

Abstract
Ischemic heart disease (IHD) is a significant cause of morbidity and mortality in Western society. Although interventions such as thrombolysis and percutaneous coronary intervention (PCI) have proven efficacious in ischemia and reperfusion (IR) injury, the underlying pathologic process of IHD, laboratory studies suggest further protection is possible, and an expansive research effort is aimed at bringing new therapeutic options to the clinic.
Mitochondrial dysfunction plays a key role in the pathogenesis of IR injury and cardiomyopathy (CM). However, despite promising mitochondria-targeted drugs emerging from the lab, very few have successfully completed clinical trials. As such, the mitochondrion is a potential untapped target for new IHD and CM therapies. Notably, there are a number of overlapping therapies for both these diseases, and as such novel therapeutic options for one condition may find use in the other. This review summarizes efforts to date in targeting mitochondria for IHD and CM therapy, and outlines emerging drug targets in this field.
Keywords: ischemia, reperfusion, therapeutics, bioenergetics, pt pore, clinical trial
1. Introduction
Ischemic heart disease (IHD) is a significant cause of morbidity and mortality in Western society.1 Although interventions such as thrombolysis and percutaneous coronary intervention (PCI) have proven efficacious in ischemia and reperfusion (IR) injury, the underlying pathologic process of IHD, laboratory studies suggest further protection is possible, and an expansive research effort is aimed at bringing new therapeutic options to the clinic.
Mitochondrial dysfunction plays a key role in the pathogenesis of IR injury and cardiomyopathy (CM).2,3 However, despite promising mitochondria-targeted drugs emerging from the lab, very few have successfully completed clinical trials. As such, the mitochondrion is a potential untapped target for new IHD and CM therapies. Notably, there are a number of overlapping therapies for both these diseases, and as such novel therapeutic options for one condition may find use in the other. This review summarizes efforts to date in targeting mitochondria for IHD and CM therapy, and outlines emerging drug targets in this field.*
1.1 Mitochondrial energetics in normal cardiac function
The main function of cardiac mitochondria is ATP synthesis via oxidative phosphorylation (Ox-Phos). Following substrate oxidation, reducing equivalents generated by the TCA cycle are utilized by the respiratory chain (RC) to generate a trans-membrane potential (ΔΨm) which drives ATP synthesis. Under normal conditions the adult heart relies mostly on fatty acids to fuel Ox-Phos, with 10–30% of total ATP derived from glucose.4 Substrate utilization does vary under normal conditions, with increased aerobic glycolysis found postprandially and during exercise, and in relation to substrate availability and circadian rhythm.5–8 In contrast, the immature heart relies predominately on glucose or lactate to provide carbon to the TCA cycle, with a transition to the mature fat-burning phenotype around 1 week of age.9
1.2 Mitochondrial dysfunction in cardiomyopathy
The major cause of acute CM, the precursor to heart failure, is myocardial ischemia (see section 1.3), while less acute forms of CM are idiopathic, inherited, or caused by chronic diseases such as hypertension and diabetes. Regardless of the cause, altered mitochondrial bioenergetics appear to play a substantial role in CM.10
Under pathologic conditions such as CM, the normal metabolic flexibility of the heart is superseded by activation of a fetal metabolic program, with a preference for glucose over fat as the substrate for Ox-Phos.11 Although such changes lower the O2 consumed per ATP produced, the yield of ATP per substrate also decreases. Such inefficient metabolism lowers ATP and phosphocreatine (PCr) levels, and decreases metabolic reserve and flexibility.5,6,8 Interestingly, adult hearts which adopt this neonatal metabolism are refractory to protection by ischemic preconditioning (IPC, see section 3)12 like the neonatal heart.13,14 However, IPC itself comprises metabolic remodeling, suggesting that altered metabolism may be an adaptive response to ischemia, becoming pathologic if the stimulus persists.
Metabolic alterations are also an important component of inherited CMs.6,15 Hypertrophic CM (the most common form with a prevalence of about 0.2% in the US population) is generally caused by contractile protein mutations which raise the energetic costs of contraction, leading to secondary energetic failure. However, around 27% of hypertrophic CM cases are caused by inborn errors of metabolism, leading to primary energetic failure.16 In addition, about 7% of dilated CM is caused by mutations in metabolism genes. Primary mitochondrial genetic disorders are also the most common etiology in non-compaction CM,17 and two recent reports suggest restrictive CM and arrhythmogenic right ventricular dysplasia may be plagued by metabolic abnormalities.18,19
1.3 Mitochondrial dysfunction in ischemic heart disease
The primary pathologic event in IHD is acute myocardial infarction (AMI), caused by coronary artery obstruction. While permanent occlusion often results in cardiac remodeling and hypertrophy, transient occlusion is also detrimental to cardiac function, and the reperfusion phase of IR injury is particularly injurious to mitochondria.
A wide spectrum of mitochondrial derangements occurs in the post-IR heart, including: (i) Inhibition of respiratory complexes20 and the adenine nucleotide translocase (ANT),21 (ii) Increased proton leak of the inner membrane,22 (iii) Oxidation of the phospholipid cardiolipin and associated membrane protein dysfunction,23 (iv) Excessive generation of reactive oxygen species (ROS),24 (v) Opening of the permeability transition (PT) pore, and release of cell-death inducing proteins including cytochrome c,25 (vi) Catastrophic nucleotide depletion,26 (vii) Mitochondrial Ca2+ overload.27 These phenomena are summarized in Figure 1, and while a brief understanding is helpful to understand the mechanisms of protection by mitochondrial therapeutics, in-depth discussions are available elsewhere.28
Figure 1. Mitochondrial pathologic events in IR injury.

During ischemia, lack of O2 inhibits Ox-Phos, diverting the glycolytic end product pyruvate to lactate, resulting in cellular acidification. Disposal of acid via the Na+/H+ exchanger (NHE) brings Na+ into the cytosol. This Na+ is exported via the Na+/Ca2+ exchanger (NCX) causing cytosolic Ca2+ overload, which is exacerbated by low ATP disabling Ca2+ export by ATPases. Inhibited Ox-Phos is the primary cause of low ATP. Mitochondrial Ca2+ uptake is minimal due to low ΔΨm, and both acidic pH and accumulation of NADH keeps the PT pore closed. At reperfusion, rapid re-establishment of Ox-Phos and ΔΨm results in mitochondrial Ca2+ overload and ROS generation. Together with normalization of pH, this triggers PT pore opening, leading to cell death. In surviving cells, ROS activates mitochondrial uncoupling (H+ leak), which inhibits ATP synthesis, exacerbating the metabolic crisis. Oxidation of cardiolipin (CL) also occurs, along with nucleotide loss (both NADH from mitochondria and ATP from the cell). ROS damage to ANT contributes to low ATP levels. For full explanation see text.(Illustration Credit: Ben Smith).
2. Current mitochondrial treatments for cardiomyopathy
Several studies have attempted to treat heart failure associated with CM by targeting bioenergetic dysfunction. The general strategy for primary mitochondrial genetic disorders is supplementation with cofactors such as co-enzyme Q (Co-Q), carnitine, riboflavin, and thiamine, or antioxidants such as ascorbate. The efficacy of such treatments for CM is unclear,16,17,29 since most mitochondrial disease trials have focused on neurologic complications of these disorders rather than CM. Several groups have tested the Co-Q analog idebenone, and in some studies this molecule slowed or prevented progression of hypertrophic CM in patients with Friedreich’s ataxia (a disease of iron-sulfur cluster assembly that affects the RC), however a more recent trial found no improvement in left ventricle (LV) mass or function.29,30
Notably, even in CMs of unknown or non-mitochondrial etiology, the mainstay clinical agents are Co-Q and carnitine,31 particularly if carnitine deficiency is evident upon blood testing. Idebenone has also been used for non-mitochondrial CM, and it improved cardiac function in a mouse model of Duchenne muscular dystrophy (DMD).32 While a subsequent phase II trial in DMD patients failed to show significant improvement in cardiac function, a promising trend was seen and a larger trial is underway.33
Another potential therapy for mitochondrial disorders is dichloroacetate (DCA), which stimulates pyruvate dehydrogenase (PDH) to shunt pyruvate into Ox-Phos. DCA has yielded mixed results in treating neurologic symptoms of mitochondrial disorders, leading to suggestions that it may only be useful in patients with PDH deficiency.29,34 The ability of DCA to treat mitochondrial CM has not been tested, but in patients with a mixture of non-ischemic and ischemic dilated CM, DCA showed mixed effects on myocardial O2 consumption and mechanical efficiency.35,36
The amino acid L-arginine has also been proposed to treat mitochondrial disease, and in patients with mitochondrial CM, arginine increased aerobic metabolism and myocardial efficiency. The mechanism appeared to be independent of myocardial blood flow (i.e. arginine as a substrate for NO• production), possibly ocurring via supplying the TCA cycle with 2-oxyglutarate.37 Another drug of interest is perhexiline, which significantly increased PCr/ATP ratio, corrected diastolic dysfunction, and increased exercise capacity in CM patients, by inhibiting fatty acid oxidation (FAO) and increasing glucose utilization.38 Other potential metabolic modulators, as well as non-pharmacologic therapies such as exercise training, may prove efficacious in treating CM.5,8 Pharmacologic options are discussed further in section 4.8.
3 Current mitochondrial treatments for ischemic heart disease
Many therapies are aimed at slowing the progression of IHD, including statins to slow atherosclerotic lesion formation, anti-anginals to improve flow to transiently ischemic tissue, and anti-platelet agents to impede clotting at plaque rupture. While these agents do impact the incidence of ischemic events, there are limited therapies to protect myocardial function once an ischemic insult occurs.
In some situations cardiac ischemia can be anticipated in advance, such as during cardiac surgery or balloon inflation during PCI. Careful quantification of cardiac damage using biomarkers can allow clinical trials to identify protective strategies. As such, in cardiac surgery, cardioplegia (controlled arrest of the heart, dropping myocardial energy requirements),39 hypothermia,40 beta blockers,41 and tight glycemic control42 are all currently used for cardioprotection.
Recently, protecting the heart with ischemic preconditioning (IPC) has begun to translate into the clinic.43 In this method, short intermittent IR cycles are administered prior to index ischemia, yielding reduced infarct size and improved post-ischemic function. IPC has completed successful clinical trials in both PCI and cardiac surgery, however it is usually accomplished by repeatedly cross-clamping the aorta, which may increase the risk of cerebral emboli or aortic vascular damage.44 In this regard, it is notable that transient occlusion at sites remote from the heart (remote ischemic preconditioning, RIPC) is also cardioprotective,45 with clinical studies showing RIPC efficacy in cardiac and non-cardiac surgery46,47 and PCI.48 However, failure of other RIPC trials suggests protocol optimization may be required.49 As shown in Figure 2, the mechanism of IPC protection is incredibly complex, with mitochondria (and particularly mitochondrial K+ channels, see section 4.3) playing a key role.50,51
Figure 2. Cardioprotective signaling: all roads lead to mitochondria.
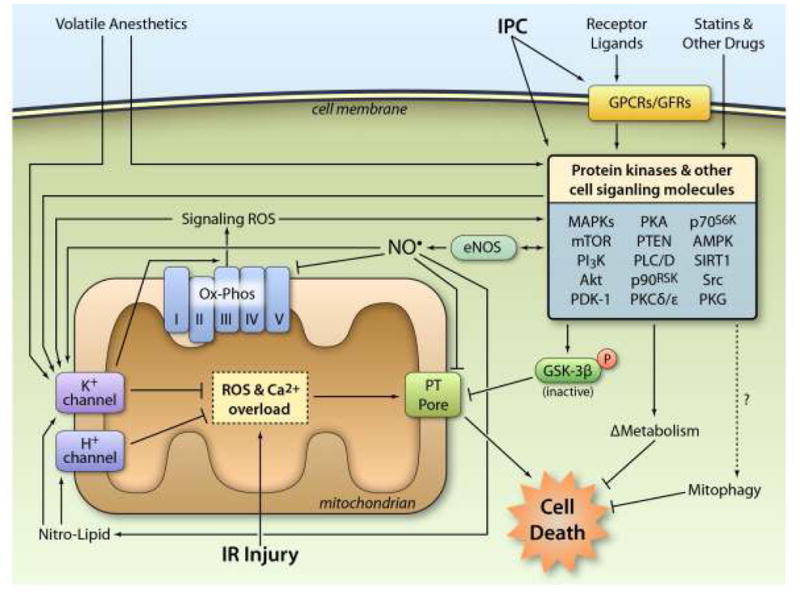
IPC either directly or indirectly (via G-protein coupled receptors or growth factor receptors) activates numerous protein kinases and other cell signaling molecules, including the generation of ROS for signaling purposes. These signals filter through a limited number of downstream mediators including: (i) eNOS generation of NO•, (ii) phosphorylation and inhibition of GSK-3β, (iii) changes in metabolism (iv) activation of mitophagy. At the mitochondrial level, many of these signals converge on the opening of mKATP and mBK channels, and the activation of H+ channels, both of which inhibit the PT pore directly, or inhibit the events leading up to its opening. Volatile anesthetics also trigger mitochondrial K+ channel opening and recruit many of the same kinase signals as IPC. Other drugs such as statins can also activate kinases in the RISK pathway. Together, these events inhibit cell death during subsequent IR injury. See text for more details. (Illustration Credit: Ben Smith).
Related to IPC, and more clinically tractable, is volatile anesthetic preconditioning (APC), in which halogenated anesthetics (iso-/des-/sevo-flurane) are known to activate many of the same intracellular signaling events as IPC. 52,53 APC is also thought to open mitochondrial K+ channels (see section 4.3).
