

Abstract
Congenital diaphragmatic hernia (CDH) is a developmental defect of the diaphragm that causes high newborn mortality. Isolated or non-syndromic CDH is considered a multifactorial disease, with strong evidence implicating genetic factors. As low heritability has been reported in isolated CDH, family-based genetic methods have yet to identify the genetic factors associated with the defect. Using the Utah Population Database, we identified distantly related patients from several extended families with a high incidence of isolated CDH. Using high-density genotyping, seven patients were analyzed by homozygosity exclusion rare allele mapping (HERAM) and phased haplotype sharing (HapShare), two methods we developed to map shared chromosome regions. Our patient cohort shared three regions not previously associated with CDH, i.e. 2q11.2-q12.1, 4p13 and 7q11.2, and two regions previously involved in CDH, i.e. 8p23.1 and 15q26.2. The latter regions contain GATA4 and NR2F2, two genes implicated in diaphragm formation in mice. Interestingly, three patients shared the 8p23.1 locus and one of them also harbored the 15q26.2 segment. No coding variants were identified in GATA4 or NR2F2, but a rare shared variant was found in intron 1 of GATA4. This work shows the role of heritability in isolated CDH. Our family-based strategy uncovers new chromosomal regions possibly associated with disease, and suggests that non-coding variants of GATA4 and NR2F2 may contribute to the development of isolated CDH. This approach could speed up the discovery of the genes and regulatory elements causing multifactorial diseases, such as isolated CDH.
Keywords: congenital diaphragmatic hernia, Utah population database, shared segment analysis, GATA4, NR2F2
INTRODUCTION
Congenital diaphragmatic hernia (CDH) is a severe neonatal defect with an incidence of 1/2,500–5,000 births and characterized by incomplete diaphragm formation. There are three major types of CDH: Bochdalek (posterolateral defect, most common), Morgagni (anterior defect), and central (rare). CDH can present either as an isolated defect, i.e. non-syndromic (~70% of cases), or in association with other defects, i.e. non-isolated or syndromic (~30% of cases) [Beurskens et al., 2009]. It results in pulmonary hypoplasia secondary to the migration of abdominal organs into the chest, and carries a mortality of about 40% despite the use of advanced therapies, including extracorporeal membrane oxygenation [Mah et al., 2009]. Mortality is mostly attributable to pulmonary vascular bed abnormalities and consequent pulmonary hypertension.
The etiology of isolated CDH remains largely unknown [Klaassens et al., 2009]. Support for a genetic contribution comes from the association of specific genetic syndromes with CDH, and the finding of copy number variants in up to 30% of both non-isolated and isolated cases [Beurskens et al., 2009; Bielinska et al., 2007; Czeizel and Kovacs, 1985; Holder et al., 2007; Klaassens et al., 2007; Norio et al., 1984; Pober, 2008; Pober et al., 2005; Slavotinek et al., 2006; Srisupundit et al., 2010; Wat et al., 2009; Wat et al., 2011]. Additionally, various genes and gene families have been implicated in diaphragm formation in mice, including Nr2f2 (aka Coup-TFII), Zfpm2 (aka Fog-2), Gata4, Sox7, Wt1, Pdgfrα, Slit3, and the sonic hedgehog signaling pathway [Ackerman et al., 2005; Beurskens et al., 2010; Beurskens et al., 2007; Bleyl et al., 2007; Jay et al., 2007; Kreidberg et al., 1993; Wat et al., 2012; You et al., 2005]. The most notable molecular pathway involved in CDH involves retinoic acid-mediated activation of NR2F2 which interacts with ZFPM2 to modulate the transcriptional activity of GATA4 [Holder et al., 2007]. In mice, diaphragmatic defects result from inhibition of retinoic acid as well as knockout of Nr2f2, Zfpm2, and Gata4 [Ackerman and Greer, 2007]. In human CDH, recurrent chromosomal abnormalities have been found in genomic regions harboring NR2F2 (15q26), ZFPM2 (8q22-q23), and GATA4/SOX7 (8p23.1), providing further evidence for involvement of genetic factors in CDH [Holder et al., 2007]. Despite the abundance of evidence, candidate gene studies have met with limited success and currently the only gene known to cause isolated human CDH is ZFPM2 [Ackerman et al., 2005; Bleyl et al., 2007; Wat et al., 2011].
Historically, isolated CDH has been considered to be a multigenic disease, postulated to arise from de novo mutational events because familial/sibling recurrence risks are low, between 0.9 and 2% [Czeizel and Kovacs, 1985; Holder et al., 2007; Pober, 2008; Pober et al., 2005]. We hypothesized that heritability plays a role in isolated CDH, and used the Utah Population Database (UPDB) [Slattery and Kerber, 1993], a unique resource that combines pedigree data with medical diagnoses, to identify a cohort of distantly related patients with the defect. The finding of distantly related CDH patients allowed us to use powerful family-based genetic methods to hone in on chromosomal regions associated with isolated CDH. Overall, our approach provides a new paradigm, not feasible in individual patients or small families, for identifying genetic variants associated with isolated CDH and other rare multigenic diseases.
MATERIALS AND METHODS
CDH Cohort Identification
The UPDB is a unique resource located at the University of Utah consisting of computerized records for over 6.5 million individuals [Slattery and Kerber, 1993]. The central component of the UPDB is an extensive set of Utah family histories in which family members are linked to demographic and medical information. The UPDB has been linked to the electronic health records in the University of Utah and Intermountain Healthcare systems and a search of the ICD-9 code for CDH was used to identify a cohort of interrelated patients with isolated CDH and no other associated developmental anomalies, including cardiovascular defects. Seven individuals were selected for genotyping because they share multiple common ancestors (Figure 1) and the UPDB calculated the disease burden within these pedigrees to be significantly higher than in the general population. By enriching our study cohort with interrelated CDH patients from families with significant disease burden, we hoped to discover non-random, disease-associated shared genomic segments. Due to the interrelatedness of the CDH patients and the possibility of additional relationships unknown to the UPDB, no single pedigree structure was assumed a priori to be CDH-associated. Therefore, we compared genotype data between all patient-parent pairs, irrespective of a known relationship, to search for shared chromosomal regions.
Figure 1. Heritability plays a role in isolated CDH.
