

Abstract
Aim
We investigated whether treatment with gliadin induces a paracellular permeability defect that enhances bacterial translocation to mesenteric lymph nodes (MLN) via resident dendritic cells (DC) expressing TLR-2 or 4 in HCD4/HLA-DQ8 transgenic mice.
Methods
HLA-DQ8 transgenic mice were sensitized and subsequently gavaged with gliadin, in the presence or absence of AT1001 (paracellular permeability inhibitor). Non-sensitized mice were gavaged with indomethacin (permeability inducer) or rice cereal. CD11c and CD103 (DC markers) and TLR-2 and 4 were investigated by immunostaining. Intestinal permeability was assessed by paracellular flux of 51Cr-EDTA in Ussing chambers. Bacterial translocation to MLN was performed by plate counting on aerobic and anaerobic conditions.
Results
In gliadin-treated mice, both 51Cr-EDTA flux in jejunal mucosa and aerobic and anaerobic bacterial counts in MLN were increased (p < 0.05) compared to indomethacin-treated mice and controls. The inhibitor AT1001 normalized 51Cr-EDTA flux, but had no effect on bacterial translocation in gliadin-treated mice. In addition, changes in mucosal DC marker distribution such as increased (p < 0.05) trans-epithelial CD103+ cells and reduction (p < 0.05) of CD11c immunostaining were detected in gliadin-treated mice. Moreover, changes in DC markers and TLR-2 or 4 immunophenotypes were not associated.
Conclusions
Pharmacological restoration of paracellular permeability was not sufficient to prevent bacterial translocation in gluten-sensitive mice. We hypothesize that transcellular mechanisms involving CD103+DC and CD11c+DC may explain in gluten-sensitive HCD4/HLA-DQ8 transgenic mice the sustained increased bacterial translocation observed in the absence of a significant inflammatory response.
Keywords: Celiac disease, Dendritic cells, Toll-like receptors, Intestinal permeability, HCD4/HLA-DQ8 transgenic mice
Introduction
Celiac disease is an autoimmune enteropathy caused by dietary gluten, and particularly gliadin, in genetically susceptible individuals [1–4]. The clinical and pathological spectrum of celiac disease is heterogeneous [5–7] and there is no current rodent model that reproduces all aspects of human celiac disease [8–10]. The transgenic HCD4/HLA-DQ8 mouse model is useful to study pathophysiological responses to gluten before the onset of severe inflammation and intestinal atrophy [8, 11–14]. This model is characterized by mild intraepithelial lymphocytosis, gluten-dependant neuromotor and intestinal barrier dysfunction in the absence of mucosal atrophy. Furthermore, the T cell response generated requires DQ8 molecules since proliferation of T cells incubated with gliadin is inhibited by anti-DQ8 monoclonal antibodies [8].
Celiac disease patients [15–17] and HCD4/HLA-DQ8 gluten-sensitive mice [12] display intestinal barrier dysfunction. Altered tight junction protein expression has been demonstrated in celiac disease patients. Moreover, increased paracellular permeability and disrupted ZO-1 [18] and claudins 2 and 3 [19] expression was reversed by AT1001, an octapeptide derived from the zonula occludens toxin secreted by Vibrio cholerae [20]. It has been hypothesized that intestinal barrier dysfunction may allow abnormal penetration of gluten-related peptides as well as luminal antigens and enteric microbes, which could amplify any subsequent immune response [12].
Dendritic cells (DC) are innate immune cells that operate as sentinels sampling antigens from peripheral tissues followed by migration to lymphoid organs where they induce T cell activation [21, 22]. DCs produce potent T cell activation when challenged with gliadin-derived peptides and they accumulate in lesions from patients with celiac disease [22–25]. However, DC are difficult to study in vitro because cell isolation may provoke changes in gene expression that mimic those occurring during inflammation [26]. Thus, investigation of DC in the mouse intestine using immunohistochemical analysis of the pan-DC marker CD11c as well as CD103 (αE2 integrin expressed by DC and intra-epithelial lymphocytes) may provide important information about the role of these cells in their microenvironment [27]. Toll-like receptors (TLR) constitute a family of pattern-recognition receptors that detect conserved microbial motifs, and are commonly expressed in DC [28, 29]. In particular, TLR-2 and 4 recognize specific antigens from Gram-positive and Gram-negative bacteria, respectively, initiating and maintaining an innate immune response. Moreover, increased protein levels as well as mRNA expression of both receptors have been detected in the duodenal mucosa from patients with celiac disease [30].
We hypothesize that, similar to findings in celiac disease patients [17], the small intestine of gluten-sensitive mice exhibits augmented paracellular permeability, which may facilitate abnormal bacterial infiltration into the mucosa. Resident DC expressing TLR-2 or 4 could uptake bacteria and migrate to mesenteric lymph nodes (MLN) inducing their translocation. Our results show that gliadin increased 51Cr-EDTA flux, which was reversed by AT1001. The enhanced bacterial translocation to MLN was not inhibited by this peptide, indicating that restoration of paracellular permeability was not sufficient to prevent bacterial translocation. Furthermore, gliadin induced a significant redistribution of DC markers (but not of TLR-2 or 4), suggesting that an increased number of trans-epithelial CD103+DC may be involved in uptake of bacteria into the mucosa while CD11c+DC in facilitating translocation to MLN.
Materials and Methods
Experimental Animals
Transgenic mice expressing HCD4/HLA-DQ8 (HLA-DQA1*0301; HLA-DQB1*0302) genes in the absence of endogenous mouse MHC class II genes were bred as previously described [8]. In brief, mice were kept in a conventional specific pathogen-free colony at McMaster University two generations before breeding and fed a gluten-free diet (Bio-Serve, Frenchtown, NJ, USA). Mice were maintained on a gluten-free diet until experiments at the age of 8–30 weeks. Age- and gender-matched animals were used in this study. We used C57BL/6 mice purchased from Taconic Farms (Germantown, NY, USA) as additional wild-type controls for bacterial translocation.