Unfortunately, the unanticipated nature of AMI makes it difficult to offer protection by preconditioning, and the current standard of care is early reperfusion via PCI or thrombolytics. Luckily it appears that intermittent reperfusion (ischemic postconditioning, IPoC) can confer some of the protective benefits of IPC.54 Clinical trials have confirmed that IPoC during PCI can decrease infarct size and improve post MI heart failure.55 Remote ischemic postconditioning (RIPoC) has also shown success in clinical trials.56 The mechanism of IPoC is thought to involve opening of mitochondrial K+ channels57 and inhibition of the mitochondrial PT pore.58
Targeting metabolism is another strategy to protect the heart in AMI. Simply reducing cardiac mitochondrial workload with β-blockers has proven effective in reducing mortality after AMI59 and coronary artery bypass graft (CABG) surgery,60 although how well this extends to non-cardiac surgery patients with IHD is debated.41,61 Another metabolic therapy is adenosine, which reduces infarct size when delivered at reperfusion in animal models.62 Early adenosine clinical trials were successful,63 but later trials (AMISTAD) showed infarct reduction only in patient subsets, with a trend toward more adverse events and no improvement in hospital outcomes.64 A large trial (AMISTAD-II) in patients undergoing PCI reported a 60% drop in infarct size, but no change in outcomes at 6 month follow-up.65 Further trials using adenosine and analogs, such as AMP579, were disappointing, and meta-analysis revealed no change in overall outcomes.66,67
Somewhat related to adenosine is acadesine (AICA riboside), an ATP metabolite which was originally thought to replenish post-IR adenine nucleotides. Although acadesine failed to elevate cellular ATP,68 it still elicited cardioprotection,69 leading to suggestions that its cardioprotective effects may result from activation of AMP dependent protein kinase (AMPK), an important regulator of metabolism (see section 4.8).70 Further study showed that acadesine increased extracellular adenosine levels and decreased myocardial stunning,71,72 leading to successful clinical trials in CABG73 and a large phase II trial (Acadesine1024), which yielded lower 2 year mortality in patients with perioperative MI.74 However, a recent multi-center phase III trial (NCT00872001†) was terminated early (Merck Inc. 2010 2nd quarter SEC filing) due to futility.
Another class of drugs that have largely failed to alleviate IR injury in the clinic, are antioxidants. Early reports on the involvement of ROS in IR pathogenesis75 (Figure 1) were shortly followed by animal studies demonstrating exogenous superoxide dismutase (SOD) and/or catalase could elicit cardioprotection.76 However, these reagents failed in large animal77 and human trials.78 Consistent with xanthine oxidase (XO) as a source of ROS in IR injury,79 the XO inhibitor allopurinol was found to be cardioprotective in animal models,80 but also failed in clinical trials. Other small molecule antioxidants (including Co-Q,81 mercaptopropionyl glycine (MPG)82 and others83) have also been tested in IR injury, but all yielded mixed results in large animal or human studies.84,85 Despite this, antioxidants remain key ingredients in cardioplegia.
Overall, numerous drugs have shown promise in the lab for protecting the heart in IHD via mitochondrial mechanisms, but none have passed clinical trials. The reasons for these failures have been discussed extensively elsewhere86,87, and may include unforeseen drug interactions, the inappropriate nature of animal models, confounding risk factors such as diabetes and aging, or poor drug delivery to ischemic tissue. Furthermore, many cardioprotective drugs may actually block endogenous cardioprotective signals – for example IPC requires ROS, such that antioxidants block IPC.88
4 Emerging mitochondrial therapeutic targets
Despite the wave of failed CM and IHD candidate therapeutics to date, the mitochondrial research community has made significant strides in deciphering mechanisms of cardioprotection (Figure 2), resulting in development of more specific and targeted molecules. Some of these newer candidates are discussed below, categorized by the mitochondrial phenomena which they target.
4.1 Mitochondrial permeability transition pore
Opening of the mitochondrial PT pore89 is a major driver of necrotic cell death in IR injury. The identity of the molecules which assemble to form the PT pore remains somewhat controversial, however transgenic animal models have demonstrated pore regulation by the ANT, the phosphate carrier,90 and cyclophilin D (CypD),91,92 with the role of the voltage dependent anion channel (VDAC) less clear.93 Other proteins implicated in the PT pore or its regulation include hexokinase II, which links the pore to cellular metabolism,94,95 and mitochondrial translocator protein (TSPO) which interacts with VDAC.96 Drugs targeting these PT pore components are candidates for cardioprotective therapeutics.
The first drug shown to inhibit PT pore opening was cyclosporine A (CsA),97 which binds to CypD. CsA protects against IR injury in myocytes98 and intact hearts,99 and a pilot clinical trial demonstrated 20% infarct reduction with CsA administration prior to PCI in MI patients,100 with benefits still evident 6 months later.101 A phase III trial (NCT01502774) is currently underway (NCT01502774). Notably, CsA is also used for immuno-suppression after organ transplant, mediated by its binding to calcineurin. Calcineurin is also a well-known hypertrophic signaling molecule, and hypertrophy due to CsA usage has been reported in transplant patients.102,103 Thus, CypD-specific CsA analogs have been developed, including sanglifehrin A,104 NIM811,105 and Debio025.106 These molecules are cardioprotective in animal models of IR injury, although none has yet progressed to clinical trials.‡ A mitochondria-targeted CsA has also been developed, exhibiting higher potency and improved CypD affinity.107 The TSPO ligands SSR180575, 4′-chlorodiazepam (Ro5-4684), and TRO40303 were also shown to reduce IR injury in various cell and animal models,108–110 and TRO40303 is entering clinical trials (NCT01374321).
4.2 Nitric oxide based mitochondrial therapies
The role of NO• in both endogenous cardioprotection and in mediating protective effects of drugs, has been reviewed extensively elsewhere.111,112 In addition to classical NO• signaling via cGMP/PKG, which feeds into RISK signaling (see section 4.6), non-classical functions of NO• are also implicated in cardioprotection, including protein S-nitrosation113,114 and the generation of nitro-lipids.115,116 Mitochondria are a critical site of action for these signals, in particular the S-nitrosation of respiratory complex I resulting in reversible inhibition.117 Furthermore cyclophilin D has been identified as a target of S-nitrosation leading to attenuation of PT pore opening.118
A series of molecules have been developed to deliver NO• to mitochondria. The first such agent was SNO-MPG, derived from the antioxidant MPG.113 SNO-MPG was actively sequestered into mitochondria, and exhibited cardioprotection in animal models of IR injury.119 Protection was associated with complex I S-nitrosation, and was lost in a complex I mutant mouse. A more recent development is MitoSNO1, an NO• donor based on the triphenylphosphonium (TPP+) mitochondrial targeting moiety, which exhibits cardioprotection when delivered at reperfusion.120 A related molecule is 2-hydroxylamine-vinyl-TPP+ (HVTP), which requires metabolism by intra-mitochondrial peroxidases to release NO•,121 although its efficacy in IR injury is not yet known.
Another set of NO•-derived molecules that target mitochondria are the nitro-lipids. These molecules are generated endogenously inside mitochondria during IPC, and when added exogenously they protect in various models of IR injury.115,116 Such protection occurred on a timescale too fast for gene-regulatory effects usually attributed to nitro-lipids (e.g., PPARγ, NFκB, Nrf2/Keap1).122 Instead, nitro-lipid protection occurred via covalent modification of ANT, eliciting a mild increase in mitochondrial uncoupling,123 which is known to confer cardioprotection.124 Nitro-lipid derivatives are currently in early stage clinical development.
Finally, significant attention has recently focused on the anti-ischemic properties of nitrite (NO2−).125,126 Several studies have implicated mitochondria, and in particular complex I, as a downstream target of nitrite in cardoprotection.126,127 Injectable sodium nitrite is FDA approved as a cyanide poisoning antidote, and several nitrite clinical trials are underway for cardiac ischemia (NCT01401517, NCT00924118, NCT01098409).
4.3 Mitochondrial potassium channels
The mitochondrial inner membrane contains numerous ion channels,128 and many studies have suggested a role for mitochondrial K+ channels in cardioprotection. The mechanism of such protection is unclear, and may involve mild uncoupling, diminishing the ΔΨm driving force for Ca2+ overload and ROS generation (see section 1.3). Mild swelling associated with mitochondrial K+ influx may also regulate the PT pore.52
A mitochondrial KATP channel (mKATP) is reported to play a critical role in cardioprotection by IPC. 129,130 One of the most widely tested mKATP openers is diazoxide, which has demonstrated protection from IR injury in multiple cell and animal models. Two small clinical trials in CABG patients reported cardioprotection in patients pretreated with diazoxide or undergoing surgery supported by diazoxide-supplemented cardioplegia.131,132 Other mKATP openers including BMS-191095, pinacidil, cromakalim, minoxidil, and nicorandil§ are cardioprotective in animal models.134–137
Although the molecular identity of mKATP is unclear, a link exists between respiratory complex II and mKATP, such that mKATP openers inhibit complex II, and complex II inhibitors open mKATP.138–140 It is hoped that definitive identification of this channel will aid in the design of mKATP specific drugs. Notably, many pharmaceuticals including fluoxetine and some sulfonylureas inhibit mKATP, abrogating cardioprotection by IPC.140,141
In addition to mKATP, mitochondria are also thought to contain a large conductance K+ channel (“BK”) encoded by the SLO gene family,142 which is implicated in cardioprotection by anesthetic preconditioning (APC).143 Until recently, consensus held that the mitochondrial BK channel (mBK) was the SLO1 isoform, based on cardioprotection by the SLO1 opener NS1619 in animal models of IR injury.144 However, NS1619 acts on several ion channels145,146 and other mitochondrial targets.147 A more specific SLO1 activator (NS11021) is also cardioprotective,143 but controversy remains as it was recently shown in C. elegans and mice that SLO1 is dispensable for APC, and SLO2 is necessary for APC in the worm.139 There are currently no SLO2 specific activators, but SLO2 may be a future therapeutic target.
4.4 Mitochondrial antioxidants
As discussed in section 3, multiple generic non-targeted antioxidants have been clinically unsuccessful in preventing IR injury, although several newer antioxidants are in clinical trials, including melatonin (NCT01172171), mangafodipir (NCT00966563), and edaravone (NCT00265239). In addition, a potentially novel therapeutic approach is the targeting of antioxidants to mitochondria.
A key chemical strategy for mitochondrial targeting is the TPP+ moiety (see section 4.2). First exploited in the 1970s as a method to measure ΔΨm,148 several TPP+-conjugated antioxidants were developed the 1990s. The most prominent of these, MitoQ, was cardioprotective in a rat model of IR injury,149 and is being developed clinically for other indications (NCT00433108). Other antioxidants conjugated to TPP+ include α-tocopherol (Mito-E), and nitroxides.150 As discussed in section 4.2, a TPP+-conjugated NO• donor confers potent cardioprotection.
Somewhat related to TPP+ conjugates are the SS peptides, which contain positively charged amino acids and a dimethyl tyrosine (DMT) residue. Originally developed as opioid peptide analgesics, following the discovery they were cardioprotective151 their mechanism was assigned to a mitochondrial antioxidant activity.152 The lead compound in this series, SS-31 (D-Arg-DMT-Asn-Phe-NH2) has been renamed “Bendavia”, and is in clinical trials for AMI (NCT01572909).
Bioactivation is another strategy to target drugs to mitochondria, an example being the omega-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids (Imz-SNAs). These inactive pro-drugs are metabolized by fatty acid β-oxidation in the mitochondrial matrix to reveal a methimizole antioxidant.153 Imz-SNAs were shown to protect isolated cardiomyocytes from IR injury, in a manner inhibited by the β-oxidation blocker etomoxir. Such molecules may be particularly efficacious in the heart, given its preference for fatty acids as a metabolic substrate (see section 1).
4.5 Respiratory Chain inhibitors
Ischemia inhibits Ox-Phos due to lack of O2 as the terminal electron acceptor for the RC. Thus, it is somewhat paradoxical that many chemical RC inhibitors are protective in ischemia. This includes inhibitors of complex I (rotenone,154 amobarbital155, S-nitrosothiols, nitrite), complex II (diazoxide,131 atpenin A5140), complex III (antimycin A156), and complex IV (CO,157 H2S,158 NO•). The mechanism of protection may involve inhibition of the large burst of ROS generation and Ca2+ overload that occurs at reperfusion, with inhibitor washout allowing a more gradual re-introduction of electron flux to the RC, somewhat akin to IPoC or slow reperfusion.159 Although neurological side effects associated with RC inhibitors (e.g., Parkinson’s disease for rotenone, Huntington’s disease for 3-NP) may preclude their clinical applicability, amobarbital is clinically available, and is protective when delivered at reperfusion.155
Related to RC inhibitors, mild uncoupling of Ox-Phos by chemicals such as FCCP can also elicit cardioprotection. The mechanism underlying this is unclear, but may include inhibition of ROS generation and Ca2+ overload.124,160 As discussed in section 4.2, cardioprotection by nitro-lipids may also proceed via mild uncoupling.
4.6 RISK Pathway
In IR injury, the PT pore remains closed during ischemia and opens early in reperfusion (Figure 1).25 Opening of the pore at reperfusion is regulated by the “reperfusion injury salvage kinase” (RISK) signaling pathway.161 This pathway is implicated in IPC and IPoC,162 and involves numerous kinases which converge on phosphorylation and inhibition of glycogen synthase kinase-3β(GSK3-β),161 which is thought to phosphorylate PT pore components (Figure 2).163 The GSK3-β inhibitors SB-216763 and lithium improve post-IR cardiac function in animal models164 (although mKATP channels may also be involved165).