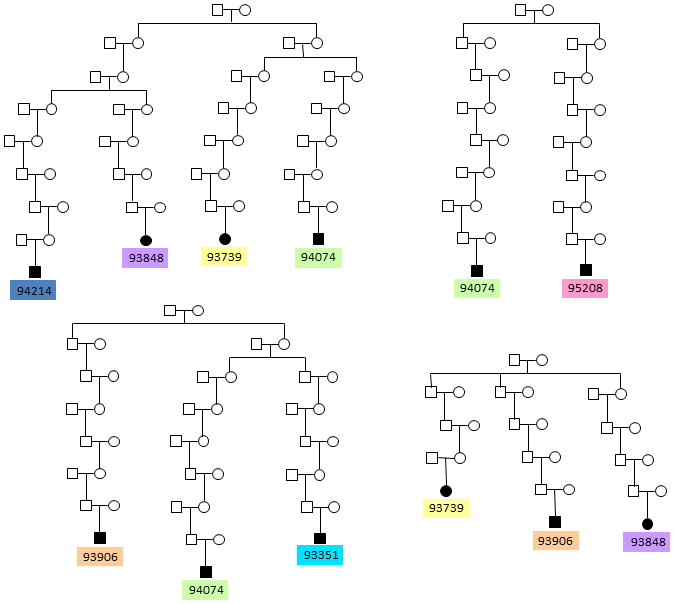
Pedigrees obtained from the UPDB showing seven interrelated isolated CDH patients that were genotyped and subjected to shared segment analysis. Note that some patients are related to each other through multiple founders. By enriching our study cohort with distantly related CDH patients, we hoped to increase the probability of finding shared, disease-associated segments. The disease burden in these pedigrees (number of affecteds/number of descendants) is significantly higher that the incidence of isolated CDH in the general population, suggesting the presence of a heritable component.
Patient Enrollment and Phenotyping
With University of Utah Institutional Review Board approval, individuals and available parents from the isolated CDH cohort were enrolled in the study after informed consent, and blood samples were collected for DNA isolation. Medical records were reviewed for demographic and clinical data.
DNA Isolation
DNA was isolated from peripheral blood samples using a Gentra Autopure LS (Qiagen, Valencia, CA). DNA samples were analyzed by agarose gel electrophoresis to confirm the integrity of the DNA and quantitated using Picogreen (Invitrogen).
SNP Genotyping, CNV Analysis and Shared Segment Analysis
High-density genotyping was performed for 1.1 million SNP markers using the Illumina Human Omni1-Quad BeadChip in the Genomics Core at the University of Utah on seven patients with CDH and available parents (for patients 94214 and 93848, only the mothers were available for genotyping; for the other five patients, genotyping was performed on both parents). An additional 61 control subjects were also genotyped, consisting of 24 children without CDH and their available parents. SNP genotypes for all 80 subjects were called by the Illumina GenomeStudio 2011.1 software using HumanOmni1-Quad_v1-0_H.bpm SNP manifest and HumanOmni1-Quad_v1-0_H.egt cluster files. Only dbSNP markers with 100% call rate were used for further analysis. To exclude copy number variation as the cause of CDH in our patient cohort, genotype data were analyzed using the CNV Package of GoldenHelix SNP & Variation Suite (SVS) 7 and predicted effects of copy number changes were assessed using DECIPHER (http://decipher.sanger.ac.uk/) and/or the Database of Genomic Variants (http://projects.tcag.ca/variation/).
Shared segments were identified using both Microsoft Excel and an automated R-script, version (available on request) of homozygosity exclusion rare allele mapping (HERAM). Genotype data for all CDH patients were compared in a pairwise manner using each parent in turn as a putative obligate carrier (total of 66 comparisons were performed). For each parent-offspring pair, the genome was apportioned into bins of 1 centimorgan (cM) and both reciprocally-homozygous (AA and BB at the same SNP) and rare-shared alleles (AB genotype, with rare B allele) were counted within each bin. In the analysis of 4 individuals (two patient-putative obligate parent pairs), rare alleles were defined as having a minor allele frequency (MAF) <0.1. MAFs were derived from CEU genotypes collected by the International HapMap Project. Using this MAF cutoff, ~650 rare shared alleles were found across the genome, a sufficient number to identify clusters of rare alleles within regions lacking reciprocally-homozygous alleles and confirm that such regions are indeed shared rather than SNP-poor. Figure 2 demonstrates the two-stage process employed by HERAM to detect shared segments. Shared segments are defined as the co-occurrence in parent-offspring pairs of gaps in reciprocally homozygous alleles and enrichment of rare alleles within the gaps where enrichment was arbitrarily defined as > 0.5 rare alleles/cM, which is more than two times the density of rare alleles across the genome assuming a uniform distribution.
Figure 2. Rationale for using HERAM to identify heritable candidate regions for a disease.
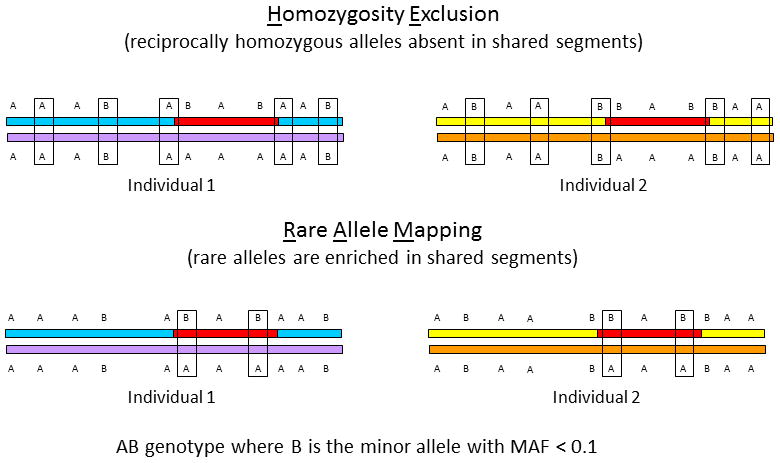
HERAM is based on two ideas. First, within a shared genomic segment (red segment), alleles will never be reciprocally homozygous (that is, AA in one individual and BB in another) at the same SNP. In the upper panel, reciprocally homozygous alleles are boxed. Note that there are no boxes in the shared segment and that boxes define the boundaries of the shared segment. Second, rare-shared alleles are enriched in shared genomic segments. These are shown in the lower diagram with boxed AB genotypes, where the MAF of the B allele is <0.1. Regions that are both devoid of exclusionary reciprocally homozygous SNPs and enriched for rare shared alleles are identical-by-state, and likely identical-by-descent when the segment is large (≥ 6 cM) and individuals are distantly related. MAF= minor allele frequency.