Overall Design
Male HCD4/HLA-DQ8 mice (n = 6–9/group) were sensitized by injecting intraperitoneally (ip) 500 ug of gliadin from wheat (Sigma-Aldrich, Ontario, Canada) dissolved in 50 μl of 0.02 M acetic acid plus 50 μl of Complete Freund’s Adjuvant (CFA, Sigma-Aldrich, Ontario, Canada). One week after sensitization, gliadin challenge was performed three times on a weekly basis by intragastric gavage, for 7 weeks, using 2 mg of gliadin from wheat dissolved in 100 μl of 0.02 M acetic acid. One control group consisted of non-sensitized mice treated ip with 50 μl of 0.02 M acetic acid plus 50 μl of CFA only, and subsequently gavaged with rice cereal (2 mg/0.02 M acetic acid). To differentiate between gliadin-induced and non-specific intestinal mucosal dysfunction, an additional control group of non-sensitized mice (treated ip as the first control group) was gavaged three times on a weekly basis, for 7 weeks, with indomethacin (Sigma–Aldrich, Ontario, Canada; 3.5 mg/kg of weight), which has previously been shown to increase intestinal permeability to a similar degree as gliadin in HLA-DQ8/HCD4 mice [12].
In experiments using AT-1001 (Alba, Baltimore, MD, USA), mice (n = 4–8/group) were orally pre-treated with 100 μl of aqueous protease inhibitor cocktail containing 9 mg/ml of bestatin-hydrochloride (Sigma-Aldrich, Ontario, Canada), 20 mg/ml of leupeptin (Sigma-Aldrich), and 9 mg/ml of captopril (Sigma-Aldrich) 1 h prior oral gavage during the 7 weeks of regime. In addition, non-sensitized rice-fed and indomethacin-treated mice were orally pre-treated with 100 μl of sterile PBS, while gliadin-treated mice received 100 μl of PBS alone or with AT1001 at doses of 0.3 mg/ml or 3 mg/ml. Groups of C57BL/6 mice (n = 4/group) non-sensitized and orally challenged with rice or indomethacin as well as sensitized and orally challenged with gliadin were used as wild-type controls to investigate bacterial translocation to MLN.
Animals were euthanized 24 h after the last oral challenge, tissue samples collected, and jejunal tissues for histochemistry fixed in 10% buffered formalin. All experiments were conducted with approval from the McMaster University Animal Care Committee. Analysis of results was performed in a blinded manner.
Reagents for Immunoreactions
Rabbit anti-mouse CD3, normal rabbit IgG for isotype control, biotinylated swine anti-rabbit IgG, antibody (Ab) diluting buffer, alkaline phosphatase-conjugated streptavidin, horseradish peroxidase (HRP)-conjugated streptavidin, 3-amino-9-ethylcarbazole, fast red substrate system, faramount aqueous mounting medium, and Mayer’s hematoxylin were obtained from Dako (Mississauga, Canada). Specific polyclonal rabbit anti-mouse CD11c (sc-28671), CD103 (sc-28662), TLR-2 (sc-10739) as well as TLR-4 (sc-10741) IgG, monoclonal hamster anti-mouse CD103 (sc-52597) IgG, polyclonal biotinylated goat anti-rabbit IgG (sc-2040), FITC-conjugated goat anti-hamster IgG (sc-2446), CY5-conjugated goat anti-rabbit IgG (sc-3844) and mounting media for fluorescence were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Immunohistochemistry
Serial sections (n = 6–9/group) were immunostained using standard protocols [31]. Samples were treated with 10% H2O2 to block endogenous peroxidase activity, then with 0.05% trypsin to unmask the antigenicity, followed by incubations with universal blocking solution (Lab Vision Corporation, Fremont, CA, USA). Subsequently, overnight incubation at 4°C was performed with the following specific polyclonal primary Ab: rabbit anti-mouse CD11c (18 μg/ml), CD103 (18 μg/ml), TLR-2 (18 μg/ml) or TLR-4 (18 μg/ml). Then, sections were incubated with biotinylated goat anti-rabbit IgG, followed by alkaline phosphatase-conjugated streptavidin and stained with fast red. In addition, CD3 immunostaining was performed according to a different methodology described elsewhere [14]. In brief, slides were incubated with anti-CD3 Ab (1:300) followed by biotinylated swine anti-rabbit IgG, then HRP-conjugated streptavidin and stained with 3-amino-9-ethylcarbazole. Negative controls were performed with either an isotype-matched Ab of inappropriate specificity or by omitting the primary Ab. Immunostaining for DC markers and TLR-2 and 4 was examined and quantified by counting stained cell number in five different fields of ×400 magnification, excluding unspecific reactions and structures lacking a nucleus. Due to the difficulties on identifying single stained CD11c+ cells caused by their network pattern, the immunoanalysis of this marker was performed quantifying % of stained area/field. Each field included lamina propria and submucosa, outside Peyer’s patches. CD3+ intraepithelial lymphocytes were counted in five randomly selected villous tips according to a method previously described [14], and results were expressed as number of CD3+ cells/100 enterocytes.
Immunofluorescence
Additional sections were processed according to standard protocols [31] and incubated overnight at 4°C with the following pair of Ab: rabbit anti-mouse TLR-2 + hamster anti-mouse CD103 (18 μg/ml each one). Then, sections were incubated in the dark with FITC-conjugated goat anti-hamster + CY5-conjugated goat anti-rabbit IgG (15 μg/ml each one) for 2 h. Negative controls as well as single immunofluorescence were performed by omitting the primary Ab or by incubating with a single primary Ab, respectively. Sections were washed, mounted in media for fluorescence and analyzed under a Carl Zeiss confocal LSM510 laser scanning microscope using a standard setting that avoid the overlap of false-positive images.
