistical analysis was performed using Mann-Whitney-Wilcoxon rank sum test and p-values were corrected for multiple comparisons using the Holm-Bonferroni method. In all cases, the significance of statistical analyses was depicted as follows: (n-s) non-significant, (*) 0.01<p<0.05, (**) 0.001<p<0.01, (***) p<0.001.
Generation of Omcg1flox/null;Rosa26CreERT2/+ ES cells
Omcg1trilox/+ ES cells (Houlard et al., 2011) were transfected with pIC-Cre plasmid (Gu et al., 1993) and plated under clonal conditions. Colonies were then analysed by PCR and Southern blot for partial Cre-loxP recombination in order to obtain Omcg1flox/+ ES cells deleted for the pgk-Neo selection cassette. Omcg1flox/+ ES cells were then electroporated with the Omcg1-βgeo targeting vector (Artus et al., 2005) to generate with high frequency (≈80% of G418R clones) Omcg1flox/null ES clones. Finally, the pRosaCreERT2 targeting vector (Hameyer et al., 2007) was linearized using SfiI restriction enzyme and electroporated into Omcg1flox/null ES cells. Southern blot analysis revealed that 70% of the puromycin resistant clones were correctly targeted. Consequences of Omcg1 loss of function were analysed in two independent Omcg1flox/null;Rosa26CreERT2/+ ES cell clones after adding 1 µM 4-OHT in culture medium for 10 hours. Omcg1flox/null;RosaCreERT2/+ ES cells were also adapted to feeder free culture conditions and transfected (Lipofectamine 2000, Invitrogen) with hygromycin resistance and H2B-mRFP expression vectors. Hygromycin-resistant clones were picked after 10 days of selection (150 µg/ml Hygromycin) and screened for mRFP fluorescence. A clone with moderate and overall homogeneous fluorescence was selected for live imaging studies. ES cells were grown in 35-mm µ-Dish chambers (Matek) coated with matrigel and treated ON with 1 µM 4-OHT. Cells were exposed at a dose of 5 Gy at a rate of 1.07 Gy/min using an IBL 637 137Cesium irradiator. Live imaging was performed using a Nikon BioStation IM. Images were captured every 3 min and processed using ImageJ.
Supplementary Material
Acknowledgments
Imaging and cell sorting analyses were performed at the Institut Pasteur Imagopole. We are grateful to E. Perret for her assistance with image and time lapse acquisition. We thank Jos Jonker and Anton Berns for the Rosa26CreERT2 targeting vector. We also thank H. Khun, P. Avé, P. Rocheteau, S. Tajbakhsh, F. Duveau, N. Arnaud, C. Deschamps, V. Marthiens, R. Basto, G. Peignon and B. Romagnolo for technical advice and helpful discussions. This work was supported by the Centre National de la Recherche Scientifique, the Institut Pasteur, the Agence Nationale de la Recherche (contract no. JC05_41835) and the Institut National du Cancer (INCa 2007-1-COL-6-IC-1 and PLBIO09-070). M.H., J.A. and T.L. received fellowships from the Institut Pasteur, the Association pour la Recherche sur le Cancer and the Ministère de l’Education Nationale, de la Recherche et de la Technologie respectively.
Footnotes
Competing interests: The authors declare that there are no competing interests.
References
- Aguilera A., Gómez-González B. (2008). Genome instability: a mechanistic view of its causes and consequences. Nat. Rev. Genet. 9, 204–217 10.1038/nrg2268 [DOI] [PubMed] [Google Scholar]
- Ahn E. Y., DeKelver R. C., Lo M. C., Nguyen T. A., Matsuura S., Boyapati A., Pandit S., Fu X. D., Zhang D. E. (2011). SON controls cell-cycle progression by coordinated regulation of RNA splicing. Mol. Cell 42, 185–198 10.1016/j.molcel.2011.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artus J., Cohen-Tannoudji M. (2008). Cell cycle regulation during early mouse embryogenesis. Mol. Cell. Endocrinol. 282, 78–86 10.1016/j.mce.2007.11.008 [DOI] [PubMed] [Google Scholar]
- Artus J., Vandormael-Pournin S., Frödin M., Nacerddine K., Babinet C., Cohen-Tannoudji M. (2005). Impaired mitotic progression and preimplantation lethality in mice lacking OMCG1, a new evolutionarily conserved nuclear protein. Mol. Cell. Biol. 25, 6289–6302 10.1128/MCB.25.14.6289-6302.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J., Lukas C., Lukas J. (2004). Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 5, 792–804 10.1038/nrm1493 [DOI] [PubMed] [Google Scholar]
- Bessonov S., Anokhina M., Will C. L., Urlaub H., Lührmann R. (2008). Isolation of an active step I spliceosome and composition of its RNP core. Nature 452, 846–850 10.1038/nature06842 [DOI] [PubMed] [Google Scholar]