Shared segments identified by HERAM were verified by a more time- and labor-intensive method that examines sharing of phased haplotype data (HapShare). This method has been used previously to identify a 5 cM shared segment containing APC in an extended colon cancer pedigree [Neklason et al., 2008]. For HapShare, markers with Mendelian errors, detected using the pedigree algorithm option of PBAT Family-Based Quality Assurance in GoldenHelix SVS 7, were excluded from the analysis. Non-polymorphic markers were also removed, and linkage disequilibrium (LD) pruning was carried out using an R2 threshold of 0.95 with the composite haplotype method (CHM) in GoldenHelix SVS 7. After culling the data, a total of 561,052 dbSNP marker genotypes remained and were used in the phased haplotype sharing analysis.
Maternal and paternal haplotypes were assigned for each marker based on a Mendelian inheritance model (Supplemental Table I, genotype-to-haplotype conversion). HapShare was carried out in 2 steps. First, each phased paternal and maternal haplotype from the seven CDH patients (total of 14 haplotypes) were compared to each other in a pair-wise manner to identify identical shared segments. Shared segments were defined as regions where at least one informative inclusion marker (haplotype 1/haplotype 2 = A/A or B/B) was flanked by exclusion markers (haplotype 1/haplotype 2 = A/B or B/A) at both ends. Shared segments were not disrupted by non-informative calls. Because paternal and maternal haplotypes can be determined even when genotypes are only available for one parent and a child, a total of 91 pair-wise combinations were analyzed using this approach. Next, when sharing was observed among CDH subjects, the shared haplotype was compared against paternal and maternal haplotypes of 24 children who did not have CDH (total of 48 haplotypes) to determine whether a shared segment might be a common haplotype.
For both HERAM and HapShare, only autosomal markers were analyzed. Also, a HapMap recombination map was required to determine the size/location of shared genomic segments. This map was generated for Omni1-Quad dbSNPs using the following website: http://hapmap.ncbi.nlm.nih.gov/downloads/recombination/2011-01_phaseII_B37/. For dbSNPs absent from the HapMap recombination data, the marker position was interpolated from the two nearest flanking HapMap SNPs.
Candidate Gene Screening
PCR primers for coding elements in GATA4, SOX7, NR2F2 and ZFPM2 were designed using the Exon Primer utility (http://ihg.gsf.de/ihg/ExonPrimer.html) and used to amplify DNA from CDH patients with shared segments containing these genes (PCR Primers are shown in Supplemental Table II). For the analysis of ZFPM2, all patient DNA samples (10 ng/sample) were amplified in duplicate by PCR using Platinum Taq DNA polymerase (Invitrogen, Carlsbad, CA) and analyzed using a plate-based Lightscanner (Idaho Technology) as previously described [Arrington et al., 2012; Arrington et al., 2008]. Samples giving abnormal profiles were purified by treatment with 4μl of Exo-SAP-IT (USB, Cleveland, OH) at 37°C for 2 hours and 80°C for 15 minutes and then submitted to the University of Utah DNA sequencing core for analysis to confirm Lightscanner results. For the analysis of GATA4, SOX7, and NR2F2, DNA from patients sharing segments containing these genes were amplified; the PCR fragments were purified and analyzed by direct DNA sequencing, as above. DNA sequence data were compared with published data.
RESULTS
To establish the involvement of heritable factors in isolated CDH, we leveraged the resources of the UPDB to identify a cohort of distantly related isolated CDH patients belonging to kindreds with significant familial standardized incidence ratios (FSIR) (2.7 to 19.9, p=0.001 to 0.0001) [Kerber, 1995]. The numbers of living affecteds in each kindred ranged from 3 to 12, with affected individuals being separated by an average of approximately 12 meioses. Within these kindreds, we focused on seven patients recruited to participate in the study (Figure 1). Each of the seven CDH patients is related to at least one other patient and, in some cases, several other CDH patients (Figure 1), making them an ideal cohort to uncover non-random, disease-associated chromosomal regions for CDH.
All patients and available parents were genotyped and the data analyzed for copy number variants. All copy number changes ≥ 0.4 Mb in size were analyzed for possible disease-association. Four duplicated regions and one deleted region were identified among the CDH cohort (Table I). None of the segments has been previously associated with CDH and all segments were predicted to be copy number polymorphisms by DECIPHER and/or the Database of Genomic Variants. A gene-by-gene analysis of these segments did not reveal any known CDH candidates.
Table I.
Copy number changes (CNC) detected in the CDH cohort.
| Patient | Type | CNC Location (GRCh37/hg19) | Size (bp) |
|---|---|---|---|
| 93906 | Duplication | chr2:89128221-91923634 | 2,795,413 |
| 95208 | Duplication | chr10:46220450-47966358 | 1,745,908 |
| 93739 | Duplication | chr14:19433398-20424184 | 990,786 |
| 93848 | Duplication | chr15:20019161-22455504 | 2,436,343 |
| 94074 | Deletion | chr15:22589211-23688944 | 1,099,733 |
Since no single ancestral relationship could be assumed to associate with disease more strongly than another, and knowledge of common ancestors could be incomplete because the UPDB only includes pedigree information from the last six-seven generations, we used a method to map shared chromosomal regions that does not require specific pedigree information. HERAM is a method we recently developed to identify shared chromosomal segments between patient-patient and/or parent-offspring pairs using both parents as putative obligate carriers. This conceptually simple method involves a two-step comparison of SNP data to identify shared regions (Figure 2). The first step borrows from the homozygosity haplotype method [Jiang et al., 2009; Thomas et al., 2008] (Figure 2, upper panel), and the second step adds information about rare alleles (Figure 2, lower panel). HERAM is based on two ideas. First, parent-offspring pairs who share a heritable genetic risk variant are never reciprocally homozygous within the shared region (e.g., AA in one individual and BB in another). Second, rare alleles are enriched in shared regions (e.g., AB genotype for SNPs with rare minor alleles). Regions that are both devoid of exclusionary, reciprocally homozygous SNPs and enriched for rare shared alleles are identical-by-state. For shared chromosomal regions extending more than a few centimorgans (cM), identity-by-state is considered equivalent to identity-by-descent.
In our patient cohort, the average number of meioses separating patients was 12.6; range: 8–16. Interestingly, recent work by Huff et al. shows that individuals separated by >7 meioses rarely share segments greater than 6 cM [Huff et al., 2011]. Therefore, we used this segment size as a general reference for potentially non-random, disease-associated segments.