Bacterial Translocation
We investigated the occurrence of enteric bacterial translocation to jejunal MLN and spleen in HCD4/HLA-DQ8 mice (n = 5 animals/group) as well as in wild-type controls (C57BL/6; n = 4/group). Samples were aseptically collected during killing (n = 2 MLN or spleens/animal), weighed and then homogenized in 0.5% of Tergitol/PBS using a Tissuelyser and sterile stainless steel for 1 min at 25 Hz as previously described [32]. Homogenates of these tissues were then plated in Columbia 5% sheep blood agar (Biomerieux, St-Laurent, Quebec, Canada) and incubated overnight at 37°C under aerobic or anaerobic conditions. CFU were normalized according to the weight of the tissues (detection limit is 103 CFU/g of MLN).
Intestinal Paracellular Permeability
Intestinal permeability was assayed in Ussing chambers testing mucosal-to-serosal flux of the paracellular probe 51Cr-EDTA [15]. In brief, jejunal segments from each mouse (n = 7–8 animals/group) were collected, placed in oxygenated ice-cold buffer, opened across the mesenteric border, stripped of the external muscle layer, and two-flat sheets without Peyer’s patches were mounted in the chambers as previously described [14]. Tissues were equilibrated for 15 min before adding 6 μCi/ml of 51Cr-EDTA (molecular mass 360 Da, Perkin-Elmer, Boston, MA, USA) to the mucosal buffer of the Ussing chambers. Samples of 100 μl were taken from the serosal side at 0, 30, 60, 90, and 120 min and compared to 100 μl of the radioactive sample (100%) obtained from the mucosal buffer at time 0. 51Cr-EDTA flux was calculated as the average of over the 2-h period and expressed as % Hot/h/cm2.
Statistical Analysis
Statistical analysis was performed with Graph Pad (Graph Pad Software, Inc., San Diego, CA, USA) using one-way ANOVA and Tukey post-test if overall p < 0.05, which is considered statistically significant. Kruskal-Wallis test was performed for non-parametric data. Data are presented as bar graphs of means ± standard error (SEM) or dot plots.
Results
Intestinal Paracellular Permeability
We observed a 2.6-fold increase (p < 0.05) in mucosal-to-serosal flux of 51Cr-EDTA in jejunal tissue samples from HCD4/HLA-DQ8 gliadin-treated mice when compared to non-sensitized rice-fed animals (Fig. 1a). Indomethacin induced a 3.9-fold increase (p < 0.05) in 51Cr-EDTA flux in non-sensitized mice compared to non-sensitized rice-fed mice (Fig. 1a). These results show that indomethacin generated a higher increase in paracellular permeability than gliadin treatment. Furthermore, administration of a high dose (3 mg/ml) of AT1001—but not of low dose (0.3 mg/ml)—restored 51Cr-EDTA flux in the jejunum from gliadin-treated mice to levels similar to those found in non-sensitized rice-fed mice (Fig. 1a).
Fig. 1.
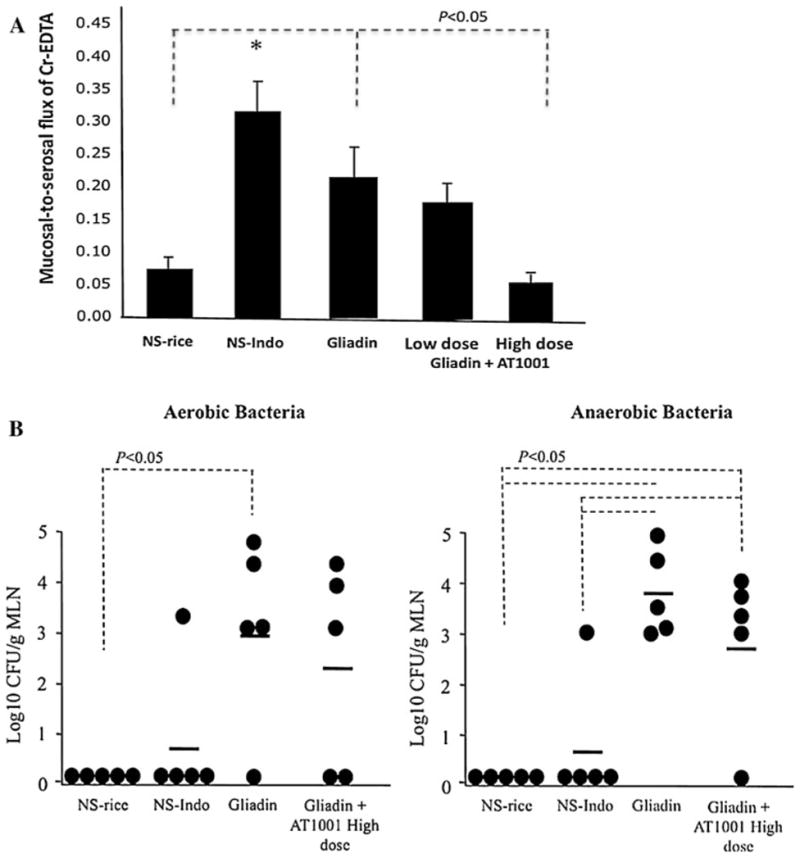
Results from mucosal-to-serosal flux of 51Cr-EDTA in jejunal tissue samples (a), and enteric bacterial translocation to jejunal mesenteric lymph nodes (b) using HCD4/HLA-DQ8 transgenic mice. We studied non-sensitized mice orally challenged with rice cereal (a: NS-rice, n = 9) or indomethacin (a: NS-Indo, n = 5) as well as animals sensitized and orally challenged with gliadin (a: Gliadin, n = 7) alone or in the presence of the paracellular permeability inhibitor AT1001 at low and high dose (a: Gliadin + AT1001, n = 8 each group). Values in black bars are mean ± SEM (A: *p < 0.05 versus NS-rice and Gliadin + AT1001 at low and high dose). Dot plots and mean values (horizontal lines) are shown in (b)
Intestinal Bacterial Translocation
Our results showed no differences in bacterial counts when jejunal MLN from indomethacin and rice-fed mice were compared (Fig. 1b). However, gliadin-treated mice exhibited a significant increase (p < 0.05) in aerobic and anaerobic enteric bacterial translocation to jejunal MLN when compared to non-sensitized mice fed with rice (Fig. 1b). Bacterial translocation to spleen was not observed in any group (data not shown). Furthermore, a high dose of AT1001 did not reduce the increased aerobic and anaerobic enteric bacterial translocation to jejunal MLN detected in gliadin-treated mice (Fig. 1b). Aerobic and anaerobic bacterial counts performed in jejunal MLN from C57BL/6 mice orally challenged with rice or indomethacin as well as gliadin-sensitized and orally challenged with gliadin, were below detection limit (< 103 CFU).