- Biggins S., Bhalla N., Chang A., Smith D. L., Murray A. W. (2001). Genes involved in sister chromatid separation and segregation in the budding yeast Saccharomyces cerevisiae. Genetics 159, 453–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns C. G., Ohi R., Mehta S., O’Toole E. T., Winey M., Clark T. A., Sugnet C. W., Ares M., Jr, Gould K. L. (2002). Removal of a single alpha-tubulin gene intron suppresses cell cycle arrest phenotypes of splicing factor mutations in Saccharomyces cerevisiae. Mol. Cell. Biol. 22, 801–815 10.1128/MCB.22.3.801-815.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castedo M., Perfettini J. L., Roumier T., Andreau K., Medema R., Kroemer G. (2004). Cell death by mitotic catastrophe: a molecular definition. Oncogene 23, 2825–2837 10.1038/sj.onc.1207528 [DOI] [PubMed] [Google Scholar]
- El Marjou F., Janssen K.-P., Chang B. H.-J., Li M., Hindie V., Chan L., Louvard D., Chambon P., Metzger D., Robine S. (2004). Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39, 186–193 10.1002/gene.20042 [DOI] [PubMed] [Google Scholar]
- Fuchs E. (2009). The tortoise and the hair: slow-cycling cells in the stem cell race. Cell 137, 811–819 10.1016/j.cell.2009.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H., Zou Y. R., Rajewsky K. (1993). Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell 73, 1155–1164 10.1016/0092-8674(93)90644-6 [DOI] [PubMed] [Google Scholar]
- Guo J., Longshore S., Nair R., Warner B. W. (2009). Retinoblastoma protein (pRb), but not p107 or p130, is required for maintenance of enterocyte quiescence and differentiation in small intestine. J. Biol. Chem. 284, 134–140 10.1074/jbc.M806133200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hameyer D., Loonstra A., Eshkind L., Schmitt S., Antunes C., Groen A., Bindels E., Jonkers J., Krimpenfort P., Meuwissen R. et al. (2007). Toxicity of ligand-dependent Cre recombinases and generation of a conditional Cre deleter mouse allowing mosaic recombination in peripheral tissues. Physiol. Genomics 31, 32–41 10.1152/physiolgenomics.00019.2007 [DOI] [PubMed] [Google Scholar]
- Heyer B. S., MacAuley A., Behrendtsen O., Werb Z. (2000). Hypersensitivity to DNA damage leads to increased apoptosis during early mouse development. Genes Dev. 14, 2072–2084 10.1101/gad.14.16.2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi A. Y., Ikawa T., Muramatsu M., Economides A. N., Niwa A., Okuda T., Murphy A. J., Rojas J., Heike T., Nakahata T. et al. (2009). Direct hematological toxicity and illegitimate chromosomal recombination caused by the systemic activation of CreERT2. J. Immunol. 182, 5633–5640 10.4049/jimmunol.0802413 [DOI] [PubMed] [Google Scholar]
- Houlard M., Artus J., Léguillier T., Vandormael-Pournin S., Cohen-Tannoudji M. (2011). DNA-RNA hybrids contribute to the replication dependent genomic instability induced by Omcg1 deficiency. Cell Cycle 10, 108–117 10.4161/cc.10.1.14379 [DOI] [PubMed] [Google Scholar]
- Imreh G., Norberg H. V., Imreh S., Zhivotovsky B. (2011). Chromosomal breaks during mitotic catastrophe trigger γH2AX-ATM-p53-mediated apoptosis. J. Cell Sci. 124, 2951–2963 10.1242/jcs.081612 [DOI] [PubMed] [Google Scholar]
- Inomata K., Aoto T., Binh N. T., Okamoto N., Tanimura S., Wakayama T., Iseki S., Hara E., Masunaga T., Shimizu H. et al. (2009). Genotoxic stress abrogates renewal of melanocyte stem cells by triggering their differentiation. Cell 137, 1088–1099 10.1016/j.cell.2009.03.037 [DOI] [PubMed] [Google Scholar]
- Jackson S. P., Bartek J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078 10.1038/nature08467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch D. G., Santiago P. M., di Tomaso E., Sullivan J. M., Hou W. S., Dayton T., Jeffords L. B., Sodha P., Mercer K. L., Cohen R. et al. (2010). p53 controls radiation-induced gastrointestinal syndrome in mice independent of apoptosis. Science 327, 593–596 10.1126/science.1166202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler R., Putz G., Pelletier L., Poser I., Heninger A. K., Drechsel D., Fischer S., Konstantinova I., Habermann B., Grabner H. et al. (2004). An endoribonuclease-prepared siRNA screen in human cells identifies genes essential for cell division. Nature 432, 1036–1040 10.1038/nature03159 [DOI] [PubMed] [Google Scholar]
- Kuraoka I., Ito S., Wada T., Hayashida M., Lee L., Saijo M., Nakatsu Y., Matsumoto M., Matsunaga T., Handa H. et al. (2008). Isolation of XAB2 complex involved in pre-mRNA splicing, transcription, and transcription-coupled repair. J. Biol. Chem. 283, 940–950 10.1074/jbc.M706647200 [DOI] [PubMed] [Google Scholar]
- Li X., Manley J. L. (2005). Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 122, 365–378 10.1016/j.cell.2005.06.008 [DOI] [PubMed] [Google Scholar]