Results of the shared segment analysis are summarized in Table II. HERAM analysis of all possible combinations of parent-offspring pairs identified two shared genomic segments larger than 6 cM, one at 8p23.1 (22.2–28.4 cM: 6.2 cM) and another at 15q26.2 (111.7–126.7 cM: 15 cM). HERAM plots depicting the shared regions on chromosomes 8 and 15 and genes within these segments are shown in Figure 3. Two additional large shared segments slightly less than 6 cM were found at 4p13 (62.5–68.3 cM: 5.8 cM) and 7q11.2 (84.3–88.5 cM: 4.2 cM). Sharing of all four segments was independently confirmed by HapShare. HapShare also identified an additional 4.7 cM segment at 2q11.2–q12.1 (119.5–124.2 cM). Retrospective analysis of HERAM data revealed that two miscalls prevented the 2q11.2–q12.1 segment from being identified. HapShare results are graphically depicted on HapMap centimorgan recombination maps (Figure 4 and Supplemental Figure 1). None of the shared segments identified among the CDH cohort were found to be common haplotypes when compared to paternal and maternal haplotypes from the 24 children who did not have CDH (Figure 4b, d and Supplemental Figure 1b,d, and f). Overall, HERAM results were validated by HapShare analysis with the caveat that Mendelian errors must be culled from the genotype data before HERAM analysis to ensure that shared regions are not missed.
Table II.
Summary of shared segments identified by HERAM and confirmed by HapShare.
| Patient_Obligate Parent | Cytoband | Shared Segment Location (GRCh37/hg19) | Size (bp) | HapMap Location (cM) | HapMap Distance (cM) |
|---|---|---|---|---|---|
| 93351_Maternal, 93906_Paternal | 2q11.2-q12.1 | chr2:100480951-106039695 | 5,558,745 | 119.5–124.1 | 4.6 |
| 93739_Paternal, 93906_Maternal | 4p14-p12 | chr4:40198905-45007208 | 4,808,304 | 62.5–68.3 | 5.8 |
| 94214_Maternal, 95208_Paternal | 7q11.22-q11.23 | chr7:69952187-73789730 | 3,837,544 | 84.3–88.5 | 4.2 |
| 93848_Paternal, 94074_Paternal | 8p23.1 | chr8:10730558-12697557 | 1,967,000 | 22.2–28.4 | 6.2 |
| 93848_Paternal, 93906_Maternal | 8p23.1 | chr8:11517977-12667357 | 1,149,381 | 24.2–28.4 | 4.2 |
| 93906_Maternal, 94074_Paternal | 8p23.1 | chr8:11541697-12667357 | 1,125,661 | 24.2–28.4 | 4.2 |
| 93739_Maternal, 93848_Paternal | 15q26.1-q26.2 | chr15:92091172-96911139 | 4,819,968 | 111.7–126.7 | 15.0 |
Figure 3. HERAM analysis in isolated CDH kindreds reveals large shared chromosomal regions.
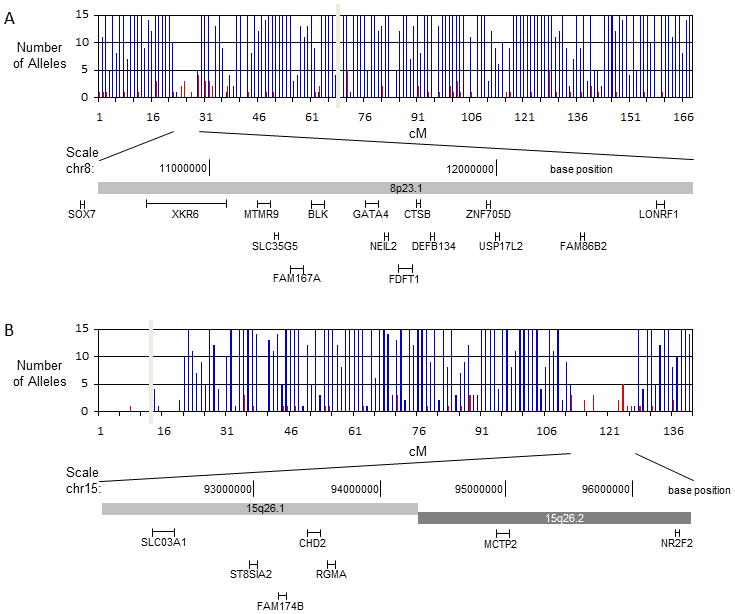
a. 8p23.1 (22.2–28.4 cM) and b. 15q26.2 (111.7–126.7 cM). HERAM plots were generated by dividing each chromosome into bins of 1 cM and counting the number of reciprocally-homozygous and rare-shared alleles within each bin (reciprocally-homozygous alleles are shown in blue and rare shared alleles in red). Shared regions are identified by the co-occurrence of gaps in the homozygous exclusion map ≥ 6 cM and enrichment of rare shared alleles within the gaps. The vertical grey bar represents the location of the centromere. The names and approximate location of genes contained within each shared region are shown below the HERAM plots. Shared regions on chromosomes 8 and 15 coincide with recurrently deleted and duplicated segments found in patients with non-isolated CDH and contain the candidate genes GATA4 and NR2F2, respectively.
Figure 4. HapShare confirms that 8p23.1 and 15q26.2 are shared and unique to isolated CDH kindreds.