Immunostaining for DC Markers and Intraepithelial Lymphocytes
As shown before [14], CD3 immunostaining (Fig. 2a–c) revealed mild enteropathy characterized by CD3+ intraepithelial lymphocytosis in gliadin-sensitized mice subsequently challenged with gliadin (p < 0.05; Fig. 3a).
Fig. 2.

CD3, CD103, and CD11c immunostainings performed in small intestinal samples from HCD4/HLA-DQ8 transgenic mice. Representative pictures from tissue samples taken from non-sensitized mice orally challenged with rice cereal (NS-rice, n = 9) or indomethacin (NS-Indo, n = 5) as well as from animals sensitized and orally challenged with gliadin (Gliadin, n = 7). CD3 immunostaining show a brown reaction (a–c) while CD103 (d–f) and CD11c (g–i) immunostaining in red (a–f, 400×; and g–i, 200×). Specific brown or red staining is highlighted by black arrows
Fig. 3.

Quantification of CD3, CD103, and CD11c immunostainings performed in small intestinal samples from HCD4/HLA-DQ8 transgenic mice. We studied non-sensitized mice orally challenged with rice cereal (NS-rice, n = 9) or indomethacin (NS-Indo, n = 5) as well as animals sensitized and orally challenged with gliadin (Gliadin, n = 7). Black bars represent number of CD3+ lymphocytes/100 enterocytes (a); number of CD103+ cells/field (b); and percentage of CD11c stained area/field (c). Values are mean ± SEM; **p < 0.05 versus NS-rice and NS-Indo; *p < 0.001 versus NS-rice; and †p < 0.01 versus NS-Indo
The CD103 immunostaining showed an intra-epithelial distribution in samples from non-sensitized as well as from gliadin-treated mice (Fig. 2d–f). CD103+ cells differed from intraepithelial lymphocytes in morphology exhibiting elongated-cylindrical cell bodies located across the epithelium (Fig. 2). Quantification of CD103+ cells demonstrated an increase (p < 0.05) in tissue samples from gliadin-treated mice compared to non-sensitized controls fed either rice or indomethacin (Fig. 3b).
CD11c immunostaining showed a unique pattern; a dense network of inter-connected cells through cytoplasmic projections across the connective tissue of villi and the submucosa in all groups (Fig. 2g–i). Non-sensitized mice treated with indomethacin exhibited decreased CD11c immunostaining compared to rice-fed controls. Quantification of the % area stained revealed a reduction of CD11c immunostaining in gliadin-treated mice compared to both non-sensitized controls fed either rice (p < 0.001) or indomethacin (p < 0.01). These results indicate that sensitization and challenge with gliadin in HCD4/HLA-DQ8 transgenic mice induces a more marked change in CD11c than the one induced by indomethacin (Fig. 3c). Due to the different morphology and distribution of CD11c versus CD103, we did not perform any further co-localization using both markers.
Immunostainings for TLR
When TLR-2 and 4 immunostaining were performed, scant numbers of intraepithelial TLR-4+ and TLR-2+ cells were detected in all groups. Furthermore, quantification of TLR-4+ and TLR-2+ cells did not show significant differences between groups (Fig. 4). Due to a similar morphology and distribution between CD103+ and TLR-2+ cells, we investigated whether the increased CD103+ cells in gluten-treated mice also expressed TLR-2 by co-localization of both markers. Results showed the presence of single and double fluorescent cells in tissues from non-sensitized rice-fed mice (Fig. 5a, b). However, only single fluorescence for CD103 and TLR-2 was detected in samples from gluten-treated mice (Fig. 5c, d).
Fig. 4.
