

Abstract
The eukaryotic cell replicates its chromosomal DNA with almost absolute fidelity in the course of every cell cycle. This accomplishment is remarkable considering that the conditions for DNA replication are rarely ideal. The replication machinery encounters a variety of obstacles on the chromosome, including damaged template DNA. In addition, a number of chromosome regions are considered to be difficult to replicate owing to DNA secondary structures and DNA binding proteins required for various transactions on the chromosome. Under these conditions, replication forks stall or break, posing grave threats to genomic integrity. How does the cell combat such stressful conditions during DNA replication? The replication fork protection complex (FPC) may help answer this question. Recent studies have demonstrated that the FPC is required for the smooth passage of replication forks at difficult-to-replicate genomic regions and plays a critical role in coordinating multiple genome maintenance processes at the replication fork.
Keywords: Timeless, Tipin, Swi1, Swi3, Tof1, Csm3, Fork protection complex, FPC, DNA replication, genome integrity, fork pausing, cohesion, checkpoint, difficult-to-replicate sites
Introduction
Eukaryotic DNA replication is rarely as easy as it looks in textbook cartoons. Genomic DNA exists in a crowded space where a cell must juggle many tasks, and where problems can easily arise. During S phase, the DNA replication machinery known as the “replisome” competes for access to template DNA with numerous DNA-binding proteins involved in various nuclear processes, including transcription, chromatin remodeling, sister chromatid cohesion (SCC) and DNA repair. Proteins involved in these nuclear transactions can block replisome progression, leading to replication fork arrest or even breakage, which, in turn, causes genomic instability, including mutations and chromosome rearrangements. To prevent these problems, DNA replication processes must accommodate multiple transactions occurring simultaneously on DNA. Such accommodation requires specialized replisome components that interact with chromatin itself as well as other chromatin-associated proteins.
One such factor is the replication fork protection complex (FPC). The FPC is comprised of two proteins: Timeless (also called Tim) and Tipin in metazoans, Swi1 and Swi3 in the fission yeast Schizosaccharomyces pombe, and Tof1 and Csm3 in the budding yeast Saccharomyces cerevisiae (Table 1). Timeless and Tipin protein levels are mutually dependent; Timeless downregulation leads to depletion of Tipin, and vice versa, indicating that Timeless and Tipin form an obligate heterodimeric complex.1-6 The FPC interacts with chromatin as well as many replisome components, including mini-chromosome maintenance (MCM) DNA helicase subunits, DNA polymerases δ and ε, replication protein A (RPA) and other ancillary factors.1-4,7-12 In both yeast and human cells, the FPC associates with replication origins at the onset of S phase and travels with the replisome during DNA replication.6,10,13 The functions of the FPC are far-reaching compared with other ancillary replisome components. Compelling evidence suggests the roles of the FPC in normal DNA replication, replication at difficult-to-replicate genomic regions, replication fork pausing/stalling, the DNA replication checkpoint and SCC. Neither protein has been characterized as an enzyme or has significant sequence homology to other proteins to inform us of their functions. Therefore, much of what is known about FPC functions comes from deletion/depletion experiments in which the consequences of FPC loss are observed. In this review, we summarize the current understanding of the roles of the FPC in the diverse pathways coordinated at the replication fork. In addition, we discuss what functions of the FPC make it central for a variety of processes at the replication fork in order to promote genome stability.
Table 1. Fork protection complex subunits.