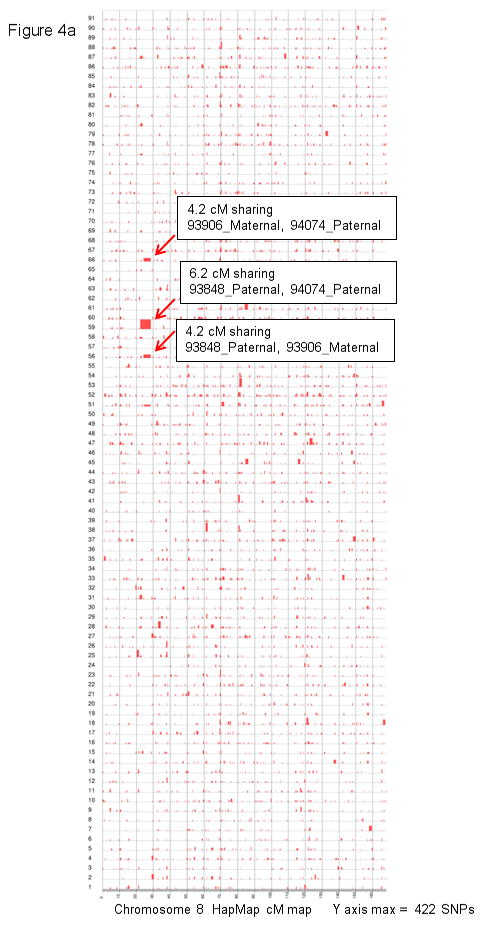
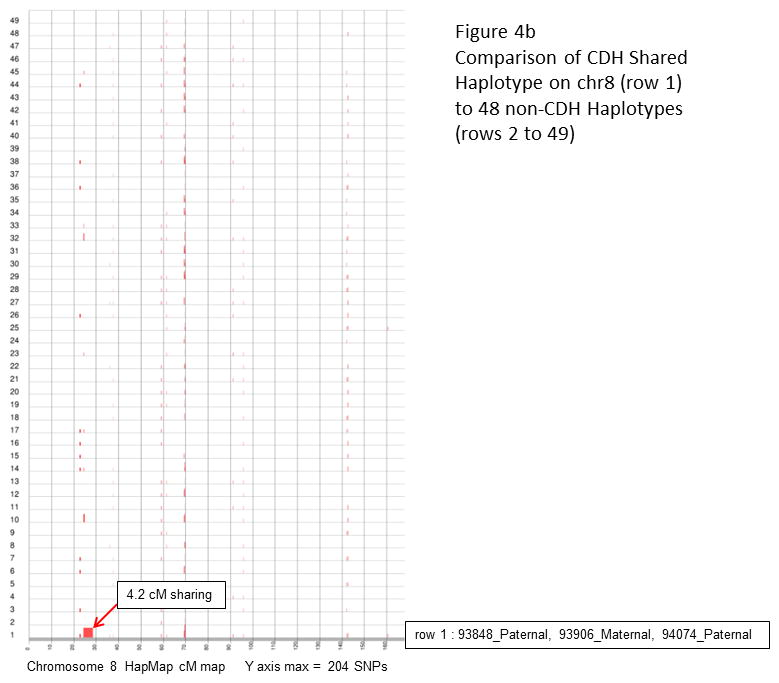
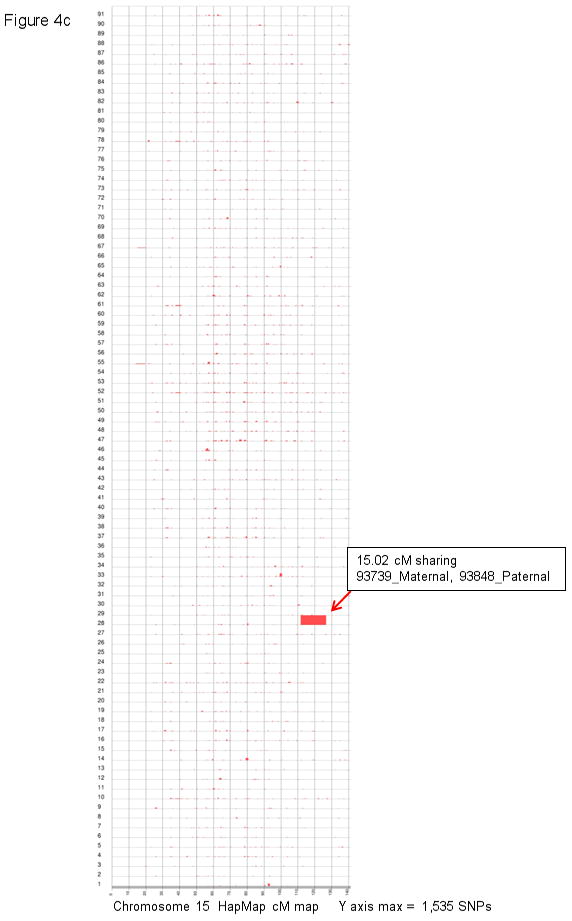
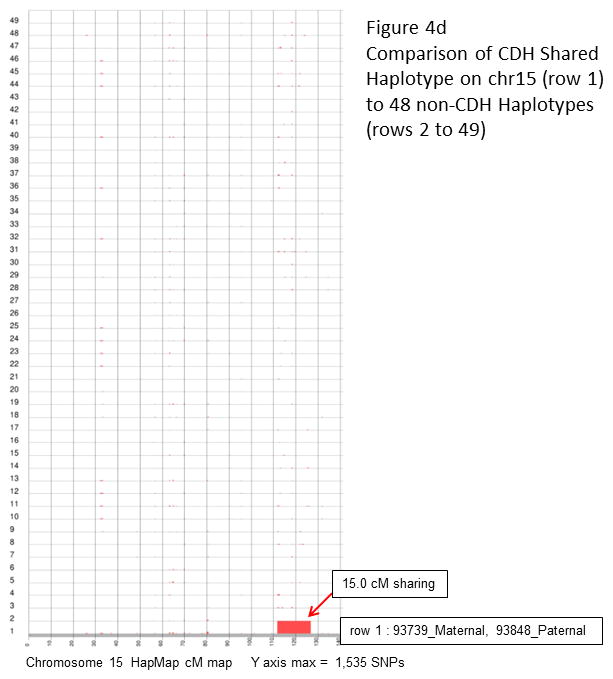
a and c: a total of 91 pair-wise combinations were evaluated for shared segments across the entire genome (7 isolated CDH patients, total of 14 haplotypes). Results for chromosome 8 (a) and 15 (c) are plotted along HapMap centimorgan (cM) recombination maps. The maximum number of consecutive inclusion markers across each chromosome is shown at the bottom of the plot and depicted as the height of a segment along the Y axis. b and d: the shared haplotypes were compared against paternal and maternal haplotypes from 24 children who did not have CDH (total of 48 haplotypes) to eliminate the possibility of common haplotypes. Neither the chromosome 8 (b) or 15 (d) shared segment was found to be a common haplotype.
The two shared segments on chromosomes 8 and 15 coincide with recurrently deleted/duplicated regions in non-isolated human CDH [Holder et al., 2007; Klaassens et al., 2005; Scott et al., 2007; Wat et al., 2009; Wat et al., 2011], and contain genes that cause CDH in mutant mice [Beurskens et al., 2007; Jay et al., 2007; You et al., 2005]. Segment 8p23.1 is the second most commonly deleted region in human CDH and its strongest candidate gene, GATA4, causes ventral, midline diaphragm defects in heterozygous mice [Jay et al., 2007]. Another strong candidate gene, SOX7, is immediately adjacent to but not within the shared segment on 8p23.1. Recent work has shown that Sox7-deficient mice also have ventral, midline diaphragm defects [Wat et al., 2012] and that SOX7 may transcriptionally regulate GATA4 [Wat et al., 2009]. The 15q26.2 locus is the most commonly deleted segment in patients with non-isolated CDH. Of the genes residing within this segment, NR2F2, is the strongest CDH candidate because its activation is regulated by retinoids, and tissue-specific ablation of Nr2f2 in mice causes posterolateral CDH. The finding of known CDH genomic regions provides strong support for the concept that large shared chromosomal regions in distantly related CDH patients likely harbor disease-causing genes. Interestingly, these susceptibility regions appear to have been inherited though multiple unaffected ‘carrier’ individuals suggesting that they are not sufficient to cause CDH, consistent with the idea that isolated CDH is multigenic.