Many extracellular signals (e.g., insulin166) can activate RISK signaling via receptors, and NO• is also implicated in RISK signaling.167,168 Several drugs also exert cardioprotective effects by activating RISK, most notably statins (simvastatin,169 mevastatin,170 atorvastatin171) via a mechanism not linked to cholesterol lowering. Multiple clinical trials (ARYMDA, NAPLES) and a meta-analysis172 have demonstrated cardioprotection with statin administration before emergent PCI. Further clinical trials are ongoing to deliver statins at reperfusion for patients presenting with MI (NCT01050348, NCT01334671, NCT00772564). Many other drugs that activate RISK signaling are reviewed elsewhere.173,174
4.7 Aldehyde dehydrogenase 2
A relatively new target for mitochondrial protection in IR injury is ALDH2, a mitochondrial enzyme which removes toxic aldehydes such as 4-hydroxy-2-nonenal (HNE). HNE has been implicated in cardiac IR injury, and can inhibit metabolic enzymes and RC complexes, and open the PT pore.175
Overexpression of ALDH2 decreases HNE levels, yielding decreased infarct size and increased post ischemic function.176 Furthermore Alda-1, a small molecule ALDH2 activator, decreases infarct size in animal models of IR injury.176 Recently α-lipoic acid, a cofactor for ALDH2, has shown cardioprotection in an ALDH2 dependent fashion,177 and is currently in clinical trials for several conditions including diabetes (NCT00398892) and atherosclerosis (NCT00765310). In addition, ALDH2 activation may be capable of delivering cardioprotection in conditions where other drugs have failed, including diabetes and nitrate tolerance.178,179 The latter is a major clinical problem, as patients taking nitrates are often at risk for MI180.
4.8 Mitochondrial metabolism
One of the earliest methods to improve mitochondrial metabolism in IR was the use of glucose-insulin-potassium (GIK) supplementation. In addition to GIK, insulin alone can drive both glucose and fatty acids into the cell to support metabolism.181 GIK has shown mixed results in CABG surgery, with a recent meta-analysis showing no improvement in the incidence of arrhythmia or mortality.182 However, with recent emphasis on tight glucose control during cardiac surgery, many patients receive intraoperative GIK anyway.183 Results in AMI have been consistently negative, although a recent trial (NCT00091507) of GIK delivery by emergency personnel in the pre-hospital setting yielded a 50% drop in in-hospital cardiac arrest and mortality.184
FAO is also an attractive drug target in ischemia, since it uses more O2 per ATP produced (vs. glucose). During IR injury, catecholamine-induced increases in serum fatty acid levels may enhance FAO,185 and indeed reperfusion with fatty acid free serum is cardioprotective.186 Inhibitors of carnitine palmitoyltransferase (CPT1, which transports fatty acids into mitochondria) such as etomoxir have also demonstrated cardioprotection in animal models of IR injury,187 and improved hemodynamics in clinical trials for heart failure.188 Another CPT1 inhibitor, perhexiline, corrected diastolic dysfunction, and increased exercise capacity in patients with hypertrophic CM.38 Oxfenicine shows similar effects but is not used in humans.189
Other drugs that inhibit FAO are the anti-anginals trimetazidine and ranolazine.190 A meta-analysis of trimetazidine in heart failure demonstrated improved ventricular function and decreased mortality, cardiovascular events and hospitalizations.191 In IR injury it exhibited mixed results in both animal models192,193 and clinical trials, demonstrating protection when administered before CABG,194 but no protection in AMI except for a cohort of patients who did not receive thrombolysis.195 Ranolazine is reported to shift cell metabolism from FAO toward glycolysis,196 although may also have effects on cellular Na+ and Ca2+ homeostasis,197 or protection of complex I.198 In animal models of heart failure ranolazine reduced diastolic dysfunction and improved LV efficiency,199 and in IR injury it decreased both infarct size and cardiac enzyme release, and improved post-ischemic ventricular function.200 Clinical trials confirmed its efficacy in treating angina, but showed no protection in AMI.201 Further ranolazine trials are in development for PCI (NCT01491061). Despite the consensus that fatty acids are toxic to the ischemic heart, it is notable that carnitine supplementation is paradoxically cardioprotective in animal models of IR injury.202 In clinical trials carnitine prevented left ventricular remodeling after STEMI (CEDIM-I), and reduced 5 day mortality, however this mortality benefit was non-significant at 6 months (CEDIM-II).203,204
Global regulators of metabolism may also be therapeutic targets in IHD. An example is AMPK, which up-regulates both FAO and glucose utilization in response to decreased ATP levels. Whether increased AMPK is helpful or harmful to myocardium after IR injury is still unclear.205 The AMPK specific activator A-769662 is cardioprotective during IR injury, 206 but activators such as AICAR or metformin may have non-specific effects. Another important metabolic regulator is SIRT1, which is required for cardioprotection by acute IPC.207 Despite controversy regarding the SIRT1 activator resveratrol,208 SIRT1 activators are in clinical development (NCT00933530).
Similarly SIRT3, which is located in the mitochondria, has also emerged as an important metabolic regulator known to deacetylate numerous mitochondrial proteins involved in FAO and Ox-Phos.209 Notably a mitochondrial acetyltransferase (GCN5L1) that is counterpart to SIRT3 has recently been described, although from a pharmacologic standpoint, specific drugs that target mitochondrial SIRT3 (vs. other SIRTs) have not yet been developed.210 Finally, it has recently emerged that mitophagy, the process by which damaged mitochondria are removed and recycled, is an important player not only in metabolic homeostasis, but is also critical for IPC.211,212
5 Conclusions & Future directions
In summary, despite numerous failures in clinical development of drugs to treat IHD and CM, the pipelines of several companies still contain IHD and CM drugs, and many of these therapies appear targeted at mitochondria. Coupled with novel compounds which have not yet reached clinical trials, these are exciting times to be involved in drug development for these debilitating conditions. However, it should be emphasized that, despite the large number of mitochondrial drugs discussed in section 4, there is no guarantee that these molecule will be more efficacious in the clinic than those tested to date. Many of the same reasons that have been invoked to explain previous clinical failures also apply to these therapeutic candidates.87
In addition to the novel compounds discussed, other significant developments also deserve brief mention. The first is CAESAR, an NIH sponsored consortium which aims to bridge the gap between pre-clinical and clinical trials for promising cardioprotective therapies.213 By adopting standardized IR protocols in several animal models, and using practices common to clinical trials (double blinding, multiple centers, placebo controls), the consortium aims to put only the most promising therapies forward into human clinical trials. This will clearly be a valuable tool for researchers in the field.
Secondly, with a few exceptions, high-throughput screening (HTS) has been under-utilized as a drug development approach for IHD and CM. This may be because clinically relevant models of IR injury often use animals and are time consuming and expensive (i.e., not amenable to HTS), while HTS-ready IR models may not be physiologically relevant. In a recent screen for molecules that alter cell metabolism, it was hypothesized that hits which promote glycolysis would be beneficial during ischemia, and indeed one such molecule (meclizine) was protective in heart and neuronal models of IR injury.214 In another recent screen, we developed an IR injury model using a 24-well metabolic phenotyping device, with post-IR cell death and metabolism as endpoints. One hit from the screen was cloxyquin, which is closely related to the mitophagy-inducing drug clioquinol, currently in clinical trials for cancer (NCT00963495). Another was ipiriflavone, which was first proposed as an anti-ischemic drug 30 years ago.215 Further phenotypic screens of this type may yield novel cardioprotective drugs which engage the mitochondrial targets discussed herein.
Table 1.
Clinical development of mitochondrial therapies for cardiomyopathy and ischemic heart disease.
| Agent | Target/MOA | Clinical development/usage | Status of cardiac clinical development | Clinical trial | Ref’s |
|---|---|---|---|---|---|
| PT pore inhibitors | |||||
| 4′-chlorodiazepam (Ro5-4684) | TSPO | 109 | |||
| CsA | CypD | In-use (immunosupression) | Phase III (AMI) | NCT01502774 | 100 |
| Debio025 | CypD | Phase II (Hepatitis C) | 106 | ||
| NIM811 | CypD | Phase II (Hepatitis C) | 216 | ||
| Sanglifehrin A | CypD | 104 | |||
| TRO40303 | TSPO | Phase II (AMI) | NCT01374321 | 110 | |
|
| |||||
| NO analogs | |||||
| Mito-SNO1 | 120 | ||||
| Nitrolipids | 123 | ||||
| Nitrite | In-use (CN antidote) | Phase II (AMI) | NCT01584453 | 127 | |
| SNO-MPG | 119 | ||||
|
| |||||
| Antioxidants | |||||
| Edaravone | In-use (Stroke, Japan) | Phase IV (AMI) | NCT00265239 | ||
| Glutathione | Nutriceutical | Cardioplegia additive | 217 | ||
| HVTP | 121 | ||||
| Imz-SNAs | 153 | ||||
| Mangafodipir | Phase II (AMI) | NCT00265239 | |||
| Melatonin | Nutriceutical | Phase II (AMI) | NCT01172171 | ||
| MitoE | 150 | ||||
| MitoQ | Phase II (Hepatitis C, NASH) | 149 | |||
| MPG | 82 | ||||
| SOD, Catalase | Various clinical trials | 78 | |||
| SS31 (Bendavia) | Phase II (AMI) | NCT01572909 | |||
|
| |||||
| Potassium Channel Openers | |||||
| 3-NP | mKATP | 152 | |||
| Atpenin A5 | mKATP | 140 | |||
| Cromakalim | mKATP | 134 | |||
| BMS-191095 | mKATP | 135 | |||
| Diazoxide | mKATP | Various clinical trials | 132 | ||
| Malonate | mKATP | Phase II (Osteoporosis) | 139 | ||
| Minoxidil | mKATP | In-use (Hypertension, Alopecia) | 136 | ||
| NS11021 | mBK | 143 | |||
| NS1619 | mBK | 145 | |||
| Pinacidil | mKATP | In-use (Hypertension) | 134 | ||
|
| |||||
| Respiratory Chain Inhibitors | |||||
| Amobarbital | Complex I | In-use (Anxiety, sedation) | 155 | ||
| Antimycin A | Complex III | 156 | |||
| H2S | Complex IV | Phase I (Renal function) | 158 | ||
| Rotenone | Complex I | 154 | |||
|
| |||||
| RISK Pathway Modulators | |||||
| Lithium | In-use (Bipolar disorder) | 164 | |||
| SB216763 | GSK3-β | 165 | |||
| Statins | HMG-CoA reductase | In-use (Hyperlipidemia) | Completed phase IV (AMI) | ARYMDA and ARYMDA-ACS | 172 |
|
| |||||
| Aldehyde Dehydrogenase 2 | |||||
| α-lipoic acid | Nutriceutical | 177 | |||
| Alda1 | 176 | ||||
|
| |||||
| Metabolic modulators | |||||
| A-769662 | AMPK | 206 | |||
| Acadesine (AICAR) | AMPK, nonspecific | Failed phase III (MI) | NCT00872001 | 74 | |
| Co-factors (carnitine, Co-Q etc.) | Nutriceutical | Various clinical trials (CM) | |||
| DCA | Pyruvate dehydrogenase | Failed clinical trials for lactic acidosis, MELAS, Multiple trials in cancer. | 36 | ||
| Etomoxir | CPT1 | 188 | |||
| GIK | Multiple trials (CABG, non-cardiac surgery, AMI) | 182 | |||
| Idebenone | Respiratory chain | In-use (mito’ myopathies) | Phase III (DMD) | NCT01027884 | 30 |
| L-Arginine | NOS & TCA cycle substrate | In-use (mito’ myopathies) | Failed phase II (MI) | NCT00051376 | 37 |
| Oxfenicine | CPT1 | 189 | |||
| Perhexiline | CPT1 | Clinical (AMI, Aus/NZ) | 38 | ||
| Ranolazine | Late Na+ channel, specific target unclear | In-use (Angina) | Failed phase III (MI) Phase IV (PCI) |
NCT00099788 NCT01491061 |
200 |
| Trimetazidine | Late Na+ channel, specific target unclear | In-use (Angina) | Multiple clinical trials (CM) | 191 | |
|
| |||||
| Other | |||||
| Adenosine | Adenosine receptors | Failed clinical trials (MI, CABG) | 65 | ||
| AMP579 | Adenosine receptors | Failed phase III (MI) | ADMIRE I & II | 66 | |
| Anesthetic preconditioning | In-use | Phase IV (cardiac surgery) | NCT00364637 | 52,53 | |
| β-blockers | β-adrenergic receptors | In-use (patients at risk of MI) | 61 | ||
| Cloxyquin/clioquinol | In-use (anti-fungal/-protozoal) | 170 | |||
| Hypothermia | In use | Phase III (AMI) | COOL-MI NCT01379261 |
40 | |
| Meclizine | Histamine receptors | In-use (Antihistamine) | 214 | ||
| Preconditioning (IPC, IPoC, RIPC) | Multiple clinical trials | ||||
Abbreviations as per master list at start of paper
Acknowledgments
Sources of funding
PSB is funded by grants from the US National Institutes of Health (HL-071158, GM-087483). AMW is supported by a grant from the University of Rochester Clinical Translational Sciences Institute and the National Institutes of Health (5 TL1 RR 24135-5). GAP is supported by the American Heart Association and the Children’s Cardiomyopathy Foundation.