| Metazoans | S. pombe | S. cerevisiae |
|---|---|---|
| Timeless |
Swi1 |
Tof1 |
| Tipin | Swi3 | Csm3 |
The FPC Supports Checkpoint Activities
The FPC proteins were first functionally characterized in fission yeast, where swi1 and swi3 were isolated in a screen for mutants unable to generate a double-strand break during mating-type switching.14 Later, budding yeast FPC protein Tof1 was implicated in processes seemingly unrelated to mating-type switching. Tof1 (topoisomerase I-interacting factor 1) was named for its interactions with topoisomerase I, both in the yeast two-hybrid assay and in vitro.15 This interaction provided clues that the FPC proteins may be involved in general genome maintenance mechanisms. The following studies highlighted the roles of the FPC proteins in the DNA replication checkpoint, the master signaling pathway responsible for recognizing problems during DNA replication at the fork, in order to arrest the cell cycle and allow time for DNA repair. To identify novel genes in the S-phase-specific checkpoint pathway, a synthetic lethal screen was performed in budding yeast, isolating mutants that conferred additional sensitivity to a rad9 mutant in response to the DNA alkylating agent, methyl methanesulfonate (MMS).16 Rad9 activates the DNA damage checkpoint throughout the cell cycle. This synthetic lethal screen was performed based on the observation that cells missing both cell cycle-wide and S-phase-specific pathways have stronger defects in checkpoint activation. Importantly, this screen identified tof1 mutations, which rendered cells highly sensitive to MMS when combined with a rad9 mutation. tof1 mutants also showed sensitivity to other S-phase stressing agents, including hydroxyurea (HU) and UV light (UV), suggesting the role of Tof1 in the S-phase-specific checkpoint pathway.16 Indeed, the same study showed that loss of both Rad9 and Tof1 (but not the single mutations) inhibits HU-dependent phosphorylation and activation of Rad53,16 the budding yeast master checkpoint kinase.17 Similarly, Swi1, an S. pombe FPC subunit, has a role in activation of the DNA replication checkpoint. In the absence of Swi1, cells are defective for full activation of the replication checkpoint kinase Cds1, the S. pombe ortholog of Rad53.18,19 Subsequently, biochemical studies identified Swi3 as a binding partner for Swi1.6,20 swi3∆ cells also have decreased Cds1 kinase activity in response to HU, indicating that both FPC proteins are required for checkpoint activation in response to replication stress.6
Although the DNA replication checkpoint pathway is somewhat different in metazoans, the role of FPC in supporting checkpoint signaling is similar to what is known in yeast systems. In metazoans, the ATR (ataxia-telangiectasia mutated and Rad3-raleted)-ATRIP (ATR-interacting protein) kinase phosphorylates and activates the Chk1 kinase to arrest the cell cycle in response to replication stress.21 In studies using human cells, cellular sensitivity to genotoxic agents (such as HU, UV, gamma irradiation and camptothecin) increases in the absence of the FPC. When Timeless or Tipin protein levels are knocked down using siRNA, Chk1 phosphorylation (and thus its activation) is reduced in response to HU or UV.1-4,13,22 In Xenopus laevis, depletion of Tipin from cell extracts also reduces Chk1 phosphorylation in response to aphidicolin, a DNA replication inhibitor.8 However, some increases of Chk1 phosphorylation occur after loss of Timeless in the absence of exogenous genotoxic agents.1,4,23 These results suggest that while FPC is required for full activation of replication stress-induced Chk1 activation; the loss of FPC results in accumulation of unusual DNA structures that promote an increase of basal ATR-dependent Chk1 activity in the absence of exogenously provoked replication stress.
These results suggest a role for the FPC in mediating checkpoint signaling in response to replication stress (Fig. 1). It is known that single-stranded DNA (ssDNA) generated at the replication fork recruits ATR-ATRIP to phosphorylate Chk1.24 It is also understood that Claspin, a checkpoint mediator, is required for Chk1 phosphorylation.25,26 Therefore, to efficiently activate checkpoint signaling, it is important to recruit all the required proteins to ssDNA at the replication fork. Biochemical studies have revealed that Tipin has such a role. Tipin binds RPA34, a subunit of the ssDNA-binding factor RPA.1,3,27 This interaction is important for localization of the FPC to ssDNA at the replication fork.28 In addition, Tipin-Claspin complexes have been detected by immunoprecipitation, and a Tipin mutant unable to bind RPA is no longer able to recruit Claspin to RPA, indicating that the Tipin also recruits Claspin to the replication fork.28 Consistently, Claspin localization to the nucleus is severely reduced in the absence of the FPC proteins in human cells, further suggesting that the FPC is required for Claspin recruitment.4 Therefore, it appears that the role of Tipin in mediating checkpoint activation is to act as a scaffold at the replication fork, recruiting all players at the RPA-ssDNA complexes to promote Chk1 activation (Fig. 1).
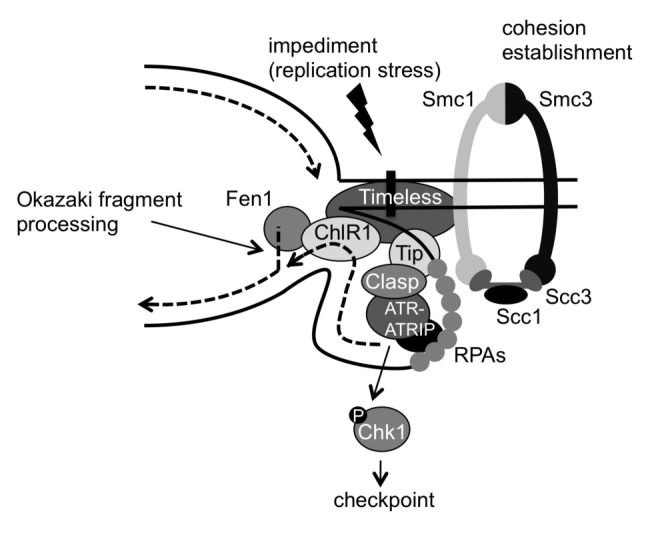
Figure 1. Model for the roles of the fork protection complex (FPC) in checkpoint activation, DNA replication and sister chromatid cohesion. The replication fork can pause or stall at a variety of impediments throughout the genome. These impediments may include DNA damage, fork barriers, DNA secondary structures, proteins bound on DNA and even cohesin complexes. ssDNA accumulated at stalled forks is coated by RPA bound by ATR-ATRIP. RPA is also bound by Tipin, which then recruits the checkpoint mediator Claspin, resulting in activation of the ATR-Chk1-dependent checkpoint. Tipin also recruits Timeless, which stabilizes cohesin subunits on chromatin. Timeless interacts with ChlR1 DNA helicase, which stimulates Fen1 activity for Okazaki fragment processing. This promotes efficient lagging-strand DNA synthesis and minimizes the size of the replication loop at the lagging strand, thereby promoting efficient progression of replication forks through cohesin rings.