The shared segments on chromosomes 2 and 4 have not been linked to isolated CDH previously. No obvious candidate genes reside within the shared segment on chromosome 2 but the segment on chromosome 4 contains SHISA3, a gene that plays a role in mesoderm maturation by attenuating FGF signaling [Pei and Grishin, 2012]. The 7q11.2 segment resides within the Williams syndrome locus and includes ELN (encoding for elastin). Though none of the genes in this locus has been directly implicated in CDH, ELN is a putative candidate gene because proper post-translational modification of elastin by lysyl oxidase (encoded by LOX) is necessary for normal diaphragm formation [Hornstra et al., 2003]. In addition, a recent case report shows a patient with Williams syndrome with a right-sided Morgagni hernia [Rashid et al., 2009].
A diagram displaying the nearest known ancestral relationships between patients with the shared genomic segments on chromosomes 8 and 15 is shown in Figure 5. Only four CDH patients are shown in this figure since patients 93351, 94214 and 95208 did not share these genomic segments. Known relationships exist between most patient-parent pairs shown in this figure (Figure 1), but for one parent-offspring pair, no common ancestor was identified by the UPDB. Because the family history data contained in the UPDB goes back six-seven generations, the unknown founders are likely very remote. Regardless, the sharing of a large genomic segment indicates that these patients are related, and because they also share a rare phenotype, the shared segments are potentially non-random and associated with disease.
Figure 5. Pedigree structure of the four patients sharing chromosomal regions 8p23.1 and 15q26.2.

Most relationships coincide with those identified by the UPDB, but one involves unknown founders that are likely very remote. Disease susceptibilities within shared segments were inherited though multiple unaffected ‘carrier’ individuals suggesting that they are not sufficient by themselves to cause CDH. Patient 93848 shares more than one segment which raises the possibility that she inherited two genetic susceptibilities, supporting a multigenic model for CDH. There is no clear phenotype-genotype relationship among patients with shared segments.
Three patients (93848, 94074 and 93906) share a segment on 8p23.1 (Figure 5). Patients 93848 and 94074 are very distantly related (separated by 13 meioses) making it highly unlikely to share a 6 cM genomic segment by chance [Huff et al., 2011], and even more unlikely to randomly share a segment previously implicated in CDH. Patient 93906 shares a smaller 8p23.1 segment (24.2–28.4 cM) with the other two patients through an unknown founder, suggesting that he may be even more distantly related. Interestingly, GATA4 is still contained within the smaller shared region.
In addition to sharing the region on 8p23.1, patient 93848 shares a genomic segment on 15q26.2 with 93739 (Figure 5). Although these two patients are only separated by 8 meioses, sharing a large 15 cM segment previously implicated in CDH [Klaassens et al., 2005; Scott et al., 2007] is not likely a random occurrence, suggesting that a CDH susceptibility variant resides within the shared region on 15q26.2. The finding of more than one shared segment in patient 93848 raises the possibility of multigenic involvement in isolated CDH.
With respect to hernia location (Table III), there was no clear genotype-phenotype correlation. Of the three patients with the 8p23.1 segment, one had a left-sided Morgagni hernia, one had a central defect and the other had a right-sided Bochdalek hernia (Figure 5). The patient with the central defect shares the 15q26.2 haplotype with a patient who was born with a left-sided Bochdalek hernia (Figure 5).
Table III.
Hernia phenotype in the patient cohort.
| Patient | Phenotype |
|---|---|
| 93351 | Left-sided Bochdalek hernia |
| 93739 | Left-sided Bochdalek hernia |
| 93848 | Central hernia |
| 93906 | Left-sided Morgagni hernia |
| 94074 | Right-sided Bochdalek hernia |
| 94214 | Agenesis of the left diaphragm |
| 95208 | Left-sided Bochdalek hernia |
Although none of the patients in our cohort were found to share the 8q23.1 segment that contains ZFPM2, we screened this gene for coding variants, but none were found. We also found no deleterious coding variants in GATA4, but we did find a rare shared variant in intron 1. The variant SNP, rs73203482 (c.-458+5G>A, NM_002052.3) (MAF 0.038), is located at the +5 position of the splice donor site of non-coding exon 1. Analysis of the variant using online algorithms produces mixed results. Netgene2 predicts no effect, Fuitfly predicts reduced confidence in the splice donor, and ExonScan predicts loss of the splice donor. Future work will be needed to determine whether this variant has a functional effect on GATA4 expression. No deleterious coding or promoter variants were found in NR2F2 or SOX7 (data not shown). Curiously, no other group has ever reported a CDH-causing mutation in GATA4, SOX7 or NR2F2. Possible explanations for the paucity of susceptibility variants within these genes include the following: 1) coding variants only cause a small fraction of CDH; 2) these genes do not cause human CDH despite strong evidence from animal models suggesting otherwise; or 3) susceptibility variants reside within non-coding, regulatory elements that alter gene expression.
DISCUSSION
A fundamental problem that has prevented researchers from discovering the genetic factors involved in isolated CDH has been the inability to identify extended families with the defect. We have done this using the UPDB, and we employed family-based genetic approaches to identify chromosomal regions strongly associated with CDH. At least two of the regions identified by HERAM and HapShare in our small CDH cohort are good candidates for harboring CDH susceptibilities. Our methods offer several advantages over the current techniques used to identify causative alleles in rare, multigenic disorders. First, they are relatively inexpensive and require few patients. In contrast, methods such as GWAS are costly and require numerous patients, a significant drawback for studying rare diseases. While CNV analysis is useful in defining disease-associated genomic regions, it precludes identification of specific gene/variant(s) responsible for disease. Although candidate gene screening can identify disease-associated genes, it is often fruitless in complex diseases such as CDH, even when there is abundant evidence pointing to a gene’s involvement in disease (as seen with GATA4 and NR2F2 in CDH). Our method overcomes these limitations by identifying shared genomic regions strongly associated with disease in a limited number of distantly related individuals. More importantly, since the genomic segments we have identified are not associated with copy number variation, future work will focus on identification of pathogenic variants within shared segments in our patient cohort using next-generation sequencing. Overall, our strategy has the potential to speed discovery of disease genes in CDH and other rare diseases.