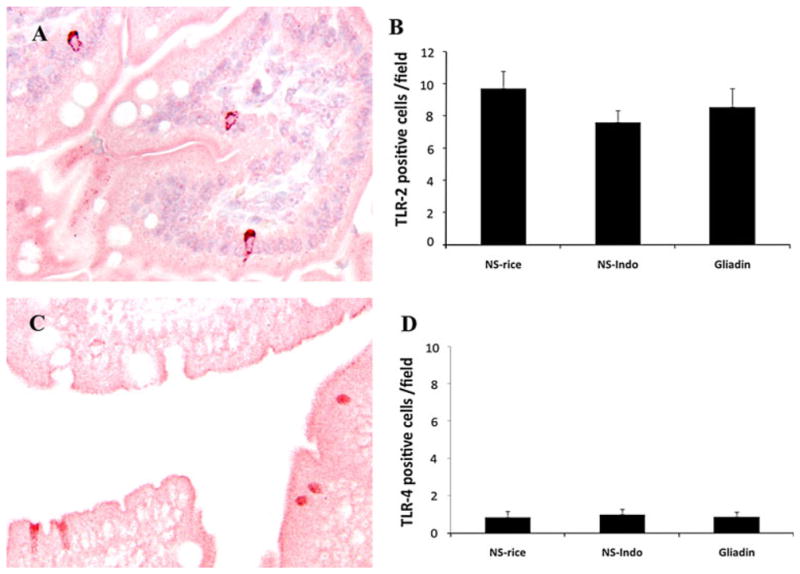
Immunohistochemistry and quantifications of Toll-like receptor (TLR)-2 and 4 performed in jejunal samples from HCD4/HLA-DQ8 transgenic mice. We studied non-sensitized mice orally challenged with rice cereal (NS-rice, n = 9) or indomethacin (NS-Indo, n = 5) as well as animals sensitized and orally challenged with gliadin (Gliadin, n = 7). Immunostaining examples of TLR-2 (a) and TLR-4 (c) from a non-sensitized mice challenged with rice cereal (NS-rice); microphotographs ×200. Number of TLR-2 (b) and TLR-4 positive cells (d). Values are represented in black bars as mean ± SEM
Fig. 5.

Two-color immunofluorescence for CD103 versus TLR-2 in jejunal samples from HCD4/HLA-DQ8 transgenic mice. White arrows show CD103+ dendritic cells (FITC-conjugated antibody) in green, TLR-2+ cells (CY5-conjugated antibody) in red, and CD103+TLR-2+ cell in yellow. Representative pictures from non-sensitized mice orally challenged with rice cereal are shown in a and b, while specimens from gliadin-sensitized and orally challenged mice are in c and d. Photomicrographs ×630
Discussion
In HCD4/HLA-DQ8 transgenic mice, gliadin induces changes in intestinal paracellular permeability associated with alterations in innate immune markers that are different from those generated by indomethacin, a non-specific trigger of mucosal dysfunction [33]. Gliadin-induced changes were characterized by an increase in trans-epithelial CD103+ cells and a major reduction of CD11c phenotype in the intestinal villus as well as by increased aerobic and anaerobic bacterial translocation to MLN, which was not inhibited by the intestinal paracellular inhibitor AT1001.
In this study we found an increased flux of the paracellular probe 51Cr-EDTA in jejunal tissue samples from gliadin-treated mice (Fig. 1a). This might be due to a direct effect on the intestinal epithelium of the gliadin orally gavaged and/or secondary to the immune response induced by gliadin sensitization [2]. However, the increased 51Cr-EDTA fluxes induced by gliadin treatment were lower than the one generated by indomethacin in non-sensitive HCD4/HLA-DQ8 mice (Fig. 1a). A defect in the paracellular pathway is relevant not only to this animal model but also to celiac disease [16, 17]. We have previously shown that in HCD4/HLA-DQ8 mice, gliadin causes an increased flux of the transcellular macromolecular probe HRP as well as an augmented short circuit current, which is not observed at this magnitude in haplotype controls (HLA-DQ6) and wild-type (C57BL/6) mice treated with gliadin [12, 14]. The alteration of various pathways involving barrier function by gliadin may allow abnormal flux of luminal antigens and/or bacteria into the mucosa via a transcellular and/or a paracellular route [12]. In this study, a defective intestinal barrier in gliadin-treated mice was associated with increased aerobic and anaerobic bacterial translocation to MLN, but not to spleen, indicating adequate containment of enteric bacteria in the mesenteric compartment, as reported in other mouse models [34]. This effect was specific to gliadin in this model because the increased 51Cr-EDTA flux induced by indomethacin in non-sensitized HCD4/HLA-DQ8 mice (Fig. 1a) was not associated with augmented bacterial translocation to MLN (Fig. 1b). Furthermore, wild-type C57BL/6 mice challenged with rice, indomethacin, or treated with gliadin had undetectable bacteria levels in MLN. Our results are in accordance with previous studies showing presence of bacteria in jejunal mucosa [35] as well as high levels of commensal-specific Ab in serum from celiac disease patients [36, 37] and from HCD4/HLA-DQ8 transgenic mice treated with gliadin [12]. In our study, enhanced enteric bacterial translocation may be related to microbial up-take via a paracellular and/or a transcellular pathway. Using the paracellular inhibitor AT1001 in gliadin-treated mice, we demonstrated that a high dose of this compound restored the paracellular flux of 51Cr-EDTA to normal levels without affecting bacterial translocation (Fig. 1a, b). Therefore, a paracellular mechanism for the increased bacterial translocation to MLN observed in gliadin-treated mice is unlikely.
An abnormal flux of luminal content into the mucosa may induce activation and migration of resident DC [31, 38]. DCs are antigen-presenting cells that can be characterized in the mouse intestine by using markers such as CD11c or CD103 [27]. In this study, the pattern of staining observed for each of the DC markers was unique. Tissue samples from all groups showed CD11c+ cells forming a network in the connective tissue from the lamina propria while CD103+ cells were distributed in the intra-epithelial compartment, exhibiting a different morphology than CD3+ intraepithelial lymphocytes (Fig. 2). These results differed from a previous study showing the presence of CD11c+CD103+DC in mouse colonic samples [27]. The differences may be explained by the fact that we investigated small intestinal samples in HCD4/HLA-DQ8 transgenic mice. The absence of endogenous MHC class II genes plus the addition of a human-derived DQ8 restriction may introduce changes in the biology and phenotype of DC, considering the fact that MHC class II is a pivotal element for their antigen-presenting role. CD11c immunostaining was markedly reduced in gliadin-treated mice when compared to both non-sensitized control groups (Fig. 2c), highlighting the fact that the network pattern generated by CD11c should not be a supportive structural element from the mucosa. Moreover, a decrease in CD11c was more marked in gliadin than in indomethacin-treated mice, indicating a unique role for gliadin and it subsequent effects as factors that may cause activation and/or migration of CD11c+ cells. Gliadin treatment resulted in increased CD103+ cells when compared to both non-sensitized control groups (Fig. 2b). This suggests an abnormal infiltration or up-regulation of CD103 in the epithelial compartment. In fact, DC can infiltrate the epithelium in order to sense for the presence of bacteria [39, 40]. Here, we hypothesized that increased number of trans-epithelial CD103+DC in the intestinal villus from gliadin-treated mice may facilitate an abnormal transcellular flux of enteric bacteria into the mucosa. Resident CD11c+DC from the mucosa could uptake the infiltrating bacteria and migrate to MLN, facilitating bacterial translocation. This could explain why inhibition of paracellular permeability did not affect the gliadin-associated bacterial translocation in our study. We cannot rule out that M cells from Peyer’s patches provide an additional route that allow transport of bacteria into the associated lymphoid tissue where uptake by resident DC and subsequent translocation to MLN may occur [41].