What, then, is the role of Timeless, the partner of Tipin? Timeless is also required for Chk1 activation in response to replication stress.1,4,22 Because Timeless depletion causes Tipin downregulation,2,4 Timeless may be required for stabilization of Tipin. However, once recruited to the replication fork, Timeless may have Tipin-independent roles, as Timeless depletion causes defects in a variety of genome maintenance processes, as discussed later in this review.
Another important activity of S-phase checkpoints is to inhibit firing of late replication origins in response to replication stress. In budding yeast, Rad53-dependent phosphorylation of Dbf4 blocks late-origin firing after DNA damage.29,30 Dbf4 interacts with the Cdc7 kinase to form the DDK complex, which is required for loading of Cdc45 and other replication factors just before an individual replication origin fires.31 In addition, DDK complex functions throughout S-phase to fire origins.32 Interestingly, the fission yeast FPC functionally interacts with the Hsk1-Dfp1 complex,11 the fission yeast ortholog of the DDK complex.33,34 Loss of functional Hsk1 sensitizes cells to S-phase-stressing agents including HU and MMS. In addition, hsk1 mutant cells have defects in arresting S-phase progression in response to MMS, and this effect is epistatic with FPC mutations.11,35,36 These data suggest that FPC-DDK interaction mediates origin inhibition in response to DNA damage, although further investigation is necessary to mechanistically understand the role of FPC in origin inhibition.
Roles of the FPC in Replisome Progression
Studies have indicated that replication fork progression is generally compromised in the absence of the FPC. One of the quantitative strategies to measure fork progression rate is the “DNA combing method,” which allows for uniformly stretching DNA fibers that are then examined by fluorescent microscopy.37 Using this method, budding yeast tof1∆ cells were found to contain shorter replication tracks when compared with wild type cells.38 In addition, tof1∆ cells take approximately 20 min longer to complete DNA replication than wild type cells, suggesting the slower rate of replication in tof1∆ cells.38 Similarly, DNA fiber analysis using human cells demonstrated that Timeless depletion also generates shorter tracks of nascent DNA synthesis.1 These results suggest that the FPC proteins promote efficient replisome progression by stabilizing replication fork structures and replisome components during DNA replication throughout the genome. However, numerous genomic regions are difficult to replicate, which causes pausing of replication forks.39 As described in the following section, the FPC has a critical role in stabilizing replication forks and suppressing DNA damage at these difficult-to-replicate regions during normal DNA replication. Therefore, we suggest that FPC depletion causes more frequent DNA breakage at difficult-to-replicate regions, resulting in the appearance of shorter replication tracks.