Through the use of a unique genealogical resource in Utah we have identified distantly related patients with isolated CDH. Several of these patients share genomic segments previously linked to CDH because they contain genes implicated in diaphragm development in animal models. Although we have yet to discover disease-causing variants, combining our family-based strategy with next-generation sequencing has the potential to efficiently and cost-effectively identify causative variants in rare, multigenic disorders such as isolated CDH.
Supplementary Material
Acknowledgments
Partial support for datasets within the Utah Population Database is provided by the Huntsman Cancer Institute, and we would like to acknowledge Richard Pimentel for his help in identifying CDH pedigrees. Partial support for C.B.A. was provided by a NICHD grant (K08HD062638). Partial support for L.B. was provided by American Heart Association grant 09BGIA2251076, and the Seed Grant Program at the University of Utah. Partial support for N.J.C. was provided by R01CA134674 and R21CA152336. This investigation was also supported by the Public Health Services research grant numbers UL1-RR025764 and C06-RR11234 from the National Center for Research Resources.
References
- Ackerman KG, Greer JJ. Development of the diaphragm and genetic mouse models of diaphragmatic defects. Am J Med Genet C Semin Med Genet. 2007;145C:109–116. doi: 10.1002/ajmg.c.30128. [DOI] [PubMed] [Google Scholar]
- Ackerman KG, Herron BJ, Vargas SO, Huang H, Tevosian SG, Kochilas L, Rao C, Pober BR, Babiuk RP, Epstein JA, Greer JJ, Beier DR. Fog2 is required for normal diaphragm and lung development in mice and humans. PLoS Genet. 2005;1:58–65. doi: 10.1371/journal.pgen.0010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrington CB, Dowse BR, Bleyl SB, Bowles NE. Non-synonymous variants in pre-B cell leukemia homeobox (PBX) genes are associated with congenital heart defects. Eur J Med Genet. 2012;55:235–237. doi: 10.1016/j.ejmg.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrington CB, Sower CT, Chuckwuk N, Stevens J, Leppert MF, Yetman AT, Bowles NE. Absence of TGFBR1 and TGFBR2 mutations in patients with bicuspid aortic valve and aortic dilation. Am J Cardiol. 2008;102:629–631. doi: 10.1016/j.amjcard.2008.04.044. [DOI] [PubMed] [Google Scholar]
- Beurskens LW, Tibboel D, Lindemans J, Duvekot JJ, Cohen-Overbeek TE, Veenma DC, de Klein A, Greer JJ, Steegers-Theunissen RP. Retinol status of newborn infants is associated with congenital diaphragmatic hernia. Pediatrics. 2010;126:712–720. doi: 10.1542/peds.2010-0521. [DOI] [PubMed] [Google Scholar]
- Beurskens LW, Tibboel D, Steegers-Theunissen RP. Role of nutrition, lifestyle factors, and genes in the pathogenesis of congenital diaphragmatic hernia: human and animal studies. Nutr Rev. 2009;67:719–730. doi: 10.1111/j.1753-4887.2009.00247.x. [DOI] [PubMed] [Google Scholar]
- Beurskens N, Klaassens M, Rottier R, de Klein A, Tibboel D. Linking animal models to human congenital diaphragmatic hernia. Birth Defects Res A Clin Mol Teratol. 2007;79:565–572. doi: 10.1002/bdra.20370. [DOI] [PubMed] [Google Scholar]
- Bielinska M, Jay PY, Erlich JM, Mannisto S, Urban Z, Heikinheimo M, Wilson DB. Molecular genetics of congenital diaphragmatic defects. Ann Med. 2007;39:261–274. doi: 10.1080/07853890701326883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleyl SB, Moshrefi A, Shaw GM, Saijoh Y, Schoenwolf GC, Pennacchio LA, Slavotinek AM. Candidate genes for congenital diaphragmatic hernia from animal models: sequencing of FOG2 and PDGFRalpha reveals rare variants in diaphragmatic hernia patients. Eur J Hum Genet. 2007;15:950–958. doi: 10.1038/sj.ejhg.5201872. [DOI] [PubMed] [Google Scholar]
- Czeizel A, Kovacs M. A family study of congenital diaphragmatic defects. Am J Med Genet. 1985;21:105–117. doi: 10.1002/ajmg.1320210115. [DOI] [PubMed] [Google Scholar]
- Holder AM, Klaassens M, Tibboel D, de Klein A, Lee B, Scott DA. Genetic factors in congenital diaphragmatic hernia. Am J Hum Genet. 2007;80:825–845. doi: 10.1086/513442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornstra IK, Birge S, Starcher B, Bailey AJ, Mecham RP, Shapiro SD. Lysyl oxidase is required for vascular and diaphragmatic development in mice. J Biol Chem. 2003;278:14387–14393. doi: 10.1074/jbc.M210144200. [DOI] [PubMed] [Google Scholar]
- Huff CD, Witherspoon DJ, Simonson TS, Xing J, Watkins WS, Zhang Y, Tuohy TM, Neklason DW, Burt RW, Guthery SL, Woodward SR, Jorde LB. Maximum-likelihood estimation of recent shared ancestry (ERSA) Genome Res. 2011;21:768–774. doi: 10.1101/gr.115972.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay PY, Bielinska M, Erlich JM, Mannisto S, Pu WT, Heikinheimo M, Wilson DB. Impaired mesenchymal cell function in Gata4 mutant mice leads to diaphragmatic hernias and primary lung defects. Dev Biol. 2007;301:602–614. doi: 10.1016/j.ydbio.2006.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Orr A, Guernsey DL, Robitaille J, Asselin G, Samuels ME, Dube MP. Application of homozygosity haplotype analysis to genetic mapping with high-density SNP genotype data. PLoS One. 2009;4:e5280. doi: 10.1371/journal.pone.0005280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerber RA. Method for calculating risk associated with family history of a disease. Genet Epidemiol. 1995;12:291–301. doi: 10.1002/gepi.1370120306. [DOI] [PubMed] [Google Scholar]
- Klaassens M, de Klein A, Tibboel D. The etiology of congenital diaphragmatic hernia: still largely unknown? Eur J Med Genet. 2009;52:281–286. doi: 10.1016/j.ejmg.2009.05.005. [DOI] [PubMed] [Google Scholar]