Abbreviations Used
*Note – many drug names contain proprietary abbreviations relating to company names (e.g. NS1619 from NeuroSearch A/S). Such abbreviations are not defined herein
- ALDH2
Aldehyde dehydrogenase isoform 2
- AMI
Acute myocardial infarction
- AMPK
Adenosine monophosphate (AMP) dependent protein kinase
- ANT
Adenine nucleotide translocase
- APC
Anesthetic preconditioning
- CABG
Coronary artery bypass graft
- CAESAR
NIH consortium for preclinicAl AssESsment of CARdioprotective therapies
- cGMP
Cyclic guanosine monophosphate
- Co-Q
C-enzyme Q10 (ubiquinone)
- CM
Cardiomyopathy
- CPT1
Carnitine palmitoyltransferase 1
- CsA
Cyclosporin A
- CypD
Cyclophilin D
- DCA
Dichloroacetate
- DMD
Duchenne muscular dystrophy
- ΔΨm
Mitochondrial membrane potential
- FAO
Fatty acid oxidation
- FCCP
Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone
- FDA
Federal drug administration
- GIK
Glucose-insulin-potassium
- GSK3-β
Glycogen synthase kinase-3β
- HTS
High throughput screening
- IHD
Ischemic heart disease
- Imz-SNA
omega-(1-methyl-1H-imidazol-2-ylthio)alkanoic acid
- IPC
Ischemic preconditioning
- IPoC
Ischemic postconditioning
- IR
Ischemia and reperfusion
- Keap-1
Kelch-like ECH-associated protein 1
- LV
Left ventricle
- MELAS
Mitochondrial encephalopathy lactic acidosis and stroke-like episodes
- MI
Myocardial infarction
- MitoQ
Mitochondrially targeted co-enzyme Q
- mKATP
Mitochondrial ATP sensitive K+ channel
- MOA
Mechanism of action
- MPG
Mercaptopropionyl glycine
- NASH
Non alcoholic steatohepatitis
- NCT #
National clinical trials registry #
- NFκB
Nuclear factor κB
- NOS
Nitric oxide synthase
- 3-NP
3-nitropropionic acid
- Nrf2
Nuclear factor erythroid 2-related factor 2
- Ox-Phos
Oxidative phosphorylation
- PCI
Percutaneous coronary intervention
- PDH
Pyruvate dehydrogenase
- PCr
Phosphocreatine
- PKG
cGMP dependent protein kinase
- PPARγ
Peroxisome proliferator activated receptor isoform gamma
- PT
Permeability transition
- RC
Respiratory chain
- RISK
Reperfusion injury salvage kinase
- ROS
Reactive oxygen species
- RIPC
Remote ischemic preconditioning
- SIRT
Silent information regulator two P homolog
- SNO-MPG
S-nitroso MPG
- SS
Szeto Schiller
- STEMI
ST segment elevation myocardial infarction
- TCA
Tricarboxylic acid
- TPP+
Triphenylphosphonium
- TSPO
mitochondrial translocator protein
- VDAC
Voltage dependent anion channel
- XO
Xanthine oxidase
Footnotes
Note: The entire field of drug development for IHD and CM is beyond the scope of this review. Several important therapeutics which have undergone clinical trials but are not necessarily linked to mitochondria, are listed in Online Supplemental Table 1.
NCT numbers in parentheses refer to national clinical trials registry (www.clinicaltrials.gov).
Debio025 (NCT00537407 and NCT00854802)and NIM811 (NCT00983060)are in clinical trials for hepatitis C.
Nicorandil failed in phase III clinical trials of acute MI (J-WIND).133
Disclosures
None
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart Disease and Stroke Statistics—2012 Update A Report From the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stanley WC, Hoppel CL. Mitochondrial Dysfunction in Heart Failure: Potential for Therapeutic Interventions? Cardiovasc Res. 2000;45:805–806. doi: 10.1016/s0008-6363(99)00419-8. [DOI] [PubMed] [Google Scholar]
- 3.Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia--reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001;33:1065–1089. doi: 10.1006/jmcc.2001.1378. [DOI] [PubMed] [Google Scholar]
- 4.Opie LH. Metabolism of the heart in health and disease. I. Am Heart J. 1968;76:685–698. doi: 10.1016/0002-8703(68)90168-3. [DOI] [PubMed] [Google Scholar]
- 5.Ashrafian H, Frenneaux MP, Opie LH. Metabolic mechanisms in heart failure. Circulation. 2007;116:434–48. doi: 10.1161/CIRCULATIONAHA.107.702795. [DOI] [PubMed] [Google Scholar]
- 6.Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res. 2009;81:412–9. doi: 10.1093/cvr/cvn301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosca MG, Hoppel CL. Mitochondria in heart failure. Cardiovasc Res. 2010;88:40–50. doi: 10.1093/cvr/cvq240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ardehali H, Sabbah HN, Burke MA, Sarma S, Liu PP, Cleland JG, Maggioni A, Fonarow GC, Abel ED, Campia U, Gheorghiade M. Targeting myocardial substrate metabolism in heart failure: potential for new therapies. Eur J Heart Fail. 2012;14:120–9. doi: 10.1093/eurjhf/hfr173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porter GA, Jr, Hom J, Hoffman D, Quintanilla R, de Mesy Bentley K, Sheu S-S. Bioenergetics, mitochondria, and cardiac myocyte differentiation. Prog Pediatr Cardiol. 2011;31:75–81. doi: 10.1016/j.ppedcard.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huss JM, Kelly DP. Mitochondrial energy metabolism in heart failure: a question of balance. J Clin Invest. 2005;115:547–555. doi: 10.1172/JCI200524405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lehman JJ, Kelly DP. Gene regulatory mechanisms governing energy metabolism during cardiac hypertrophic growth. Heart Fail Rev. 2002;7:175–85. doi: 10.1023/a:1015332726303. [DOI] [PubMed] [Google Scholar]
- 12.Opie LH, Sack MN. Metabolic plasticity and the promotion of cardiac protection in ischemia and ischemic preconditioning. J Mol Cell Cardiol. 2002;34:1077–1089. doi: 10.1006/jmcc.2002.2066. [DOI] [PubMed] [Google Scholar]
- 13.Awad WI, Shattock MJ, Chambers DJ. Ischemic preconditioning in immature myocardium. Circulation. 1998;98:II206–13. [PubMed] [Google Scholar]
- 14.Ostadalova I, Ostadal B, Kolar F, Parratt JR, Wilson S. Tolerance to ischaemia and ischaemic preconditioning in neonatal rat heart. J Mol Cell Cardiol. 1998;30:857–65. doi: 10.1006/jmcc.1998.0653. [DOI] [PubMed] [Google Scholar]
- 15.Watkins H, Ashrafian H, Redwood C. Inherited cardiomyopathies. N Engl J Med. 2011;364:1643–56. doi: 10.1056/NEJMra0902923. [DOI] [PubMed] [Google Scholar]
- 16.Cox GF. Diagnostic Approaches to Pediatric Cardiomyopathy of Metabolic Genetic Etiologies and Their Relation to Therapy. Prog Pediatr Cardiol. 2007;24:15–25. doi: 10.1016/j.ppedcard.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finsterer J. Cardiogenetics, neurogenetics, and pathogenetics of left ventricular hypertrabeculation/noncompaction. Pediatr Cardiol. 2009;30:659–81. doi: 10.1007/s00246-008-9359-0. [DOI] [PubMed] [Google Scholar]
- 18.Kant S, Krull P, Eisner S, Leube RE, Krusche CA. Histological and ultrastructural abnormalities in murine desmoglein 2-mutant hearts. Cell Tissue Res. 2012;384:249–259. doi: 10.1007/s00441-011-1322-3. [DOI] [PubMed] [Google Scholar]
- 19.Thebault C, Ollivier R, Leurent G, Marcorelles P, Langella B, Donal E. Mitochondriopathy: a rare aetiology of restrictive cardiomyopathy. Eur J Echocardiogr. 2008;9:840–5. doi: 10.1093/ejechocard/jen189. [DOI] [PubMed] [Google Scholar]
- 20.Rouslin W. Mitochondrial complexes I, II, III, IV, and V in myocardial ischemia and autolysis. Am J Physiol. 1983;244:H743–748. doi: 10.1152/ajpheart.1983.244.6.H743. [DOI] [PubMed] [Google Scholar]
- 21.Asimakis GK, Conti VR. Myocardial ischemia: correlation of mitochondrial adenine nucleotide and respiratory function. J Mol Cell Cardiol. 1984;16:439–447. doi: 10.1016/s0022-2828(84)80615-x. [DOI] [PubMed] [Google Scholar]
- 22.Borutaite V, Mildaziene V, Brown GC, Brand MD. Control and kinetic analysis of ischemia-damaged heart mitochondria: which parts of the oxidative phosphorylation system are affected by ischemia? Biochim Biophys Acta. 1995;1272:154–158. doi: 10.1016/0925-4439(95)00080-1. [DOI] [PubMed] [Google Scholar]
- 23.Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Serena D, Ruggiero FM. Lipid peroxidation and alterations to oxidative metabolism in mitochondria isolated from rat heart subjected to ischemia and reperfusion. Free Radic Biol Med. 1999;27:42–50. doi: 10.1016/s0891-5849(99)00032-5. [DOI] [PubMed] [Google Scholar]
- 24.Turrens JF, Beconi M, Barilla J, Chavez UB, McCord JM. Mitochondrial generation of oxygen radicals during reoxygenation of ischemic tissues. Free Radic Res Commun. 1991;12–13(Pt 2):681–689. doi: 10.3109/10715769109145847. [DOI] [PubMed] [Google Scholar]
- 25.Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. 1995;307:93–98. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Lisa F, Menabò R, Canton M, Barile M, Bernardi P. Opening of the Mitochondrial Permeability Transition Pore Causes Depletion of Mitochondrial and Cytosolic NAD+ and Is a Causative Event in the Death of Myocytes in Postischemic Reperfusion of the Heart. J Biol Chem. 2001;276:2571–2575. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- 27.Stone D, Darley-Usmar V, Smith DR, O’Leary V. Hypoxia-reoxygenation induced increase in cellular Ca2+ in myocytes and perfused hearts: the role of mitochondria. J Mol Cell Cardiol. 1989;21:963–973. doi: 10.1016/0022-2828(89)90795-5. [DOI] [PubMed] [Google Scholar]
- 28.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu S-S. Calcium, ATP, and ROS: A Mitochondrial Love-Hate Triangle. Am J Physiol. 2004;287:C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 29.Kerr DS. Treatment of mitochondrial electron transport chain disorders: a review of clinical trials over the past decade. Mol Genet Metab. 2010;99:246–55. doi: 10.1016/j.ymgme.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 30.Lagedrost SJ, Sutton MS, Cohen MS, Satou GM, Kaufman BD, Perlman SL, Rummey C, Meier T, Lynch DR. Idebenone in Friedreich ataxia cardiomyopathy-results from a 6-month phase III study (IONIA) Am Heart J. 2011;161:639–645. e1. doi: 10.1016/j.ahj.2010.10.038. [DOI] [PubMed] [Google Scholar]
- 31.Gürlek A, Tutar E, Akçil E, Dinçer İ, Erol Ç, Kocatürk PA, Oral D. The Effects of L-Carnitine Treatment on Left Ventricular Function and Erythrocyte Superoxide Dismutase Activity in Patients with Ischemic Cardiomyopathy. Eur J Heart Fail. 2000;2:189–193. doi: 10.1016/s1388-9842(00)00064-7. [DOI] [PubMed] [Google Scholar]
- 32.Buyse GM, Van der Mieren G, Erb M, D’Hooge J, Herijgers P, Verbeken E, Jara A, Van Den Bergh A, Mertens L, Courdier-Fruh I, Barzaghi P, Meier T. Long-term blinded placebo-controlled study of SNT-MC17/idebenone in the dystrophin deficient mdx mouse: cardiac protection and improved exercise performance. Eur Heart J. 2009;30:116–24. doi: 10.1093/eurheartj/ehn406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buyse GM, Goemans N, van den Hauwe M, Thijs D, de Groot IJ, Schara U, Ceulemans B, Meier T, Mertens L. Idebenone as a novel, therapeutic approach for Duchenne muscular dystrophy: results from a 12 month, double-blind, randomized placebo-controlled trial. Neuromuscul Disord. 2011;21:396–405. doi: 10.1016/j.nmd.2011.02.016. [DOI] [PubMed] [Google Scholar]
- 34.Stacpoole PW, Kurtz TL, Han Z, Langaee T. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev. 2008;60:1478–1487. doi: 10.1016/j.addr.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bersin RM, Wolfe C, Kwasman M, Lau D, Klinski C, Tanaka K, Khorrami P, Henderson GN, de Marco T, Chatterjee K. Improved hemodynamic function and mechanical efficiency in congestive heart failure with sodium dichloroacetate. J Am Coll Cardiol. 1994;23:1617–24. doi: 10.1016/0735-1097(94)90665-3. [DOI] [PubMed] [Google Scholar]
- 36.Lewis JF, DaCosta M, Wargowich T, Stacpoole P. Effects of dichloroacetate in patients with congestive heart failure. Clin Cardiol. 1998;21:888–92. doi: 10.1002/clc.4960211206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arakawa K, Kudo T, Ikawa M, Morikawa N, Kawai Y, Sahashi K, Lee JD, Kuriyama M, Miyamori I, Okazawa H, Yoneda M. Abnormal myocardial energy-production state in mitochondrial cardiomyopathy and acute response to L-arginine infusion. C-11 acetate kinetics revealed by positron emission tomography. Circ J. 2010;74:2702–11. doi: 10.1253/circj.cj-10-0044. [DOI] [PubMed] [Google Scholar]
- 38.Abozguia K, Elliott P, McKenna W, Phan TT, Nallur-Shivu G, Ahmed I, Maher AR, Kaur K, Taylor J, Henning A, Ashrafian H, Watkins H, Frenneaux M. Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation. 2010;122:1562–9. doi: 10.1161/CIRCULATIONAHA.109.934059. [DOI] [PubMed] [Google Scholar]