Indeed, the FPC is required for stable fork pausing at rDNA fork block sites in budding yeast (Fig. 2B). Tof1 and Csm3 remain with the replisome at the fork block sites, maintaining the replisome complex.7 FPC-dependent fork pausing and stabilization is independent of checkpoints, as Rad53 (Chk1/Chk2) and Mec1 (ATR) are dispensable for replisome pausing.7 These data suggest that continuous FPC association with the replisome during replication fork pausing is required to stabilize the replisome. The data also suggest that the FPC stabilizes the forked DNA to prevent the formation of unusual DNA structures that induce DNA repair pathways for resolution. Pausing defects could lead to a temporal increase of replication fork velocity in the presence of fork-blocking agents including HU. Indeed, ChIP-on-chip analysis (chromatin immunoprecipitation with microarray analysis) of replication proteins suggests that the replication fork migrates faster in FPC-deficient cells than in wild type cells upon HU treatment.10 This study showed that Cdc45 localization was coupled with replicated regions represented by BrdU incorporation in HU-treated wild type budding yeast cells. However, in the absence of Tof1, Cdc45 moved 2.5 to 3.0 kb further from replicated regions in response to HU.10 Cdc45 complexes with GINS (go ichi ni san) and MCM subunits (Mcm2 to 7), which unwinds duplex DNA at the replication fork.9 Therefore, the ChIP-on-chip results indicate that DNA unwinding is “uncoupled” from the site of DNA synthesis in tof1∆ cells. In addition, this uncoupling leads to extensive exposure of ssDNA at the replication fork and subsequent activation of DNA damage checkpoints. Accordingly, a checkpoint mediator Rad9 was recruited to replicating chromatin in tof1∆ cells but not in wild type cells.10
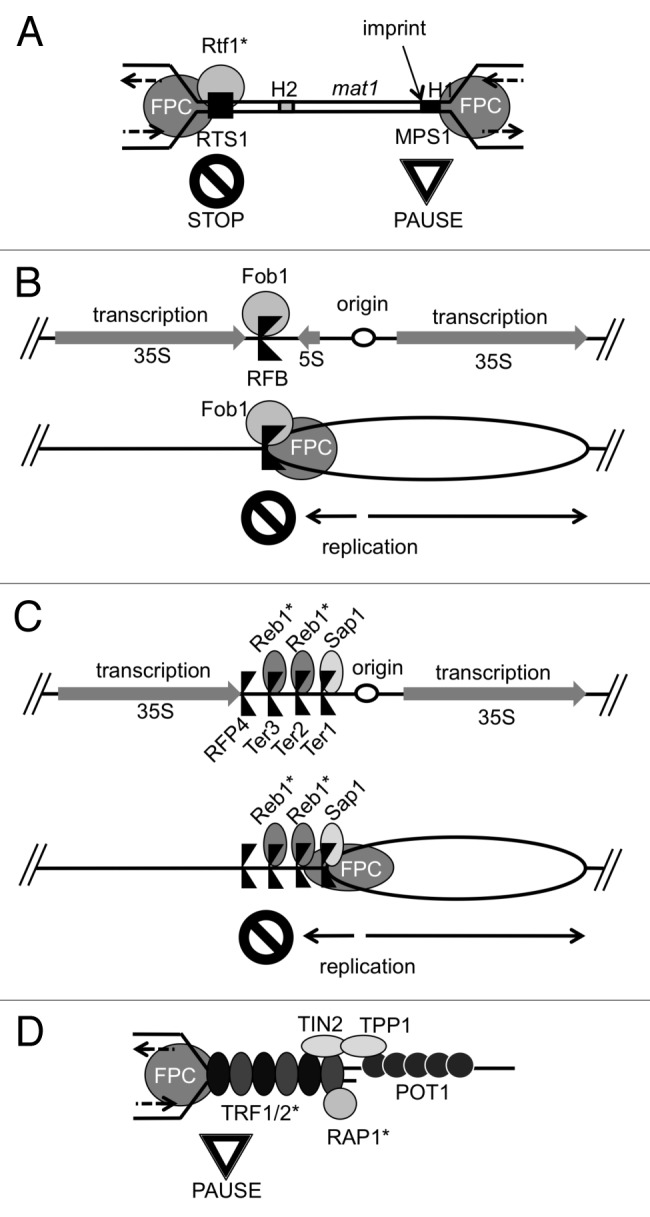
Figure 2. Roles of the fork protection complex (FPC) at various fork barriers in the genome. (A) The S. pombe mating-type (mat1) locus contains two distinct polar fork block sites. A Myb factor, Rtf1 cooperates with the FPC to promote polar fork arrest at RTS1. FPC-dependent fork pausing at MPS1 is required to generate an imprint to initiate a gene conversion event that leads to mating-type switching. (B) The S. cerevisiae rDNA locus contains an RFB that is bound by Fob1. Fob1 functions together with the FPC to inhibit fork progression in the direction that is opposite to the transcriptional direction of the 35S rRNA gene. This polar fork block allows the replication fork to move only in the direction of transcription. (C) The S. pombe rDNA locus contains three fork block sites that are dependent on the FPC. The Myb factor Reb1 binds Ter2 and Ter3 to promote polar fork arrest, while the Sap1 protein binds Ter1 for fork arrest. Fork arrest at RFP4 is not dependent on the FPC and is regulated by transcription.60 (D) Metazoan telomeres contain Myb factors, TRF1 and TRF2, which protect telomeres. Timeless interacts with TRF1 and TRF2 to promote efficient DNA replication through telomeres in human cells. TRF2 binds RAP1, which also contains a Myb DNA-binding domain. *Asterisks indicate proteins containing the Myb DNA-binding domains.