- Klaassens M, Galjaard RJ, Scott DA, Bruggenwirth HT, van Opstal D, Fox MV, Higgins RR, Cohen-Overbeek TE, Schoonderwaldt EM, Lee B, Tibboel D, de Klein A. Prenatal detection and outcome of congenital diaphragmatic hernia (CDH) associated with deletion of chromosome 15q26: two patients and review of the literature. Am J Med Genet A. 2007;143A:2204–2212. doi: 10.1002/ajmg.a.31892. [DOI] [PubMed] [Google Scholar]
- Klaassens M, van Dooren M, Eussen HJ, Douben H, den Dekker AT, Lee C, Donahoe PK, Galjaard RJ, Goemaere N, de Krijger RR, Wouters C, Wauters J, Oostra BA, Tibboel D, de Klein A. Congenital diaphragmatic hernia and chromosome 15q26: determination of a candidate region by use of fluorescent in situ hybridization and array-based comparative genomic hybridization. Am J Hum Genet. 2005;76:877–882. doi: 10.1086/429842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J, Housman D, Jaenisch R. WT-1 is required for early kidney development. Cell. 1993;74:679–691. doi: 10.1016/0092-8674(93)90515-r. [DOI] [PubMed] [Google Scholar]
- Mah VK, Zamakhshary M, Mah DY, Cameron B, Bass J, Bohn D, Scott L, Himidan S, Walker M, Kim PC. Absolute vs relative improvements in congenital diaphragmatic hernia survival: what happened to “hidden mortality”. J Pediatr Surg. 2009;44:877–882. doi: 10.1016/j.jpedsurg.2009.01.046. [DOI] [PubMed] [Google Scholar]
- Neklason DW, Stevens J, Boucher KM, Kerber RA, Matsunami N, Barlow J, Mineau G, Leppert MF, Burt RW. American founder mutation for attenuated familial adenomatous polyposis. Clin Gastroenterol Hepatol. 2008;6:46–52. doi: 10.1016/j.cgh.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norio R, Kaariainen H, Rapola J, Herva R, Kekomaki M. Familial congenital diaphragmatic defects: aspects of etiology, prenatal diagnosis, and treatment. Am J Med Genet. 1984;17:471–483. doi: 10.1002/ajmg.1320170210. [DOI] [PubMed] [Google Scholar]
- Pei J, Grishin NV. Unexpected diversity in Shisa-like proteins suggests the importance of their roles as transmembrane adaptors. Cell Signal. 2012;24:758–769. doi: 10.1016/j.cellsig.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pober BR. Genetic aspects of human congenital diaphragmatic hernia. Clin Genet. 2008;74:1–15. doi: 10.1111/j.1399-0004.2008.01031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pober BR, Lin A, Russell M, Ackerman KG, Chakravorty S, Strauss B, Westgate MN, Wilson J, Donahoe PK, Holmes LB. Infants with Bochdalek diaphragmatic hernia: sibling precurrence and monozygotic twin discordance in a hospital-based malformation surveillance program. Am J Med Genet A. 2005;138A:81–88. doi: 10.1002/ajmg.a.30904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid F, Chaparala R, Ahmed J, Iftikhar SY. Atypical right diaphragmatic hernia (hernia of Morgagni), spigelian hernia and epigastric hernia in a patient with Williams syndrome: a case report. J Med Case Reports. 2009;3:7. doi: 10.1186/1752-1947-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DA, Klaassens M, Holder AM, Lally KP, Fernandes CJ, Galjaard RJ, Tibboel D, de Klein A, Lee B. Genome-wide oligonucleotide-based array comparative genome hybridization analysis of non-isolated congenital diaphragmatic hernia. Hum Mol Genet. 2007;16:424–430. doi: 10.1093/hmg/ddl475. [DOI] [PubMed] [Google Scholar]
- Slattery ML, Kerber RA. A comprehensive evaluation of family history and breast cancer risk. The Utah Population Database. JAMA. 1993;270:1563–1568. [PubMed] [Google Scholar]
- Slavotinek AM, Moshrefi A, Davis R, Leeth E, Schaeffer GB, Burchard GE, Shaw GM, James B, Ptacek L, Pennacchio LA. Array comparative genomic hybridization in patients with congenital diaphragmatic hernia: mapping of four CDH-critical regions and sequencing of candidate genes at 15q26.1–15q26.2. Eur J Hum Genet. 2006;14:999–1008. doi: 10.1038/sj.ejhg.5201652. [DOI] [PubMed] [Google Scholar]
- Srisupundit K, Brady PD, Devriendt K, Fryns JP, Cruz-Martinez R, Gratacos E, Deprest JA, Vermeesch JR. Targeted array comparative genomic hybridisation (array CGH) identifies genomic imbalances associated with isolated congenital diaphragmatic hernia (CDH) Prenat Diagn. 2010;30:1198–1206. doi: 10.1002/pd.2651. [DOI] [PubMed] [Google Scholar]
- Thomas A, Camp NJ, Farnham JM, Allen-Brady K, Cannon-Albright LA. Shared genomic segment analysis. Mapping disease predisposition genes in extended pedigrees using SNP genotype assays. Ann Hum Genet. 2008;72:279–287. doi: 10.1111/j.1469-1809.2007.00406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wat MJ, Beck TF, Hernandez-Garcia A, Yu Z, Veenma D, Garcia M, Holder AM, Wat JJ, Chen Y, Mohila CA, Lally KP, Dickinson M, Tibboel D, de Klein A, Lee B, Scott DA. Hum Mol Genet. 2012. Mouse model reveals the role of SOX7 in the development of congenital diaphragmatic hernia associated with recurrent deletions of 8p23.1. Epub Jul 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wat MJ, Shchelochkov OA, Holder AM, Breman AM, Dagli A, Bacino C, Scaglia F, Zori RT, Cheung SW, Scott DA, Kang SH. Chromosome 8p23.1 deletions as a cause of complex congenital heart defects and diaphragmatic hernia. Am J Med Genet A. 2009;149A:1661–1677. doi: 10.1002/ajmg.a.32896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wat MJ, Veenma D, Hogue J, Holder AM, Yu Z, Wat JJ, Hanchard N, Shchelochkov OA, Fernandes CJ, Johnson A, Lally KP, Slavotinek A, Danhaive O, Schaible T, Cheung SW, Rauen KA, Tonk VS, Tibboel D, de Klein A, Scott DA. Genomic alterations that contribute to the development of isolated and non-isolated congenital diaphragmatic hernia. J Med Genet. 2011;48:299–307. doi: 10.1136/jmg.2011.089680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You LR, Takamoto N, Yu CT, Tanaka T, Kodama T, Demayo FJ, Tsai SY, Tsai MJ. Mouse lacking COUP-TFII as an animal model of Bochdalek-type congenital diaphragmatic hernia. Proc Natl Acad Sci U S A. 2005;102:16351–16356. doi: 10.1073/pnas.0507832102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.