- 39.Follette DM, Fey KH, Steed DL, Foglia RP, Buckberg GD. Reducing reperfusion injury with hypocalcemic, hyperkalemic, alkalotic blood during reoxygenation. Surg Forum. 1978;29:284–286. [PubMed] [Google Scholar]
- 40.Spencer FC, Bahnson HT. The Present Role of Hypothermia in Cardiac Surgery. Circulation. 1962;26:292–300. doi: 10.1161/01.cir.26.2.292. [DOI] [PubMed] [Google Scholar]
- 41.Wallace AW, Au S, Cason BA. Association of the pattern of use of perioperative β-blockade and postoperative mortality. Anesthesiology. 2010;113:794–805. doi: 10.1097/ALN.0b013e3181f1c061. [DOI] [PubMed] [Google Scholar]
- 42.Lazar HL, Chipkin SR, Fitzgerald CA, Bao Y, Cabral H, Apstein CS. Tight Glycemic Control in Diabetic Coronary Artery Bypass Graft Patients Improves Perioperative Outcomes and Decreases Recurrent Ischemic Events. Circulation. 2004;109:1497–1502. doi: 10.1161/01.CIR.0000121747.71054.79. [DOI] [PubMed] [Google Scholar]
- 43.Murry CE, Jennings RB, Reimer KA. Preconditioning with Ischemia: A Delay of Lethal Cell Injury in Ischemic Myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 44.Kloner RA. Clinical Application of Remote Ischemic Preconditioning. Circulation. 2009;119:776–778. doi: 10.1161/CIRCULATIONAHA.108.832832. [DOI] [PubMed] [Google Scholar]
- 45.Kharbanda RK, Mortensen UM, White PA, Kristiansen SB, Schmidt MR, Hoschtitzky JA, Vogel M, Sorensen K, Redington AN, MacAllister R. Transient Limb Ischemia Induces Remote Ischemic Preconditioning In Vivo. Circulation. 2002;106:2881–2883. doi: 10.1161/01.cir.0000043806.51912.9b. [DOI] [PubMed] [Google Scholar]
- 46.Hausenloy DJ, Mwamure PK, Venugopal V, Harris J, Barnard M, Grundy E, Ashley E, Vichare S, Di Salvo C, Kolvekar S, Hayward M, Keogh B, MacAllister RJ, Yellon DM. Effect of remote ischaemic preconditioning on myocardial injury in patients undergoing coronary artery bypass graft surgery: a randomised controlled trial. Lancet. 2007;370:575–579. doi: 10.1016/S0140-6736(07)61296-3. [DOI] [PubMed] [Google Scholar]
- 47.Ali ZA, Callaghan CJ, Lim E, Ali AA, Reza Nouraei SA, Akthar AM, Boyle JR, Varty K, Kharbanda RK, Dutka DP, Gaunt ME. Remote Ischemic Preconditioning Reduces Myocardial and Renal Injury After Elective Abdominal Aortic Aneurysm Repair A Randomized Controlled Trial. Circulation. 2007;116:I98–I105. doi: 10.1161/circulationaha.106.679167. [DOI] [PubMed] [Google Scholar]
- 48.Takagi H, Manabe H, Kawai N, Goto S-N, Umemoto T. Review and meta-analysis of randomized controlled clinical trials of remote ischemic preconditioning in cardiovascular surgery. Am J Cardiol. 2008;102:1487–1488. doi: 10.1016/j.amjcard.2008.07.036. [DOI] [PubMed] [Google Scholar]
- 49.Candilio L, Hausenloy DJ, Yellon DM. Remote ischemic conditioning: a clinical trial’s update. J Cardiovasc Pharmacol Ther. 2011;16:304–312. doi: 10.1177/1074248411411711. [DOI] [PubMed] [Google Scholar]
- 50.Crestanello JA, Doliba NM, Babsky AM, Doliba NM, Niibori K, Osbakken MD, Whitman GJ. Mitochondrial function during ischemic preconditioning. Surgery. 2002;131:172–178. doi: 10.1067/msy.2002.119490. [DOI] [PubMed] [Google Scholar]
- 51.Yang X, Cohen MV, Downey JM. Mechanism of Cardioprotection by Early Ischemic Preconditioning. Cardiovasc Drugs Ther. 2010;24:225–234. doi: 10.1007/s10557-010-6236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zaugg M, Lucchinetti E, Uecker M, Pasch T, Schaub MC. Anaesthetics and Cardiac Preconditioning. Part I. Signalling and Cytoprotective Mechanisms. Br J Anaesth. 2003;91:551–565. doi: 10.1093/bja/aeg205. [DOI] [PubMed] [Google Scholar]
- 53.Zaugg M, Lucchinetti E, Garcia C, Pasch T, Spahn DR, Schaub MC. Anaesthetics and Cardiac Preconditioning. Part II. Clinical Implications. Br J Anaesth. 2003;91:566–576. doi: 10.1093/bja/aeg206. [DOI] [PubMed] [Google Scholar]
- 54.Zhao Z-Q, Corvera JS, Halkos ME, Kerendi F, Wang N-P, Guyton RA, Vinten-Johansen J. Inhibition of Myocardial Injury by Ischemic Postconditioning During Reperfusion: Comparison with Ischemic Preconditioning. Am J Physiol. 2003;285:H579–H588. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]
- 55.Hansen PR, Thibault H, Abdulla J. Postconditioning during primary percutaneous coronary intervention: a review and meta-analysis. Int J Cardiol. 2010;144:22–25. doi: 10.1016/j.ijcard.2009.03.118. [DOI] [PubMed] [Google Scholar]
- 56.Bøtker HE, Kharbanda R, Schmidt MR, Bøttcher M, Kaltoft AK, Terkelsen CJ, Munk K, Andersen NH, Hansen TM, Trautner S, Lassen JF, Christiansen EH, Krusell LR, Kristensen SD, Thuesen L, Nielsen SS, Rehling M, Sørensen HT, Redington AN, Nielsen TT. Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: a randomised trial. Lancet. 2010;375:727–734. doi: 10.1016/S0140-6736(09)62001-8. [DOI] [PubMed] [Google Scholar]
- 57.Okorie MI, Bhavsar DD, Ridout D, Charakida M, Deanfield JE, Loukogeorgakis SP, MacAllister RJ. Postconditioning Protects Against Human Endothelial Ischaemia–Reperfusion Injury Via Subtype-Specific KATP Channel Activation and Is Mimicked by Inhibition of the Mitochondrial Permeability Transition Pore. Eur Heart J. 2011;32:1266–1274. doi: 10.1093/eurheartj/ehr041. [DOI] [PubMed] [Google Scholar]
- 58.Argaud L, Gateau-Roesch O, Raisky O, Loufouat J, Robert D, Ovize M. Postconditioning Inhibits Mitochondrial Permeability Transition. Circulation. 2005;111:194–197. doi: 10.1161/01.CIR.0000151290.04952.3B. [DOI] [PubMed] [Google Scholar]
- 59.Freemantle N, Cleland J, Young P, Mason J, Harrison J. β Blockade after myocardial infarction: systematic review and meta regression analysis. Brit Med J. 1999;318:1730–1737. doi: 10.1136/bmj.318.7200.1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferguson TB, Coombs LP, Peterson ED. Preoperative B-Blocker Use and Mortality and Morbidity Following CABG Surgery in North America. J Am Med Assoc. 2002;287:2221–2227. doi: 10.1001/jama.287.17.2221. [DOI] [PubMed] [Google Scholar]
- 61.Bangalore S, Wetterslev J, Pranesh S, Sawhney S, Gluud C, Messerli FH. Perioperative beta blockers in patients having non-cardiac surgery: a meta-analysis. Lancet. 2008;372:1962–1976. doi: 10.1016/S0140-6736(08)61560-3. [DOI] [PubMed] [Google Scholar]
- 62.Pitarys CJ, Virmani R, Vildibill HD, Jackson EK, Forman MB. Reduction of Myocardial Reperfusion Injury by Intravenous Adenosine Administered During the Early Reperfusion Period. Circulation. 1991;83:237–247. doi: 10.1161/01.cir.83.1.237. [DOI] [PubMed] [Google Scholar]
- 63.Garratt KN, Holmes DR, Jr, Molina-Viamonte V, Reeder GS, Hodge DO, Bailey KR, Lobl JK, Laudon DA, Gibbons RJ. Intravenous adenosine and lidocaine in patients with acute mycocardial infarction. Am Heart J. 1998;136:196–204. doi: 10.1053/hj.1998.v136.89910. [DOI] [PubMed] [Google Scholar]
- 64.Mahaffey KW, Puma JA, Barbagelata NA, DiCarli MF, Leesar MA, Browne KF, Eisenberg PR, Bolli R, Casas AC, Molina-Viamonte V, Orlandi C, Blevins R, Gibbons RJ, Califf RM, Granger CB. Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of a multicenter, randomized, placebo-controlled trial: the Acute Myocardial Infarction STudy of ADenosine (AMISTAD) trial. J Am Coll Cardiol. 1999;34:1711–1720. doi: 10.1016/s0735-1097(99)00418-0. [DOI] [PubMed] [Google Scholar]
- 65.Ross AM, Gibbons RJ, Stone GW, Kloner RA, Alexander RW. A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II) J Am Coll Cardiol. 2005;45:1775–1780. doi: 10.1016/j.jacc.2005.02.061. [DOI] [PubMed] [Google Scholar]
- 66.Kopecky SL, Aviles RJ, Bell MR, Lobl JK, Tipping D, Frommell G, Ramsey K, Holland AE, Midei M, Jain A, Kellett M, Gibbons RJ. A randomized, double-blinded, placebo-controlled, dose-ranging study measuring the effect of an adenosine agonist on infarct size reduction in patients undergoing primary percutaneous transluminal coronary angioplasty: the ADMIRE (AmP579 Delivery for Myocardial Infarction REduction) study. Am Heart J. 2003;146:146–152. doi: 10.1016/S0002-8703(03)00172-8. [DOI] [PubMed] [Google Scholar]
- 67.Navarese EP, Buffon A, Andreotti F, Gurbel PA, Kozinski M, Kubica A, Musumeci G, Cremonesi A, Tavazzi L, Kubica J, Castriota F. Adenosine improves post-procedural coronary flow but not clinical outcomes in patients with acute coronary syndrome: A meta-analysis of randomized trials. Atherosclerosis. 2012;222:1–7. doi: 10.1016/j.atherosclerosis.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 68.Glower DD, Spratt JA, Newton JR, Wolfe JA, Rankin JS, Swain JL. Dissociation Between Early Recovery of Regional Function and Purine Nucleotide Content in Postischaemic Myocardium in the Conscious Dog. Cardiovasc Res. 1987;21:328–336. doi: 10.1093/cvr/21.5.328. [DOI] [PubMed] [Google Scholar]
- 69.Zhao Z-Q, Williams MW, Sato H, Hudspeth DA, McGee DS, Vinten-Johansen J, Van Wylen DG. Acadesine Reduces Myocardial Infarct Size by an Adenosine Mediated Mechanism. Cardiovasc Res. 1995;29:495–505. [PubMed] [Google Scholar]
- 70.Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- 71.Gruber HE, Hoffer ME, McAllister DR, Laikind PK, Lane TA, Schmid-Schoenbein GW, Engler RL. Increased Adenosine Concentration in Blood from Ischemic Myocardium by AICA Riboside. Effects on Flow, Granulocytes, and Injury. Circulation. 1989;80:1400–1411. doi: 10.1161/01.cir.80.5.1400. [DOI] [PubMed] [Google Scholar]
- 72.Young MA, Mullane KM. Progressive Cardiac Dysfunction with Repeated Pacing-Induced Ischemia: Protection by AICA-Riboside. Am J Physiol. 1991;261:H1570–H1577. doi: 10.1152/ajpheart.1991.261.5.H1570. [DOI] [PubMed] [Google Scholar]
- 73.Mangano DT. Effects of Acadesine on Myocardial Infarction, Stroke, and Death Following Surgery A Meta-Analysis of the 5 International Randomized Trials. J Am Med Assoc. 1997;277:325–332. doi: 10.1001/jama.277.4.325. [DOI] [PubMed] [Google Scholar]
- 74.Mangano DT, Miao Y, Tudor IC, Dietzel C. Post-reperfusion myocardial infarction: long- term survival improvement using adenosine regulation with acadesine. J Am Coll Cardiol. 2006;48:206–214. doi: 10.1016/j.jacc.2006.04.044. [DOI] [PubMed] [Google Scholar]
- 75.Shlafer M, Kane PF, Wiggins VY, Kirsh MM. Possible role for cytotoxic oxygen metabolites in the pathogenesis of cardiac ischemic injury. Circulation. 1982;66:I85–92. [PubMed] [Google Scholar]
- 76.Gardner TJ, Stewart JR, Casale AS, Downey JM, Chambers DE. Reduction of myocardial ischemic injury with oxygen-derived free radical scavengers. Surgery. 1983;94:423–427. [PubMed] [Google Scholar]
- 77.Klein HH, Pich S, Lindert S, Buchwald A, Nebendahl K, Kreuzer H. Intracoronary superoxide dismutase for the treatment of “reperfusion injury”, A blind randomized placebo-controlled trial in ischemic, reperfused porcine hearts. Basic Res Cardiol. 1988;83:141–148. doi: 10.1007/BF01907268. [DOI] [PubMed] [Google Scholar]
- 78.Flaherty JT, Pitt B, Gruber JW, Heuser RR, Rothbaum DA, Burwell LR, George BS, Kereiakes DJ, Deitchman D, Gustafson N. Recombinant Human Superoxide Dismutase (h-SOD) Fails to Improve Recovery of Ventricular Function in Patients Undergoing Coronary Angioplasty for Acute Myocardial Infarction. Circulation. 1994;89:1982–1991. doi: 10.1161/01.cir.89.5.1982. [DOI] [PubMed] [Google Scholar]
- 79.Chambers DE, Parks DA, Patterson G, Roy R, McCord JM, Yoshida S, Parmley LF, Downey JM. Xanthine oxidase as a source of free radical damage in myocardial ischemia. J Mol Cell Cardiol. 1985;17:145–152. doi: 10.1016/s0022-2828(85)80017-1. [DOI] [PubMed] [Google Scholar]
- 80.Stewart JR, Crute SL, Loughlin V, Hess ML, Greenfield LJ. Prevention of free radical-induced myocardial reperfusion injury with allopurinol. J Thorac Cardiovasc Surg. 1985;90:68–72. [PubMed] [Google Scholar]
- 81.Zhou M, Zhi Q, Tang Y, Yu D, Han J. Effects of coenzyme Q10 on myocardial protection during cardiac valve replacement and scavenging free radical activity in vitro. J Cardiovasc Surg (Torino) 1999;40:355–361. [PubMed] [Google Scholar]
- 82.Mitsos SE, Askew TE, Fantone JC, Kunkel SL, Abrams GD, Schork A, Lucchesi BR. Protective Effects of N-2-Mercaptopropionyl Glycine Against Myocardial Reperfusion Injury After Neutrophil Depletion in the Dog: Evidence for the Role of Intracellular-Derived Free Radicals. Circulation. 1986;73:1077–1086. doi: 10.1161/01.cir.73.5.1077. [DOI] [PubMed] [Google Scholar]