TLR immunophenotypes were also analyzed. We found no changes between gliadin-treated and non-sensitized mice were detected in the number of TLR-2+ and TLR-4+ cells (Fig. 4). A previous study performed in vitro challenging bone marrow-derived CD11c+DC from HCD4/HLA-DQ8 transgenic mice with gliadin derived-peptide has shown that these cells expressed high levels of TLR-4 and a low T cell proliferative response [42]. However, bone marrow-derived DC may be functionally and phenotypically different to intestinal DC, in particular when grown in the absence of microenvironment components that have the capacity to control their phenotype [38]. TLR-4 is over expressed in different inflammatory conditions of the gastrointestinal tract such as inflammatory bowel diseases, celiac disease, and animal models of intestinal inflammation [30, 31, 43]. A lack of TLR-4 induction in our study could be due to the fact that gliadin treatment in HCD4/HLA-DQ8 transgenic mice only causes mild enteropathy (Fig. 2c). Our results are consistent with previous studies in vitro demonstrating that the effect of gliadin is TLR-2 and 4 independent [44]. Thus, TLR-2 and 4 do not seem to play a role in this model of gliadin sensitivity, although the role of other pattern recognition receptors cannot be ruled out. Over-expression of TLR-2 and 4 in samples from patients with active celiac disease may represent a consequence of inflammation rather than a role in the initiation of gluten-induced pathology. Finally, due to the similar distribution and morphology between TLR-2+ and CD103+ cells, we performed a confocal evaluation using both markers. As a result, CD103+TLR-2+ cells were only detected in the non-sensitized controls fed with rice (Fig. 4b), indicating that this particular DC sub-population was equally affected by gliadin or indomethacin treatment. This effect might be a consequence of an increase in intestinal permeability induced by both treatments [12]. Additionally, the absence of CD103+TLR-2+ cells in the gliadin-treated group indicates that TLR-2 was not involved in the specific induction of CD103 mediated by gliadin in this model.
In conclusion, gliadin sensitization and oral challenge in HCD4/HLA-DQ8 transgenic mice is associated with increased paracellular permeability and bacterial translocation to MLN and augmented number of trans-epithelial CD103+ cells. The paracellular modulator AT-1001 normalized permeability, but did not prevent bacterial translocation induced by gliadin. Therefore, restoration of paracellular permeability alone was not sufficient to prevent bacterial translocation. We propose that trans-epithelial CD103+DC located in the intestinal villus may handle enteric bacteria into the mucosa while resident CD11c+DC could up-take them and have migrated to MLN, inducing their translocation. Our findings may help understand innate pathophysiological mechanisms that occur both at the initiation of the celiac lesion or in patients with gluten sensitivity without celiac disease.
Acknowledgments
This work was supported by grants from the Canadian Association of Gastroenterology (CAG)/Canadian Institute of Health Research (CIHR) (GN2-114709), the Canadian Celiac Association New Investigator Award, ALBA Therapeutics (E. Verdú), the National Institute of Health grant R01 DK048373-13 (A. Fasano), and AGL2008-01440/ALI and Consolider Fun-C-Food CSD2007-00063 from Ministry of Science and Innovation (Spain, Y. Sanz). E. Verdú holds a McMaster University Department of Medicine Internal Career Research Award. Drs. David and Murray were supported in part by R01 DK71003.
Contributor Information
Manuel A. Silva, Email: msilvamu@hotmail.com, Department of Medicine, Health Sciences Centre, Farncombe Family Digestive Health Research Institute, McMaster University, 1200 Main Street West, Hamilton, ON L8N 3Z5, Canada
Jennifer Jury, Department of Medicine, Health Sciences Centre, Farncombe Family Digestive Health Research Institute, McMaster University, 1200 Main Street West, Hamilton, ON L8N 3Z5, Canada.
Yolanda Sanz, Institute of Agrochemistry and Food Technology (IATA), Spanish National Research Council (CSIC), Valencia, Spain.
Michelle Wiepjes, Department of Medicine, Health Sciences Centre, Farncombe Family Digestive Health Research Institute, McMaster University, 1200 Main Street West, Hamilton, ON L8N 3Z5, Canada.
Xianxi Huang, Department of Medicine, Health Sciences Centre, Farncombe Family Digestive Health Research Institute, McMaster University, 1200 Main Street West, Hamilton, ON L8N 3Z5, Canada.
Joseph A. Murray, Division of Gastroenterology, Mayo Clinic, Rochester, MN, USA
Chella S. David, Department of Immunology, Mayo Clinic, Rochester, MN, USA
Alessio Fasano, Center for Celiac Research and Mucosal Biology Research Center, University of Maryland School of Medicine, Baltimore, MD, USA.