Such replication fork uncoupling gives rise to deleterious consequences at the replication fork. In fission yeast, swi1 deletion results in replication fork collapse near the rDNA replication pausing site, leading to accumulation of recombination structures, including Holliday junctions, as visualized by two-dimensional gel electrophoresis (2D-gel) of chromosomal DNA.6,19 The formation of Holliday junction structures are indicative of replication fork collapse and rearrangement, and these collapsed replication structures correlate with an increase in Rad22 DNA repair foci in swi1Δ and swi3Δ nuclei, even in the absence of exogenous DNA-damaging agents.6,19 Rad22 is the S. pombe ortholog of Rad52 recombinase that localizes to sites of DNA damage.40,41 Elevated levels of Rad22 foci indicate a higher level of DNA damage, indicating that Swi1 prevents replication fork collapse. Similarly, Timeless-depleted mouse cells accumulate ssDNA at the fork and DNA damage foci containing Rad51 and Rad52 recombinases. The increase of DNA damage foci correlated with elevated levels of sister chromatid exchange, which is an indication of a DNA repair process that utilizes sister-chromatids for homologous recombination.42 Taken together, these finding suggest that the FPC has a conserved role in preventing recombinogenic DNA damage at the replication fork during genotoxic stress and unperturbed DNA replication.
Roles of the FPC at Replication Fork Barriers
Replication checkpoint studies have typically used chemical agents to stall replication forks. However, there are a number of chromosome regions that present obstacles for DNA replication. These include replication fork blocking sites, DNA secondary structures caused by repeat sequences (such as telomeres) and DNA-binding proteins (such as the transcription machinery). These sites are considered to be difficult to replicate, causing replication fork arrest or even breakage during normal DNA replication.39,43-45 The difficult-to-replicate genome regions can be divided into two classes. The first class includes protein-DNA barriers, where the replication fork pauses to control the direction of fork progression and to coordinate replication pausing with other processes on chromatin. These can be considered “programmed” pausing sites. Pausing at these sites is often regulated by protein-protein interactions.46,47 The second type of replication stoppage occurs at repetitive DNA, where fork stalling occurs as a result of the inherent properties of DNA repeats. Stalling at these sites is induced by the higher propensity of short repeat sequences to form hairpins and other secondary structures. Such DNA structures can cause repeat contraction and expansion, resulting in negative consequences, including human neurodegenerative diseases.48,49 Therefore, cells must possess mechanisms to ensure proper replication fork pausing and the smooth passage of replication forks through difficult-to-replicate regions. Interestingly, the FPC appears to play a critical role in these processes.
One well-characterized fork-pausing event involves mating-type switching in fission yeast (Fig. 2A). Two genetically programmed fork pausing sites are found near the mating-type (mat1) locus: the mat1 replication pause site 1 (MPS1) and the replication termination site 1 (RTS1). A strong polar replication pausing at RTS1 prevents one replication fork from migrating into the mat1 locus. This allows only the opposing replication fork to enter the mat1 locus.50 This fork pauses at MPS1 and generates an imprint, probably a DNA strand discontinuity, which initiates a specific recombination event, resulting in mating-type switching (Fig. 2A).51,52 In the absence of Swi1 or Swi3, the replication fork fails to pause at these sites; consequently, swi1∆ and swi3∆ cells are defective for mating-type switching.50 Interestingly, RTS1 is bound by Rtf1, a protein containing the Myb DNA-binding domain, and rtf1 deletion also causes loss of fork pausing at RTS1 (Fig. 2A),53 suggesting that the FPC works together with Rtf1 to pause the fork at RTS1.
Another well-studied replication pausing occurs at rDNA arrays and is dependent on the FPC (Fig. 2B). Budding yeast chromosome XII contains approximately 150 repeats of the rDNA unit, each of which contains two transcribed regions: 35S and 5S rRNA genes. Each repeat also contains a replication origin and a replication fork barrier (RFB) upstream and downstream of the 35S rRNA gene, respectively. A site-specific DNA-binding protein Fob1 binds RFB and blocks the fork that moves in the direction opposite to 35S rRNA transcription; thus, Fob1 causes polar fork arrest (Fig. 2B). In addition, FPC subunits Tof1 and Csm3 allows for stable replication fork pausing at RFBs by protecting Fob1 from Rrm3 helicase, which displaces Fob1 to promote fork progression.54 This polar fork block arrangement prevents replication fork progression in the direction opposite to the direction of transcription, preventing head-on collisions of the replisome and the transcription machinery (Fig. 2B).55,56
In S. pombe, a Fob1-like protein has not been identified. However, similar fork blocking strategies are used to coordinate the directionality of replication and transcription at rDNA repeats (Fig. 2C). The S. pombe rDNA unit has four polar fork barriers, three of which are dependent on the FPC. The Sap1 protein, which recognizes the switch-activating site (SAS1) near the mating-type locus, binds the Ter1 sequence to cause polar fork arrest in collaboration with the FPC.57,58 Interestingly, the Ter1 sequence is highly similar to the SAS1 sequence, indicating that Sap1 is a sequence-specific DNA-binding protein. Two additional FPC-dependent fork barriers within the S. pombe rDNA repeat are Ter2 and Ter3. Reb1, which is the S. pombe ortholog of the mammalian transcription termination factor-1 (TTF-1), binds Ter2 and Ter3 and causes polar fork arrest (Fig. 2C).59,60 Reb1 contains the Myb DNA-binding domain, and a similar fork block mechanism is reported in mammalian cells; HeLa cell extracts depleted of TTF-1 fail to exhibit fork block activity at the Sal boxes of the rDNA repeat.61 Interestingly, mammalian TTF-1 and fission yeast Reb1 also mediate termination of transcription by RNA polymerase I (RNA Pol I) at the Sal boxes and Ter2/3 sites, respectively, although budding yeast Reb1 has no apparent role in fork blocking activity.61-67 Nevertheless, replication arrest and transcription termination appear to share similar mechanisms. Although whether the TTF-1-dependent fork block requires the FPC is unknown, fork arrest at the S. pombe Ter2 and Ter3 sites is dependent on the FPC proteins (Fig. 2C). Therefore, cooperation of the FPC and Myb factors likely promotes replication fork pausing at specific sites in the genome.