- 83.Marczin N, El-Habashi N, Hoare GS, Bundy RE, Yacoub M. Antioxidants in myocardial ischemia-reperfusion injury: therapeutic potential and basic mechanisms. Arch Biochem Biophys. 2003;420:222–236. doi: 10.1016/j.abb.2003.08.037. [DOI] [PubMed] [Google Scholar]
- 84.Horwitz LD, Fennessey PV, Shikes RH, Kong Y. Marked Reduction in Myocardial Infarct Size Due to Prolonged Infusion of an Antioxidant During Reperfusion. Circulation. 1994;89:1792–1801. doi: 10.1161/01.cir.89.4.1792. [DOI] [PubMed] [Google Scholar]
- 85.Venturini CM, Flickinger AG, Womack CR, Smith ME, McMahon EG. The Antioxidant, N-(2-mercaptopropionyl)-glycine (MPG), Does Not Reduce Myocardial Infarct Size in an Acute Canine Model of Myocardial Ischemia and Reperfusion. J Thromb Thrombolysis. 1998;5:135–141. doi: 10.1023/A:1008830129106. [DOI] [PubMed] [Google Scholar]
- 86.Dirksen MT, Laarman GJ, Simoons ML, Duncker DJG. Reperfusion Injury in Humans: A Review of Clinical Trials on Reperfusion Injury Inhibitory Strategies. Cardiovasc Res. 2007;74:343–355. doi: 10.1016/j.cardiores.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 87.Downey JM, Cohen MV. Why Do We Still Not Have Cardioprotective Drugs? Circulation Journal. 2009;73:1171–1177. doi: 10.1253/circj.cj-09-0338. [DOI] [PubMed] [Google Scholar]
- 88.Tang X-L, Takano H, Rizvi A, Turrens JF, Qiu Y, Wu W-J, Zhang Q, Bolli R. Oxidant Species Trigger Late Preconditioning Against Myocardial Stunning in Conscious Rabbits. Am J Physiol. 2002;282:H281–H291. doi: 10.1152/ajpheart.2002.282.1.H281. [DOI] [PubMed] [Google Scholar]
- 89.Crompton M, Costi A, Hayat L. Evidence for the presence of a reversible Ca2+-dependent pore activated by oxidative stress in heart mitochondria. Biochem J. 1987;245:915–918. doi: 10.1042/bj2450915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 92.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 93.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Smeele KMA, Southworth R, Wu R, Xie C, Nederlof R, Warley A, Nelson JK, van Horssen P, van den Wijngaard JP, Heikkinen S, Laakso M, Koeman A, Siebes M, Eerbeek O, Akar FG, Ardehali H, Hollmann MW, Zuurbier CJ. Disruption of hexokinase II-mitochondrial binding blocks ischemic preconditioning and causes rapid cardiac necrosis. Circ Res. 2011;108:1165–1169. doi: 10.1161/CIRCRESAHA.111.244962. [DOI] [PubMed] [Google Scholar]
- 95.Mergenthaler P, Kahl A, Kamitz A, van Laak V, Stohlmann K, Thomsen S, Klawitter H, Przesdzing I, Neeb L, Freyer D, Priller J, Collins TJ, Megow D, Dirnagl U, Andrews DW, Meisel A. Mitochondrial hexokinase II (HKII) and phosphoprotein enriched in astrocytes (PEA15) form a molecular switch governing cellular fate depending on the metabolic state. Proc Natl Acad Sci USA. 2012;109:1518–1523. doi: 10.1073/pnas.1108225109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McEnery MW, Snowman AM, Trifiletti RR, Snyder SH. Isolation of the Mitochondrial Benzodiazepine Receptor: Association with the Voltage-Dependent Anion Channel and the Adenine Nucleotide Carrier. Proc Natl Acad Sci USA. 1992;89:3170–3174. doi: 10.1073/pnas.89.8.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fournier N, Ducet G, Crevat A. Action of cyclosporine on mitochondrial calcium fluxes. J Bioenerg Biomembr. 1987;19:297–303. doi: 10.1007/BF00762419. [DOI] [PubMed] [Google Scholar]
- 98.Griffiths EJ, Ocampo CJ, Savage JS, Stern MD, Silverman HS. Protective effects of low and high doses of cyclosporin A against reoxygenation injury in isolated rat cardiomyocytes are associated with differential effects on mitochondrial calcium levels. Cell Calcium. 2000;27:87–95. doi: 10.1054/ceca.1999.0094. [DOI] [PubMed] [Google Scholar]
- 99.Griffiths EJ, Halestrap AP. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J Mol Cell Cardiol. 1993;25:1461–1469. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- 100.Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, Macia C, Raczka F, Sportouch C, Gahide G, Finet G, André-Fouët X, Revel D, Kirkorian G, Monassier J-P, Derumeaux G, Ovize M. Effect of Cyclosporine on Reperfusion Injury in Acute Myocardial Infarction. N Engl J Med. 2008;359:473–481. doi: 10.1056/NEJMoa071142. [DOI] [PubMed] [Google Scholar]
- 101.Mewton N, Croisille P, Gahide G, Rioufol G, Bonnefoy E, Sanchez I, Cung TT, Sportouch C, Angoulvant D, Finet G, André-Fouët X, Derumeaux G, Piot C, Vernhet H, Revel D, Ovize M. Effect of cyclosporine on left ventricular remodeling after reperfused myocardial infarction. J Am Coll Cardiol. 2010;55:1200–1205. doi: 10.1016/j.jacc.2009.10.052. [DOI] [PubMed] [Google Scholar]
- 102.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Roberts CA, Stern DL, Radio SJ. Asymmetric cardiac hypertrophy at autopsy in patients who received FK506 (tacrolimus) or cyclosporine A after liver transplant. Transplantation. 2002;74:817–821. doi: 10.1097/00007890-200209270-00015. [DOI] [PubMed] [Google Scholar]
- 104.Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting Mitochondrial Permeability Transition Pore Opening at Reperfusion Protects Against Ischaemia Reperfusion Injury. Cardiovasc Res. 2003;60:617–625. doi: 10.1016/j.cardiores.2003.09.025. [DOI] [PubMed] [Google Scholar]
- 105.Argaud L, Gateau-Roesch O, Muntean D, Chalabreysse L, Loufouat J, Robert D, Ovize M. Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J Mol Cell Cardiol. 2005;38:367–374. doi: 10.1016/j.yjmcc.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 106.Gomez L, Thibault H, Gharib A, Dumont J-M, Vuagniaux G, Scalfaro P, Derumeaux G, Ovize M. Inhibition of Mitochondrial Permeability Transition Improves Functional Recovery and Reduces Mortality Following Acute Myocardial Infarction in Mice. Am J Physiol. 2007;293:H1654–H1661. doi: 10.1152/ajpheart.01378.2006. [DOI] [PubMed] [Google Scholar]
- 107.Dube H, Selwood D, Malouitre S, Capano M, Simone MI, Crompton M. A mitochondrial-targeted cyclosporin A with high binding affinity for cyclophilin D yields improved cytoprotection of cardiomyocytes. Biochem J. 2012;441:901–907. doi: 10.1042/BJ20111301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Leducq N, Bono F, Sulpice T, Vin VÉ, Janiak P, Fur GL, O’Connor SE, Herbert J-M. Role of Peripheral Benzodiazepine Receptors in Mitochondrial, Cellular, and Cardiac Damage Induced by Oxidative Stress and Ischemia-Reperfusion. J Pharmacol Exp Ther. 2003;306:828–837. doi: 10.1124/jpet.103.052068. [DOI] [PubMed] [Google Scholar]
- 109.Obame FN, Zini R, Souktani R, Berdeaux A, Morin D. Peripheral Benzodiazepine Receptor-Induced Myocardial Protection Is Mediated by Inhibition of Mitochondrial Membrane Permeabilization. J Pharmacol Exp Ther. 2007;323:336–345. doi: 10.1124/jpet.107.124255. [DOI] [PubMed] [Google Scholar]
- 110.Schaller S, Paradis S, Ngoh GA, Assaly R, Buisson B, Drouot C, Ostuni MA, Lacapere J-J, Bassissi F, Bordet T, Berdeaux A, Jones SP, Morin D, Pruss RM. TRO40303, a New Cardioprotective Compound, Inhibits Mitochondrial Permeability Transition. J Pharmacol Exp Ther. 2010;333:696–706. doi: 10.1124/jpet.110.167486. [DOI] [PubMed] [Google Scholar]
- 111.Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006;40:16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 112.Cohen MV, Yang X-M, Downey JM. Nitric Oxide Is a Preconditioning Mimetic and Cardioprotectant and Is the Basis of Many Available Infarct-Sparing Strategies. Cardiovasc Res. 2006;70:231–239. doi: 10.1016/j.cardiores.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 113.Nadtochiy SM, Burwell LS, Brookes PS. Cardioprotection and mitochondrial S-nitrosation: effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury. J Mol Cell Cardiol. 2007;42:812–825. doi: 10.1016/j.yjmcc.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sun J, Murphy E. Protein S-Nitrosylation and Cardioprotection. Circ Res. 2010;106:285–296. doi: 10.1161/CIRCRESAHA.109.209452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rudolph V, Rudolph TK, Schopfer FJ, Bonacci G, Woodcock SR, Cole MP, Baker PRS, Ramani R, Freeman BA. Endogenous generation and protective effects of nitro-fatty acids in a murine model of focal cardiac ischaemia and reperfusion. Cardiovasc Res. 2010;85:155–166. doi: 10.1093/cvr/cvp275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nadtochiy SM, Baker PRS, Freeman BA, Brookes PS. Mitochondrial nitroalkene formation and mild uncoupling in ischaemic preconditioning: implications for cardioprotection. Cardiovasc Res. 2009;82:333–340. doi: 10.1093/cvr/cvn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Burwell LS, Brookes PS. Mitochondria as a target for the cardioprotective effects of nitric oxide in ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10:579–599. doi: 10.1089/ars.2007.1845. [DOI] [PubMed] [Google Scholar]
- 118.Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN, Murphy E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J Biol Chem. 2011;286:40184–40192. doi: 10.1074/jbc.M111.243469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nadtochiy SM, Burwell LS, Ingraham CA, Spencer CM, Friedman AE, Pinkert CA, Brookes PS. In vivo cardioprotection by S-nitroso-2-mercaptopropionyl glycine. J Mol Cell Cardiol. 2009;46:960–968. doi: 10.1016/j.yjmcc.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Prime TA, Blaikie FH, Evans C, Nadtochiy SM, James AM, Dahm CC, Vitturi DA, Patel RP, Hiley CR, Abakumova I, Requejo R, Chouchani ET, Hurd TR, Garvey JF, Taylor CT, Brookes PS, Smith RAJ, Murphy MP. A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2009;106:10764–10769. doi: 10.1073/pnas.0903250106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stoyanovsky DA, Vlasova II, Belikova NA, Kapralov A, Tyurin V, Kagan VE. Activation of NO Donors in Mitochondria: Peroxidase Metabolism of (2-hydroxyamino-vinyl)-triphenyl-phosphonium by Cytochrome c Releases NO and Protects Cells Against Apoptosis. FEBS Lett. 2008;582:725–728. doi: 10.1016/j.febslet.2008.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Freeman BA, Baker PRS, Schopfer FJ, Woodcock SR, Napolitano A, d’ Ischia M. Nitro-fatty Acid Formation and Signaling. J Biol Chem. 2008;283:15515–15519. doi: 10.1074/jbc.R800004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nadtochiy SM, Zhu Q, Urciuoli W, Rafikov R, Black SM, Brookes PS. Nitroalkenes Confer Acute Cardioprotection via Adenine Nucleotide Translocase 1. J Biol Chem. 2012;287:3573–3580. doi: 10.1074/jbc.M111.298406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Brennan JP, Southworth R, Medina RA, Davidson SM, Duchen MR, Shattock MJ. Mitochondrial Uncoupling, with Low Concentration FCCP, Induces ROS-Dependent Cardioprotection Independent of KATP Channel Activation. Cardiovasc Res. 2006;72:313–321. doi: 10.1016/j.cardiores.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 125.Duranski MR, Greer JJM, Dejam A, Jaganmohan S, Hogg N, Langston W, Patel RP, Yet S-F, Wang X, Kevil CG, Gladwin MT, Lefer DJ. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest. 2005;115:1232–1240. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, Burwell L, Wang X, MacArthur PH, Shoja A, Raghavachari N, Calvert JW, Brookes PS, Lefer DJ, Gladwin MT. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med. 2007;204:2089–2102. doi: 10.1084/jem.20070198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dezfulian C, Shiva S, Alekseyenko A, Pendyal A, Beiser D, Munasinghe JP, Anderson SA, Chesley CF, Hoek TV, Gladwin MT. Nitrite therapy after cardiac arrest reduces ROS generation, improves cardiac and neurological function and enhances survival via reversible inhibition of mitochondrial complex I. Circulation. 2009;120:897–905. doi: 10.1161/CIRCULATIONAHA.109.853267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.O’Rourke B. Mitochondrial Ion Channels. Annu Rev Physiol. 2007;69:19–49. doi: 10.1146/annurev.physiol.69.031905.163804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D’Alonzo AJ, Lodge NJ, Smith MA, Grover GJ. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ Res. 1997;81:1072–1082. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- 130.Murata M, Akao M, O’Rourke B, Marbán E. Mitochondrial ATP-sensitive potassium channels attenuate matrix Ca2+ overload during simulated ischemia and reperfusion: possible mechanism of cardioprotection. Circ Res. 2001;89:891–898. doi: 10.1161/hh2201.100205. [DOI] [PubMed] [Google Scholar]