Elena F. Verdú, Email: verdue@mcmaster.ca, Department of Medicine, Health Sciences Centre, Farncombe Family Digestive Health Research Institute, McMaster University, 1200 Main Street West, Hamilton, ON L8N 3Z5, Canada
References
- 1.Kagnoff MF. Celiac disease: pathogenesis of a model immunogenetic disease. J Clin Invest. 2007;117:41–49. doi: 10.1172/JCI30253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silva MA, Guiraldes E, Perdue MH. The adverse effect of eating wheat and related grasses for some individuals: coeliac disease and gluten sensitivity. AgroFOOD Ind Hi-Tech. 2007;18:18–20. [Google Scholar]
- 3.Sollid LM, Lie BA. Celiac disease genetics: current concepts and practical applications. Clin Gastroenterol Hepatol. 2005;3:843–851. doi: 10.1016/s1542-3565(05)00532-x. [DOI] [PubMed] [Google Scholar]
- 4.Verdú EF, Armstrong D, Murray JA. Between celiac disease and irritable bowel syndrome: the “no man’s land” of gluten sensitivity. Am J Gastroenterol. 2009;104:1587–1594. doi: 10.1038/ajg.2009.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaukinen K, Collin P, Holm K, Karvonen AL, Pikkarainem P, Mäki M. Small-bowel mucosal inflammation in reticulin or gliadin antibody-positive patients without villous atrophy. Scand J Gastroenterol. 1998;33:944–949. doi: 10.1080/003655298750026967. [DOI] [PubMed] [Google Scholar]
- 6.Niveloni S, et al. Gluten sensitivity in patients with primary biliary cirrhosis. Am J Gastroenterol. 1998;93:404–408. doi: 10.1111/j.1572-0241.1998.00404.x. [DOI] [PubMed] [Google Scholar]
- 7.Troncone R, et al. Gluten sensitivity in a subset of children with insulin dependent diabetes mellitus. Am J Gastroenterol. 2003;98:590–595. doi: 10.1111/j.1572-0241.2003.07301.x. [DOI] [PubMed] [Google Scholar]
- 8.Black KE, Murray JA, David CS. HLA-DQ determines the response to exogenous wheat proteins: a model of gluten sensitivity in transgenic knockout mice. J Immunol. 2002;169:5595–5600. doi: 10.4049/jimmunol.169.10.5595. [DOI] [PubMed] [Google Scholar]
- 9.Stepánková R, Tlaskalová-Hogenová H, Sinkora J, Jodl J, Fric P. Changes in jejunal mucosa after long-term feeding of germfree rats with gluten. Scan J Gastroenterol. 1996;31:551–557. doi: 10.3109/00365529609009127. [DOI] [PubMed] [Google Scholar]
- 10.de Kauwe AL, et al. Resistance to celiac disease in humanized HLA-DR3-DQ2-transgenic mice expressing specific anti-gliadin CD4+ T cells. J Immunol. 2009;182:7440–7450. doi: 10.4049/jimmunol.0900233. [DOI] [PubMed] [Google Scholar]
- 11.Marietta E, et al. A new model for dermatitis herpetiformis that uses HLA-DQ8 transgenic NOD mice. J Clin Invest. 2004;114:1090–1097. doi: 10.1172/JCI21055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Natividad J, et al. Host responses to intestinal microbial antigens in gluten-sensitive mice. PLoS ONE. 2009;4:e6472. doi: 10.1371/journal.pone.0006472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinier M, et al. Polymeric binders suppress gliadin-induced toxicity in the intestinal epithelium. Gastroenterology. 2009;136:288–298. doi: 10.1053/j.gastro.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 14.Verdú EF, et al. Gliadin-dependent neuromuscular and epithelial secretory responses in gluten-sensitive HLA-DQ8 transgenic mice. Am J Physiol Gastrointest Liver Physiol. 2008;294:G217–G225. doi: 10.1152/ajpgi.00225.2007. [DOI] [PubMed] [Google Scholar]
- 15.Arrieta MC, Bistritz L, Meddings JB. Alterations in intestinal permeability. Gut. 2006;55:1512–1520. doi: 10.1136/gut.2005.085373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cox MA, Lewis KO, Cooper BT. Measurements of small intestinal permeability markers, lactulose, and mannitol in serum: results in celiac disease. Dig Dis Sci. 1999;44:402–406. doi: 10.1023/a:1026679123148. [DOI] [PubMed] [Google Scholar]
- 17.Drago S, et al. Gliadin, zonulin and gut permeability: effect on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand J Gastroenterol. 2006;41:408–419. doi: 10.1080/00365520500235334. [DOI] [PubMed] [Google Scholar]
- 18.Montalto M, Cuoco L, Ricci R, Maggiano N, Vecchio FM, Gasbarrini G. Immunohistochemical analysis of ZO-1 in the duodenal mucosa of patients with untreated and treated celiac disease. Digestion. 2002;65:227–233. doi: 10.1159/000063817. [DOI] [PubMed] [Google Scholar]
- 19.Szakál DN, Gyorffy H, Arató A, et al. Mucosal expression of claudins 2, 3 and 4 in proximal and distal part of duodenum in children with coeliac disease. Virchows Arch. 2010;456:245–250. doi: 10.1007/s00428-009-0879-7. [DOI] [PubMed] [Google Scholar]
- 20.Paterson BM, Lammers KM, Arrieta MC, Fasano A, Meddings JB. The safety, tolerance, pharmacokinetic and pharmacodynamic effects of single doses of AT-1001 in coeliac disease subjects: a proof of concept study. Aliment Pharmacol Ther. 2007;26:757–766. doi: 10.1111/j.1365-2036.2007.03413.x. [DOI] [PubMed] [Google Scholar]
- 21.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 22.Steinman RM. Dendritic cells: understanding immunogenicity. Eur J Immunol. 2007;37(Suppl 1):S53–S60. doi: 10.1002/eji.200737400. [DOI] [PubMed] [Google Scholar]