Other FPC-mediated replication fork pausing events, which are also associated with protein-DNA interaction, also occur frequently during DNA replication. In S. cerevisiae, replication forks pause at tRNA and highly transcribed RNA polymerase II (Pol II) genes.68,69 Such pausing events appeared to be dependent on the FPC, because 2D-gel analyses detected pausing events at tRNA sites only in the presence of Tof1.70 Researchers have suggested that high rates of transcription and the presence of the transcription machinery increase the chances of the replisome encountering an RNA transcription fork. Analyses of replication through budding yeast tRNA genes indicate that replication forks pause at these sites for approximately 10 sec, roughly four times longer than replisome transit of a similarly sized region.71 Owing to the abundance of pausing sites, replication forks must be constantly stabilized throughout the genome (Fig. 3). In fission yeast, the Pfh1 DNA helicase is required for the replisome to replicate through highly transcribed RNA Pol II and RNA Pol III genes.72 In pfh1 mutants, paused replication forks accumulate at these genes, which would then require the function of the FPC to stabilize them. Indeed, swi1 deletion is synthetically lethal with pfh1 mutations.72 These results indicate that highly transcribed genomic regions are difficult to replicate, and that the FPC plays a critical role in stabilization of paused replication forks at these genomic regions (Fig. 3).

Figure 3. Local requirements of the fork protection complex (FPC) throughout the genome. The FPC travels with the replication fork, monitoring and detecting problems during DNA replication. The FPC coordinates DNA replication at a variety of difficult-to-replicate regions, including cohesin sites, highly transcribed regions, fork block sites, secondary structures and telomeres. Many difficult-to-replicate regions may be bound by Myb factors.
The FPC Stabilizes Replication Forks at DNA Repeats
In addition to DNA-protein complexes, DNA structures themselves can cause fork pausing or arrest. It is widely thought that repeat DNA sequences form secondary structures that are associated with genomic instability.49 Emerging evidence indicates that FPC proteins prevent genomic instability at these “non-programmed” sites.73 However, at these sites, the FPC proteins play a different role in regulating stalled forks. Instead of allowing for stable pausing, the FPC appears to counteract replication fork stalling mediated by DNA structures, such as hairpins. When inverted Alu repeats are inserted into the budding yeast genome, replication fork stalling at the Alu repeats nearly doubles in the absence of Tof1.74 This result is consistent with the notion that the FPC helps to move replication forks forward by minimizing stalling events at DNA structure-mediated fork block sites.
Increases in replication fork stalling is thought to contribute to expansion of trinucleotide repeats,75 and a number of human diseases are associated with abnormal replication of repeat regions. In human cells, CGG repeat expansion leads to an increased propensity to form secondary structures, which is thought to lead to DNA replication mediated breakage, causing fragile X syndrome.49,76 In budding yeast tof1Δ cells, the rate of fork stalling at inserted CGG repeats increases compared with wild type strains. The increase in fork stalling could lead to repeat expansion. Importantly, the lagging-strand folds back to form a loop structure at the fork (Fig. 1). Owing to the presence of repeat DNA sequences, once the replisome stalls at repeat regions, the replisome might continue DNA synthesis by switching the template to the lagging-strand loop, which is located upstream of the stalled site. This results in over-replication and expansion of the repeat region.77 Consistently, when GAA repeats were inserted into the budding yeast genome, the repeats expanded at a rate corresponding to their original length in a manner dependent on DNA replication (the longer the repeats, the faster the expansion). Importantly, this expansion is accelerated in the absence of Tof1.78
Similar genomic instability in the absence of the FPC is reported in human cells. In humans, the expansion of a particular (CTG)n·(CAG)n repeat tract on chromosome 19 is associated with myotonic dystrophy type 1 (DM1), a form of muscular dystrophy. In a tissue culture model, Timeless or Tipin depletion enhances DM1-linked repeat expansion.79 FPC proteins appear to prevent or reduce replication fork stalling when the replication fork is blocked by DNA secondary structure. Fork stalling could result in elevated levels of ssDNA exposed at the replication fork upon FPC dysfunction.10,23 Existence of longer ssDNA may further increase the risk of secondary structure formation and fork stalling. Currently, whether the FPC promotes replication of repeat regions by interacting with other proteins is unknown. However, the mechanism of the FPC-dependent fork regulation at repeat regions appeared to be different from that at protein-DNA complex-mediated fork barriers.