- 131.Wang X, Wei M, Kuukasjärvi P, Laurikka J, Järvinen O, Rinne T, Honkonen E-L, Tarkka M. Novel Pharmacological Preconditioning with Diazoxide Attenuates Myocardial Stunning in Coronary Artery Bypass Grafting. Eur J Cardiothorac Surg. 2003;24:967–973. doi: 10.1016/s1010-7940(03)00438-x. [DOI] [PubMed] [Google Scholar]
- 132.Deja MA, Malinowski M, Gołba KS, Kajor M, Lebda-Wyborny T, Hudziak D, Domaradzki W, Szurlej D, Bończyk A, Biernat J, Woś S. Diazoxide protects myocardial mitochondria metabolism and function during cardiac surgery: a double-blind randomized feasibility study of diazoxide-supplemented cardioplegia. J Thorac Cardiovasc Surg. 2009;137:997–1004. 1004e1–2. doi: 10.1016/j.jtcvs.2008.08.068. [DOI] [PubMed] [Google Scholar]
- 133.Kitakaze M, Asakura M, Kim J, Shintani Y, Asanuma H, Hamasaki T, Seguchi O, Myoishi M, Minamino T, Ohara T, Nagai Y, Nanto S, Watanabe K, Fukuzawa S, Hirayama A, Nakamura N, Kimura K, Fujii K, Ishihara M, Saito Y, Tomoike H, Kitamura S. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): two randomised trials. Lancet. 2007;370:1483–1493. doi: 10.1016/S0140-6736(07)61634-1. [DOI] [PubMed] [Google Scholar]
- 134.Grover GJ, McCullough JR, Henry DE, Conder ML, Sleph PG. Anti-Ischemic Effects of the Potassium Channel Activators Pinacidil and Cromakalim and the Reversal of These Effects with the Potassium Channel Blocker Glyburide. J PharmacolExp Ther. 1989;251:98–104. [PubMed] [Google Scholar]
- 135.Grover GJ, Atwal KS. Pharmacologic profile of the selective mitochondrial-K(ATP) opener BMS-191095 for treatment of acute myocardial ischemia. Cardiovasc Drug Rev. 2002;20:121–136. doi: 10.1111/j.1527-3466.2002.tb00187.x. [DOI] [PubMed] [Google Scholar]
- 136.Sato T, Li Y, Saito T, Nakaya H. Minoxidil opens mitochondrial KATP channels and confers cardioprotection. Br J Pharmacol. 2004;141:360–366. doi: 10.1038/sj.bjp.0705613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Auchampach JA, Cavero I, Gross GJ. Nicorandil attenuates myocardial dysfunction associated with transient ischemia by opening ATP-dependent potassium channels. J Cardiovasc Pharmacol. 1992;20:765–771. [PubMed] [Google Scholar]
- 138.Ardehali H, Chen Z, Ko Y, Mejía-Alvarez R, Marbán E. Multiprotein complex containing succinate dehydrogenase confers mitochondrial ATP-sensitive K+ channel activity. Proc Natl Acad Sci USA. 2004;101:11880–11885. doi: 10.1073/pnas.0401703101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wojtovich AP, Brookes PS. The endogenous mitochondrial complex II inhibitor malonate regulates mitochondrial ATP-sensitive potassium channels: implications for ischemic preconditioning. Biochim Biophys Acta. 2008;1777:882–889. doi: 10.1016/j.bbabio.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wojtovich AP, Brookes PS. The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res Cardiol. 2009;104:121–129. doi: 10.1007/s00395-009-0001-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Engler RL, Yellon DM. Sulfonylurea KATP Blockade in Type II Diabetes and Preconditioning in Cardiovascular Disease: Time for Reconsideration. Circulation. 1996;94:2297–2301. doi: 10.1161/01.cir.94.9.2297. [DOI] [PubMed] [Google Scholar]
- 142.Salkoff L, Butler A, Ferreira G, Santi C, Wei A. High-conductance potassium channels of the SLO family. NatRev Neurosci. 2006;7:921–931. doi: 10.1038/nrn1992. [DOI] [PubMed] [Google Scholar]
- 143.Bentzen BH, Osadchii O, Jespersen T, Hansen RS, Olesen S-P, Grunnet M. Activation of big conductance Ca2+-activated K+ channels (BK) protects the heart against ischemia-reperfusion injury. Pflugers Arch. 2009;457:979–988. doi: 10.1007/s00424-008-0583-5. [DOI] [PubMed] [Google Scholar]
- 144.Sakamoto K, Ohya S, Muraki K, Imaizumi Y. A novel opener of large-conductance Ca2+- activated K+ (BK) channel reduces ischemic injury in rat cardiac myocytes by activating mitochondrial KCa channel. J Pharmacol Sci. 2008;108:135–139. doi: 10.1254/jphs.08150sc. [DOI] [PubMed] [Google Scholar]
- 145.Park WS, Kang SH, Son YK, Kim N, Ko J-H, Kim HK, Ko EA, Kim CD, Han J. The mitochondrial Ca2+-activated K+ channel activator, NS 1619 inhibits L-type Ca2+ channels in rat ventricular myocytes. Biochem Biophys Res Commun. 2007;362:31–36. doi: 10.1016/j.bbrc.2007.07.057. [DOI] [PubMed] [Google Scholar]
- 146.Saleh SN, Angermann JE, Sones WR, Leblanc N, Greenwood IA. Stimulation of Ca2+-gated Cl- currents by the calcium-dependent K+ channel modulators NS1619 [1,3-dihydro-1-[2-hydroxy-5-(trifluoromethyl)phenyl]-5-(trifluoromethyl)-2H-benzimidazol-2-one] and isopimaric acid. JPharmacol Exp Ther. 2007;321:1075–1084. doi: 10.1124/jpet.106.118786. [DOI] [PubMed] [Google Scholar]
- 147.Cancherini DV, Queliconi BB, Kowaltowski AJ. Pharmacological and Physiological Stimuli Do Not Promote Ca2+-Sensitive K+ Channel Activity in Isolated Heart Mitochondria. Cardiovasc Res. 2007;73:720–728. doi: 10.1016/j.cardiores.2006.11.035. [DOI] [PubMed] [Google Scholar]
- 148.Liberman EA, Skulachev VP. Conversion of biomembrane-produced energy into electric form. IV. General discussion. Biochim Biophys Acta. 1970;216:30–42. doi: 10.1016/0005-2728(70)90156-8. [DOI] [PubMed] [Google Scholar]
- 149.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RAJ, Murphy MP, Sammut IA. Targeting an Antioxidant to Mitochondria Decreases Cardiac Ischemia-Reperfusion Injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 150.Smith RAJ, Adlam VJ, Blaikie FH, Manas A-RB, Porteous CM, James AM, Ross MF, Logan A, Cochemé HM, Trnka J, Prime TA, Abakumova I, Jones BA, Filipovska A, Murphy MP. Mitochondria-targeted antioxidants in the treatment of disease. Ann NY Acad Sci. 2008;1147:105–111. doi: 10.1196/annals.1427.003. [DOI] [PubMed] [Google Scholar]
- 151.Wu D, Soong Y, Zhao G-M, Szeto HH. A Highly Potent Peptide Analgesic That Protects Against Ischemia-Reperfusion-Induced Myocardial Stunning. Am J Physiol. 2002;283:H783–H791. doi: 10.1152/ajpheart.00193.2002. [DOI] [PubMed] [Google Scholar]
- 152.Zhao K, Zhao G-M, Wu D, Soong Y, Birk AV, Schiller PW, Szeto HH. Cell-Permeable Peptide Antioxidants Targeted to Inner Mitochondrial Membrane Inhibit Mitochondrial Swelling, Oxidative Cell Death, and Reperfusion Injury. J Biol Chem. 2004;279:34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 153.Roser KS, Brookes PS, Wojtovich AP, Olson LP, Shojaie J, Parton RL, Anders MW. Mitochondrial biotransformation of ω-(phenoxy)alkanoic acids, 3-(phenoxy)acrylic acids, and ω-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids: A prodrug strategy for targeting cytoprotective antioxidants to mitochondria. Bioorg Med Chem. 2010;18:1441–1448. doi: 10.1016/j.bmc.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lesnefsky EJ, Chen Q, Moghaddas S, Hassan MO, Tandler B, Hoppel CL. Blockade of Electron Transport During Ischemia Protects Cardiac Mitochondria. J Biol Chem. 2004;279:47961–47967. doi: 10.1074/jbc.M409720200. [DOI] [PubMed] [Google Scholar]
- 155.Stewart S, Lesnefsky EJ, Chen Q. Reversible blockade of electron transport with amobarbital at the onset of reperfusion attenuates cardiac injury. Transl Res. 2009;153:224–231. doi: 10.1016/j.trsl.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 156.Kabir AMN, Cao X, Gorog DA, Tanno M, Bassi R, Bellahcene M, Quinlan RA, Davis RJ, Flavell RA, Shattock MJ, Marber MS. Antimycin A induced cardioprotection is dependent on pre-ischemic p38-MAPK activation but independent of MKK3. J Mol Cell Cardiol. 2005;39:709–717. doi: 10.1016/j.yjmcc.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 157.Clark JE, Naughton P, Shurey S, Green CJ, Johnson TR, Mann BE, Foresti R, Motterlini R. Cardioprotective actions by a water-soluble carbon monoxide-releasing molecule. Circ Res. 2003;93:e2–8. doi: 10.1161/01.RES.0000084381.86567.08. [DOI] [PubMed] [Google Scholar]
- 158.Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, Kimura H, Chow C-W, Lefer DJ. Hydrogen Sulfide Attenuates Myocardial Ischemia-Reperfusion Injury by Preservation of Mitochondrial Function. Proc Natl Acad Sci USA. 2007;104:15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Burwell LS, Nadtochiy SM, Brookes PS. Cardioprotection by metabolic shut-down and gradual wake-up. J Mol Cell Cardiol. 2009;46:804–810. doi: 10.1016/j.yjmcc.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Nadtochiy SM, Tompkins AJ, Brookes PS. Different mechanisms of mitochondrial proton leak in ischaemia/reperfusion injury and preconditioning: implications for pathology and cardioprotection. Biochem J. 2006;395:611–618. doi: 10.1042/BJ20051927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hausenloy DJ, Yellon DM. New Directions for Protecting the Heart Against Ischaemia Reperfusion Injury: Targeting the Reperfusion Injury Salvage Kinase (RISK)-Pathway. Cardiovasc Res. 2004;61:448–460. doi: 10.1016/j.cardiores.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 162.Hausenloy DJ, Tsang A, Yellon DM. The reperfusion injury salvage kinase pathway: a common target for both ischemic preconditioning and postconditioning. Trends Cardiovasc Med. 2005;15:69–75. doi: 10.1016/j.tcm.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 163.Davidson SM, Hausenloy D, Duchen MR, Yellon DM. Signalling via the reperfusion injury signalling kinase (RISK) pathway links closure of the mitochondrial permeability transition pore to cardioprotection. Int J Biochem Cell Biol. 2006;38:414–419. doi: 10.1016/j.biocel.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 164.Tong H, Imahashi K, Steenbergen C, Murphy E. Phosphorylation of glycogen synthase kinase-3beta during preconditioning through a phosphatidylinositol-3-kinase--dependent pathway is cardioprotective. Circ Res. 2002;90:377–379. doi: 10.1161/01.res.0000012567.95445.55. [DOI] [PubMed] [Google Scholar]
- 165.Gross ER, Hsu AK, Gross GJ. Delayed Cardioprotection Afforded by the Glycogen Synthase Kinase 3 Inhibitor SB-216763 Occurs Via a KATP- and MPTP-Dependent Mechanism at Reperfusion. Am J Physiol. 2008;294:H1497–H1500. doi: 10.1152/ajpheart.01381.2007. [DOI] [PubMed] [Google Scholar]
- 166.Sack MN, Yellon DM. Insulin therapy as an adjunct to reperfusion after acute coronary ischemia: a proposed direct myocardial cell survival effect independent of metabolic modulation. J Am Coll Cardiol. 2003;41:1404–1407. doi: 10.1016/s0735-1097(03)00164-5. [DOI] [PubMed] [Google Scholar]
- 167.Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of Endothelial Nitric Oxide Synthase by HMG CoA Reductase Inhibitors. Circulation. 1998;97:1129–1135. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- 168.Kukreja RC. Synergistic effects of atorvastatin and sildenafil in cardioprotection-role of NO. Cardiovasc Drugs Ther. 2006;20:5–8. doi: 10.1007/s10557-006-7298-7. [DOI] [PubMed] [Google Scholar]
- 169.Lefer AM, Campbell B, Shin Y-K, Scalia R, Hayward R, Lefer DJ. Simvastatin Preserves the Ischemic-Reperfused Myocardium in Normocholesterolemic Rat Hearts. Circulation. 1999;100:178–184. doi: 10.1161/01.cir.100.2.178. [DOI] [PubMed] [Google Scholar]
- 170.Guo S, Olm-Shipman A, Walters A, Urciuoli WR, Devito S, Nadtochiy SM, Wojtovich AP, Brookes PS. A cell-based phenotypic assay to identify cardioprotective agents. Circ Res. 2012;110:948–957. doi: 10.1161/CIRCRESAHA.111.263715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bell RM, Yellon DM. Atorvastatin, administered at the onset of reperfusion, and independent oflipid lowering, protects the myocardiumby up-regulating a pro-survival pathway. J Am Coll Cardiol. 2003;41:508–515. doi: 10.1016/s0735-1097(02)02816-4. [DOI] [PubMed] [Google Scholar]
- 172.Fonarow GC, Wright RS, Spencer FA, Fredrick PD, Dong W, Every N, French WJ. Effect of statin use within the first 24 hours of admission for acute myocardial infarction on early morbidity and mortality. Am J Cardiol. 2005;96:611–616. doi: 10.1016/j.amjcard.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 173.Hausenloy DJ, Yellon DM. Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection. Heart Fail Rev. 2007;12:217–234. doi: 10.1007/s10741-007-9026-1. [DOI] [PubMed] [Google Scholar]