- 23.Di Sabatino A, et al. Evidence for the role of interferon-alfa production by dendritic cells in the Th1 response in celiac disease. Gastroenterology. 2007;133:1175–1187. doi: 10.1053/j.gastro.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 24.Qiao SW, et al. Antigen presentation to celiac lesion-derived T cells of a 33-mer gliadin peptide naturally formed by gastrointestinal digestion. J Immunol. 2004;173:1757–1762. doi: 10.4049/jimmunol.173.3.1757. [DOI] [PubMed] [Google Scholar]
- 25.Ráki M, Tollefsen S, Molberg Ø, Lundin KE, Sollid LM, Jahnsen FL. A unique dendritic cell subset accumulates in the celiac lesion and efficiently activates gluten-reactive T cells. Gastroenterology. 2006;131:428–438. doi: 10.1053/j.gastro.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 26.Liu L, MacPherson G. Rat intestinal dendritic cells: immunostimulatory potency and phenotypic characterization. Immunology. 1995;85:88–93. [PMC free article] [PubMed] [Google Scholar]
- 27.Annacker O, et al. Essential role for CD103 in the T cell-mediated regulation of experimental colitis. J Exp Med. 2005;202:1051–1061. doi: 10.1084/jem.20040662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parker L, Prince L, Sabroe I. Translational mini-review series on Toll-Like receptors: networks regulated by Toll-like receptors mediate innate and adaptive immunity. Clin Exp Immunol. 2007;147:199–207. doi: 10.1111/j.1365-2249.2006.03203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takeda K, Kaisho T, Akira S. Toll-like receptors. Ann Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 30.Szebeni B, et al. Increased mucosal expression of Toll-like receptor (TLR)2 and TLR4 in coeliac disease. J Pediatr Gastroenterol Nutr. 2007;45:187–193. doi: 10.1097/MPG.0b013e318064514a. [DOI] [PubMed] [Google Scholar]
- 31.Silva MA, Jury J, Porras M, Vergara P, Perdue MH. Intestinal epithelial barrier dysfunction and dendritic cell redistribution during early stages of inflammation in the rat: role for TLR-2 and -4 Blockage. Inflamm Bowel Dis. 2008;14:632–644. doi: 10.1002/ibd.20379. [DOI] [PubMed] [Google Scholar]
- 32.Hapfelmeier S, et al. The salmonella pathogenicity island (SPI)-2 and SPI-1 type III secretion systems allow Salmonella serovar Typhimurium to trigger colitis via MyD88-dependent and MyD88-independent mechanisms. J Immunol. 2005;174:1675–1685. doi: 10.4049/jimmunol.174.3.1675. [DOI] [PubMed] [Google Scholar]
- 33.Sigthorsson G, et al. COX-1 and 2, intestinal integrity, and pathogenesis of nonsteroidal anti-inflammatory drug enteropathy in mice. Gastroenterology. 2002;122:1913–1923. doi: 10.1053/gast.2002.33647. [DOI] [PubMed] [Google Scholar]
- 34.Slack E, et al. Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science. 2009;325:617–620. doi: 10.1126/science.1172747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forsberg G, Fahlgren A, Hörstedt P, Hammarström S, Hernell O, Hammarström ML. Presence of bacteria and innate immunity of intestinal epithelium in childhood celiac disease. Am J Gastroenterol. 2004;99:894–904. doi: 10.1111/j.1572-0241.2004.04157.x. [DOI] [PubMed] [Google Scholar]
- 36.Ashorn S, et al. Serological responses to microbial antigens in celiac disease patients during a gluten-free diet. J Clin Immunol. 2009;29:190–195. doi: 10.1007/s10875-008-9255-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ashorn S, et al. Elevated serum anti-Saccharomyces cerevisiae, anti-I2 and anti-OmpW antibody levels in patients with suspicion of celiac disease. J Clin Immunol. 2008;28:486–494. doi: 10.1007/s10875-008-9200-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Silva MA. Intestinal dendritic cells and epithelial barrier dysfunction in Crohn’s disease. Inflamm Bowel Dis. 2009;15:436–453. doi: 10.1002/ibd.20660. [DOI] [PubMed] [Google Scholar]
- 39.Chieppa M, Rescigno M, Huang AY, Germain RN. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J Exp Med. 2006;203:2841–2852. doi: 10.1084/jem.20061884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rescigno M, et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2:361–367. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 41.Pickard JM, Chervonsky AV. Sampling of the intestinal micro-biota by epithelial M cells. Curr Gastroenterol Rep. 2010;5:331–339. doi: 10.1007/s11894-010-0128-x. [DOI] [PubMed] [Google Scholar]
- 42.Ciccocioppo R, et al. Effects of gliadin stimulation on bone marrow-derived dendritic cells from HLA-DQ8 transgenic mice. Dig Liver Dis. 2008;40:927–935. doi: 10.1016/j.dld.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 43.Silva MA, Quera R, Valenzuela J, Salim SY, Söderholm JD, Perdue MH. Dendritic cells and Toll-like receptor 2 and 4 in the ileum from Crohn’s disease patients. Dig Dis Sci. 2008;53:1917–1928. doi: 10.1007/s10620-007-0105-x. [DOI] [PubMed] [Google Scholar]
- 44.Thomas KE, Sapone A, Fasano A, Vogel SN. Gliadin stimulation of murine macrophage inflammatory gene expression and intestinal permeability are MyD88-dependent: role of the innate immune response in Celiac disease. J Immunol. 2006;176:2512–2521. doi: 10.4049/jimmunol.176.4.2512. [DOI] [PubMed] [Google Scholar]