Telomere Replication and the FPC
An interesting example of FPC-dependent fork protection against site-specific DNA damage is the telomere. At the telomere, the replication fork encounters protein-DNA complexes as well as repetitive DNA regions. For that reason, the replisome must deal with both protein-DNA complex-mediated fork barriers and DNA structure-based impediments. Indeed, telomeres are difficult to replicate, and replication forks stall at telomere repeats.80,81 Interestingly, telomeres also recruit proteins that contain the Myb DNA-binding domain. In fission yeast, a Myb factor, Taz1, binds specifically to telomere repeats and promotes efficient replication through telomeres.81 In metazoans, Taz1 orthologs, TRF1 and TRF2, also contain the Myb DNA-binding domain and bind telomere repeat DNA sequences (Fig. 2D).82 In mouse cells, TRF1 prevents telomere damage foci generated during S-phase, and TRF1 deletion lowers the efficiency of DNA replication, as visualized by DNA combing.83 Conversely, Ohki and Ishikawa showed that overexpression of TRF1 or TRF2 increases fork stalling specifically at telomere repeats in vivo and in vitro.84 Although these results contradict each other, they suggest that TRF1 and TRF2 prevent replication fork collapse at telomeres, and that maintaining the quantity of these proteins at telomeres is required for efficient telomere replication. Considering that Taz1 and TRF1/2 are Myb-related proteins, molecular mechanisms required for replication of telomeres may be similar to those required for replication through other protein-DNA barriers, including the fission yeast mat1 locus and rDNA barriers in fission yeast and mammalian cells (Fig. 2A and C). In many cases, Myb-related proteins associate tightly with barrier regions, and the FPC is required for stable fork pausing and prevention of fork collapse at the barrier regions.
Compelling evidence suggests that the FPC is required for efficient replication of telomeres. In yeast, loss of Timeless orthologs leads to contrasting telomere length phenotypes; budding yeast tof1Δ cells contain extended telomeres while swi1∆ cells in fission yeast undergo telomere shortening.85-87 The differences may be attributed to the fact that fission yeasts express a Myb-DNA binding domain-containing protein, Taz1, whereas S. cerevisiae has no apparent Taz1 ortholog.88 Indeed, in human cells, in which Taz1 orthologs (TRF1/2) exist, long-term knockdown of Timeless causes telomere shortening.86 These findings suggest that the FPC regulates replication of telomeric DNA sequences that are bound by Myb factors. Consistently, immunoprecipitation studies in human cells demonstrated that Timeless interacts with TRF1 and TRF2.86 Studies have also shown that Timeless-depleted cells fail to maintain TRF1-mediated replication fork arrest.86 These results suggest that Timeless works together with TRF1 to promote efficient replication of telomeres. However, Timeless-TRF1 interaction may not be the only FPC role in telomere replication. In the same study, in vitro replication assays also demonstrated that the loss of Timeless reduces replication efficiency on naked telomeric DNA, suggesting that the FPC may be involved in replisome stability through repeats independent of interaction with TRF1.86 Therefore, Timeless may have two roles during telomere replication: (1) stabilization of the replisome through DNA structure-based impediments, and (2) specific interaction with TRF1 to promote proper fork pausing where TRF1 binds telomeres. Therefore, telomeres may be unique loci where two distinct FPC-dependent stabilization mechanisms are required.