- 174.Hausenloy DJ, Yellon DM. Clinical translation of cardioprotective strategies: report and recommendations of the Hatter Institute 5th International Workshop on Cardioprotection. Basic Res Cardiol. 2008;103:493–500. doi: 10.1007/s00395-008-0736-x. [DOI] [PubMed] [Google Scholar]
- 175.Kristal BS, Park BK, Yu BP. 4-Hydroxyhexenal Is a Potent Inducer of the Mitochondrial Permeability Transition. J Biol Chem. 1996;271:6033–6038. doi: 10.1074/jbc.271.11.6033. [DOI] [PubMed] [Google Scholar]
- 176.Chen C-H, Budas GR, Churchill EN, Disatnik M-H, Hurley TD, Mochly-Rosen D. Activation of Aldehyde Dehydrogenase-2 Reduces Ischemic Damage to the Heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.He L, Liu B, Dai Z, Zhang H-F, Zhang Y-S, Luo X-J, Ma Q-L, Peng J. Alpha lipoic acid protects heart against myocardial ischemia-reperfusion injury through a mechanism involving aldehyde dehydrogenase 2 activation. Eur J Pharmacol. 2012;678:32–38. doi: 10.1016/j.ejphar.2011.12.042. [DOI] [PubMed] [Google Scholar]
- 178.Wang H-J, Kang P-F, Ye H-W, Yu Y, Wang X-M, Gao Q. Anti-apoptotic role of mitochondrial aldehyde dehydrogenase 2 in myocardial ischemia/reperfusion injury in diabetic rats. Nan Fang Yi Ke Da Xue Xue Bao. 2012;32:345–348. [PubMed] [Google Scholar]
- 179.Szöcs K, Lassègue B, Wenzel P, Wendt M, Daiber A, Oelze M, Meinertz T, Münzel T, Baldus S. Increased superoxide production in nitrate tolerance is associated with NAD(P)H oxidase and aldehyde dehydrogenase 2 downregulation. J Mol Cell Cardiol. 2007;42:1111–1118. doi: 10.1016/j.yjmcc.2007.03.904. [DOI] [PubMed] [Google Scholar]
- 180.Oelze M, Schuhmacher S, Daiber A. Organic nitrates and nitrate resistance in diabetes: the role of vascular dysfunction and oxidative stress with emphasis on antioxidant properties of pentaerithrityl tetranitrate. Exp Diabetes Res. 2010;2010:213176. doi: 10.1155/2010/213176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lazar HL. The Insulin Cardioplegia Trial. J Thorac Cardiovasc Surg. 2002;123:842–844. doi: 10.1067/mtc.2002.121041. [DOI] [PubMed] [Google Scholar]
- 182.Rabi D, Clement F, McAlister F, Majumdar S, Sauve R, Johnson J, Ghali W. Effect of perioperative glucose-insulin-potassium infusions on mortality and atrial fibrillation after coronary artery bypass grafting: a systematic review and meta-analysis. Can J Cardiol. 2010;26:178–184. doi: 10.1016/s0828-282x(10)70394-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kersten JR, Warltier DC, Pagel PS. Aggressive control of intraoperative blood glucose concentration: a shifting paradigm? Anesthesiology. 2005;103:677–678. doi: 10.1097/00000542-200510000-00002. [DOI] [PubMed] [Google Scholar]
- 184.Selker HP, Beshansky JR, Sheehan PR, Massaro JM, Griffith JL, D’Agostino RB, Ruthazer R, Atkins JM, Sayah AJ, Levy MK, Richards ME, Aufderheide TP, Braude DA, Pirrallo RG, Doyle DD, Frascone RJ, Kosiak DJ, Leaming JM, Van Gelder CM, Walter G-P, Wayne MA, Woolard RH, Opie LH, Rackley CE, Apstein CS, Udelson JE. Out-of-hospital administration of intravenous glucose-insulin-potassium in patients with suspected acute coronary syndromes: the IMMEDIATE randomized controlled trial. J Am Med Assoc. 2012;307:1925–1933. doi: 10.1001/jama.2012.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Oliver MF, Opie LH. Effects of glucose and fatty acids on myocardial ischaemia and arrhythmias. Lancet. 1994;343:155–158. doi: 10.1016/s0140-6736(94)90939-3. [DOI] [PubMed] [Google Scholar]
- 186.Lopaschuk GD, Spafford MA, Davies NJ, Wall SR. Glucose and palmitate oxidation in isolated working rat hearts reperfused after a period of transient global ischemia. Circ Res. 1990;66:546–553. doi: 10.1161/01.res.66.2.546. [DOI] [PubMed] [Google Scholar]
- 187.Lopaschuk GD, McNeil GF, McVeigh JJ. Glucose oxidation is stimulated in reperfused ischemic hearts with the carnitine palmitoyltransferase 1 inhibitor, Etomoxir. Mol Cell Biochem. 1989;88:175–179. doi: 10.1007/BF00223440. [DOI] [PubMed] [Google Scholar]
- 188.Schmidt-Schweda S, Holubarsch C. First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin Sci. 2000;99:27–35. [PubMed] [Google Scholar]
- 189.Lionetti V, Linke A, Chandler MP, Young ME, Penn MS, Gupte S, d’ Agostino C, Hintze TH, Stanley WC, Recchia FA. Carnitine Palmitoyl Transferase-I Inhibition Prevents Ventricular Remodeling and Delays Decompensation in Pacing-Induced Heart Failure. Cardiovasc Res. 2005;66:454–461. doi: 10.1016/j.cardiores.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 190.Lee L, Horowitz J, Frenneaux M. Metabolic Manipulation in Ischaemic Heart Disease, a Novel Approach to Treatment. Eur Heart J. 2004;25:634–641. doi: 10.1016/j.ehj.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 191.Gao D, Ning N, Niu X, Hao G, Meng Z. Trimetazidine: A Meta-Analysis of Randomised Controlled Trials in Heart Failure. Heart. 2011;97:278–286. doi: 10.1136/hrt.2010.208751. [DOI] [PubMed] [Google Scholar]
- 192.Khan M, Meduru S, Mostafa M, Khan S, Hideg K, Kuppusamy P. Trimetazidine, Administered at the Onset of Reperfusion, Ameliorates Myocardial Dysfunction and Injury by Activation of P38 Mitogen-Activated Protein Kinase and Akt Signaling. J Pharmacol Exp Ther. 2010;333:421–429. doi: 10.1124/jpet.109.165175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Mouquet F, Rousseau D, Domergue-Dupont V, Grynberg A, Liao R. Effects of trimetazidine, a partial inhibitor of fatty acid oxidation, on ventricular function and survival after myocardial infarction and reperfusion in the rat. Fundam Clin Pharmacol. 2010;24:469–476. doi: 10.1111/j.1472-8206.2009.00802.x. [DOI] [PubMed] [Google Scholar]
- 194.Martins GF, Siqueira Filho AG, de Santos JB, de F, Assunção CRC, Bottino F, Carvalho KG, de Valência A. Trimetazidine on ischemic injury and reperfusion in coronary artery bypass grafting. Arq Bras Cardiol. 2011;97:209–216. doi: 10.1590/s0066-782x2011005000079. [DOI] [PubMed] [Google Scholar]
- 195.Effect of 48-H Intravenous Trimetazidine on Short- and Long-Term Outcomes of Patients with Acute Myocardial Infarction, with and Without Thrombolytic Therapy. A Double-Blind, Placebo-Controlled, Randomized Trial. Eur Heart J. 2000;21:1537–1546. doi: 10.1053/euhj.1999.2439. [DOI] [PubMed] [Google Scholar]
- 196.Clarke B, Wyatt KM, McCormack JG. Ranolazine increases active pyruvate dehydrogenase in perfused normoxic rat hearts: evidence for an indirect mechanism. J Mol Cell Cardiol. 1996;28:341–350. doi: 10.1006/jmcc.1996.0032. [DOI] [PubMed] [Google Scholar]
- 197.Belardinelli L. Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart. 2006;92:iv6–iv14. doi: 10.1136/hrt.2005.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Gadicherla AK, Stowe DF, Antholine WE, Yang M, Camara AKS. Damage to mitochondrial complex I during cardiac ischemia reperfusion injury is reduced indirectly by anti-anginal drug ranolazine. Biochim Biophys Acta. 2012;1817:419–429. doi: 10.1016/j.bbabio.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Chandler MP, Stanley WC, Morita H, Suzuki G, Roth BA, Blackburn B, Wolff A, Sabbah HN. Short-term treatment with ranolazine improves mechanical efficiency in dogs with chronic heart failure. Circ Res. 2002;91:278–280. doi: 10.1161/01.res.0000031151.21145.59. [DOI] [PubMed] [Google Scholar]
- 200.Hale SL, Leeka JA, Kloner RA. Improved Left Ventricular Function and Reduced Necrosis After Myocardial Ischemia/Reperfusion in Rabbits Treated with Ranolazine, an Inhibitor of the Late Sodium Channel. J Pharmacol Exp Ther. 2006;318:418–423. doi: 10.1124/jpet.106.103242. [DOI] [PubMed] [Google Scholar]
- 201.Morrow DA, Scirica BM, Karwatowska-Prokopczuk E, Murphy SA, Budaj A, Varshavsky S, Wolff AA, Skene A, McCabe CH, Braunwald E. Effects of Ranolazine on Recurrent Cardiovascular Events in Patients With Non ST-Elevation Acute Coronary Syndromes The MERLIN-TIMI 36 Randomized Trial. J Am Med Assoc. 2007;297:1775–1783. doi: 10.1001/jama.297.16.1775. [DOI] [PubMed] [Google Scholar]
- 202.Liedtke AJ, Nellis SH, Whitesell LF. Effects of carnitine isomers on fatty acid metabolism in ischemic swine hearts. Circ Res. 1981;48:859–866. doi: 10.1161/01.res.48.6.859. [DOI] [PubMed] [Google Scholar]
- 203.Iliceto S, Scrutinio D, Bruzzi P, D’Ambrosio G, Boni L, Di Biase M, Biasco G, Hugenholtz PG, Rizzon P. Effects of L-carnitine administration on left ventricular remodeling after acute anterior myocardial infarction: the L-Carnitine Ecocardiografia Digitalizzata Infarto Miocardico (CEDIM) Trial. J Am Coll Cardiol. 1995;26:380–387. doi: 10.1016/0735-1097(95)80010-e. [DOI] [PubMed] [Google Scholar]
- 204.Tarantini G, Scrutinio D, Bruzzi P, Boni L, Rizzon P, Iliceto S. Metabolic Treatment with L-Carnitine in Acute Anterior ST Segment Elevation Myocardial Infarction. A Randomized Controlled Trial. Cardiology. 2006;106:215–23. doi: 10.1159/000093131. [DOI] [PubMed] [Google Scholar]
- 205.Dyck JRB, Lopaschuk GD. AMPK alterations in cardiac physiology and pathology: enemy or ally? J Physiol. 2006;574:95–112. doi: 10.1113/jphysiol.2006.109389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kim AS, Miller EJ, Wright TM, Li J, Qi D, Atsina K, Zaha V, Sakamoto K, Young LH. A small molecule AMPK activator protects the heart against ischemia-reperfusion injury. J Mol Cell Cardiol. 2011;51:24–32. doi: 10.1016/j.yjmcc.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Nadtochiy SM, Redman E, Rahman I, Brookes PS. Lysine Deacetylation in Ischaemic Preconditioning: The Role of SIRT1. Cardiovasc Res. 2011;89:643–649. doi: 10.1093/cvr/cvq287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Couzin-Frankel J. Aging Genes: The Sirtuin Story Unravels. Science. 2011;334:1194–1198. doi: 10.1126/science.334.6060.1194. [DOI] [PubMed] [Google Scholar]
- 209.Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, RVF, Alt FW, Kahn CR, Verdin E. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Scott I, Webster BR, Li JH, Sack MN. Identification of a molecular component of the mitochondrial acetyltransferase programme: a novel role for GCN5L1. Biochem J. 2012;443:655–661. doi: 10.1042/BJ20120118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Gustafsson ÅB, Gottlieb RA. Autophagy in Ischemic Heart Disease. Circ Res. 2009;104:150–158. doi: 10.1161/CIRCRESAHA.108.187427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning Involves Selective Mitophagy Mediated by Parkin and p62/SQSTM1. PLoS ONE. 2011;6:e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lefer DJ, Bolli R. Development of an NIH consortium for preclinicAl AssESsment of CARdioprotective therapies (CAESAR): a paradigm shift in studies of infarct size limitation. J Cardiovasc Pharmacol Ther. 2011;16:332–339. doi: 10.1177/1074248411414155. [DOI] [PubMed] [Google Scholar]
- 214.Gohil VM, Sheth SA, Nilsson R, Wojtovich AP, Lee JH, Perocchi F, Chen W, Clish CB, Ayata C, Brookes PS, Mootha VK. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol. 2010;28:249–255. doi: 10.1038/nbt.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Feuer L, Baráth P, Strauss I, Kékes E. Experimental studies on the cardiological effects of ipriflavone on the isolated rabbit heart and in rat and dog. Arzneimittelforschung. 1981;31:953–958. [PubMed] [Google Scholar]
- 216.Hansson MJ, Mattiasson G, Månsson R, Karlsson J, Keep MF, Waldmeier P, Ruegg UT, Dumont J-M, Besseghir K, Elmér E. The nonimmunosuppressive cyclosporin analogs NIM811 and UNIL025 display nanomolar potencies on permeability transition in brain-derived mitochondria. J Bioenerg Biomembr. 2004;36:407–413. doi: 10.1023/B:JOBB.0000041776.31885.45. [DOI] [PubMed] [Google Scholar]
- 217.Chambers DJ, Astras G, Takahashi A, Manning AS, Braimbridge MV, Hearse DJ. Free radicals and cardioplegia: organic anti-oxidants as additives to the St Thomas’ Hospital cardioplegic solution. Cardiovasc Res. 1989;23:351–358. doi: 10.1093/cvr/23.4.351. [DOI] [PubMed] [Google Scholar]