- Matsuoka S., Ballif B. A., Smogorzewska A., McDonald E. R., 3rd, Hurov K. E., Luo J., Bakalarski C. E., Zhao Z., Solimini N., Lerenthal Y. et al. (2007). ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 10.1126/science.1140321 [DOI] [PubMed] [Google Scholar]
- Méchali M. (2010). Eukaryotic DNA replication origins: many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 11, 728–738 10.1038/nrm2976 [DOI] [PubMed] [Google Scholar]
- Merritt A. J., Allen T. D., Potten C. S., Hickman J. A. (1997). Apoptosis in small intestinal epithelial from p53-null mice: evidence for a delayed, p53-independent G2/M-associated cell death after gamma-irradiation. Oncogene 14, 2759–2766 10.1038/sj.onc.1201126 [DOI] [PubMed] [Google Scholar]
- Mohrin M., Bourke E., Alexander D., Warr M. R., Barry-Holson K., Le Beau M. M., Morrison C. G., Passegué E. (2010). Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell 7, 174–185 10.1016/j.stem.2010.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore N., Lyle S. (2011). Quiescent, slow-cycling stem cell populations in cancer: a review of the evidence and discussion of significance. J. Oncol. 2011. pii 396076 10.1155/2011/396076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naiche L. A., Papaioannou V. E. (2007). Cre activity causes widespread apoptosis and lethal anemia during embryonic development. Genesis 45, 768–775 10.1002/dvg.20353 [DOI] [PubMed] [Google Scholar]
- Neumann B., Walter T., Hériché J. K., Bulkescher J., Erfle H., Conrad C., Rogers P., Poser I., Held M., Liebel U. et al. (2010). Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464, 721–727 10.1038/nature08869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen R. D., Soni D. V., Wollman R., Hahn A. T., Yee M. C., Guan A., Hesley J. A., Miller S. C., Cromwell E. F., Solow-Cordero D. E. et al. (2009). A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol. Cell 35, 228–239 10.1016/j.molcel.2009.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potten C. S. (1990). A comprehensive study of the radiobiological response of the murine (BDF1) small intestine. Int. J. Radiat. Biol. 58, 925–973 10.1080/09553009014552281 [DOI] [PubMed] [Google Scholar]
- Potten C. S. (2004). Radiation, the ideal cytotoxic agent for studying the cell biology of tissues such as the small intestine. Radiat. Res. 161, 123–136 10.1667/RR3104 [DOI] [PubMed] [Google Scholar]
- Qiu W., Carson-Walter E. B., Liu H., Epperly M., Greenberger J. S., Zambetti G. P., Zhang L., Yu J. (2008). PUMA regulates intestinal progenitor cell radiosensitivity and gastrointestinal syndrome. Cell Stem Cell 2, 576–583 10.1016/j.stem.2008.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder C. L., Maiato H. (2004). Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev. Cell 7, 637–651 10.1016/j.devcel.2004.09.002 [DOI] [PubMed] [Google Scholar]
- Rogakou E. P., Nieves-Neira W., Boon C., Pommier Y., Bonner W. M. (2000). Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem. 275, 9390–9395 10.1074/jbc.275.13.9390 [DOI] [PubMed] [Google Scholar]
- Ruzankina Y., Pinzon-Guzman C., Asare A., Ong T., Pontano L., Cotsarelis G., Zediak V. P., Velez M., Bhandoola A., Brown E. J. (2007). Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 1, 113–126 10.1016/j.stem.2007.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiropoulou P. A., Candi A., Mascré G., De Clercq S., Youssef K. K., Lapouge G., Dahl E., Semeraro C., Denecker G., Marine J. C. et al. (2010). Bcl-2 and accelerated DNA repair mediates resistance of hair follicle bulge stem cells to DNA-damage-induced cell death. Nat. Cell Biol. 12, 572–582 10.1038/ncb2059 [DOI] [PubMed] [Google Scholar]
- Tichy E. D., Stambrook P. J. (2008). DNA repair in murine embryonic stem cells and differentiated cells. Exp. Cell Res. 314, 1929–1936 10.1016/j.yexcr.2008.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till J. E., McCulloch E. A. (2011). A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. 1961. Radiat. Res. 175, 145–149 10.1667/RRXX28.1 [DOI] [PubMed] [Google Scholar]
- Tuduri S., Crabbé L., Conti C., Tourrière H., Holtgreve-Grez H., Jauch A., Pantesco V., De Vos J., Thomas A., Theillet C. et al. (2009). Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 11, 1315–1324 10.1038/ncb1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetake Y., Sluder G. (2010). Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr. Biol. 20, 1666–1671 10.1016/j.cub.2010.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakifahmetoglu H., Olsson M., Zhivotovsky B. (2008). Death through a tragedy: mitotic catastrophe. Cell Death Differ. 15, 1153–1162 10.1038/cdd.2008.47 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}