Coordinating Cohesion Establishment with Replication
Seemingly disparate from its role in checkpoint activation, the FPC plays a role in the establishment of SCC during DNA replication. The establishment of SCC requires DNA replication in S-phase.89,90 During DNA replication, newly copied chromosomes are held in close association by a functional complex of proteins known as cohesins. The cohesin complex ensures that sister chromosomes are paired until they can be separated equally during mitosis. Smc1 and Smc3 (structural maintenance of chromosomes 1 and 3) are conserved cohesin proteins with long flexible linker domains thought to encircle one or both copies of DNA.91-93 They are held in a ring-like complex by the non-SMC subunits, Scc1 and Scc3 proteins (Fig. 1).94-96 Although the mechanism by which the cohesins promote SCC is unclear, stable loading of cohesins onto DNA evidently requires acetylation of Smc3 by the conserved Eco1/Ctf7 acetyltransferase (known as ESCO1/2 in humans).97-99 Proper SCC is important for cellular health and viability, as mutations in cohesion pathways cause cohesinopathies such as Cornelia de Lange and Roberts syndromes characterized by multisystem developmental defects.100-102
Earlier studies implicated the role of FPC proteins in chromosome cohesion and segregation. In an effort to understand the mechanism of meiotic chromosome segregation, 301 meiotic gene-deletion mutants were screened for chromosome missegregation. This screen identified Csm3, the budding yeast Tipin ortholog, as a factor required for proper chromosome segregation in meiosis. csm3 deletion caused mild chromosome missegregation and reduced spore viability, suggesting a possible role of Csm3 in chromatid pairing.103 In Caenorhabditis elegans, Meyer and colleagues used a biochemical approach to understand the cohesion mechanism.104 They performed immunoprecipitation of SMC-1 (an Smc1 ortholog) using C. elegans embryonic extracts. Subsequent mass spectrometry analysis identified TIM-1 (Timeless ortholog) as an SMC-1-interacting factor. TIM-1 depletion or mutation was found to cause defects in meiotic chromosome cohesion and segregation. This study also reported that TIM-1 is required for the loading or stability of non-SMC subunits of the cohesin complex. These findings suggested the involvement of TIM-1 in meiotic SCC and segregation.104
How does the FPC promote cohesion processes? Yeast genetic studies demonstrated a strong link between DNA replication and cohesion processes. Several replication factors including Ctf4 (DNA polymerase α-binding factor) and RFCCtf18 (alternative replication factor C-like complex) are involved in efficient SCC in budding yeast.105-108 To identify additional cohesion factors, synthetic lethal screens were performed using ctf4 and ctf8 (a subunit of RFCCtf18) mutations. These studies revealed that both tof1∆ and csm3∆ were synthetically lethal with ctf4∆ or ctf8∆. They also found that Tof1 and Csm3 were required for SCC and that Tof1 co-purifies with Csm3 from budding yeast cell extracts.5,109 In fission yeast, a synthetic lethal screen was performed using swi1 mutation to identify additional factors involved in replication fork maintenance. This screen identified RFCCtf18. Consistently, swi1∆ and swi3∆ cells displayed premature centromere dissociation during metaphase.110 Considering that FPC subunits (Tof1/Swi1–Csm3/Swi3) are replication fork constituents,3,6,9,10 these genetic data confirmed the role of replication fork processing in the establishment of SCC. Furthermore, because the FPC is involved in stabilization of replication forks,6,13 it is possible that FPC proteins coordinate DNA synthesis with SCC established at the replication fork.
Using molecular biology approaches in vertebrate model systems, investigators have also detected physical defects in SCC upon FPC disruption. Xenopus egg extracts depleted of Timeless or Tipin fail to pair sister chromatids in a manner similar to Smc3 depleted extracts, indicating that loss of Xenopus Timeless-Tipin induces an SCC defect.12,111 Interestingly, in Xenopus egg extracts, Timeless interacts with And1, a Ctf4 homolog and supports stable chromatin binding of DNA polymerase α and DNA replication under minimal licensing conditions.111 In human epithelial cells, Timeless or Tipin depletion via RNAi also causes a substantial increase in SCC defects.13,112 A recent study using human fibroblasts found that SCC defects caused by Timeless depletion are 10 times greater than those caused by Tipin deletion, suggesting that Timeless but not Tipin has a specific role in SCC establishment.113 Mechanistically, cohesin subunits, including Smc1 and Smc3, coimmunoprecipitate with Timeless or Tipin in human cell extracts. Timeless-cohesins interaction was maintained after DNase treatment, whereas Tipin-cohesins interaction was disrupted. This is consistent with the notion that Timeless is closely associated with the cohesin complex (Fig. 1). Moreover, Timeless depletion led to dissociation of cohesin subunits from chromatin, suggesting that Timeless is required for stable association of cohesins with chromatin.13
Additional evidence supports the notion that the FPC facilitates SCC through protein-protein interactions. The aforementioned synthetic lethal screens revealed that deletion of the Chl1 DNA helicase is synthetically lethal with ctf8 deletion.5 Chl1 (ChlR1 in humans) is a DEAH/DEAD box containing DNA helicase belonging to the FANCJ-like DNA helicase family.114 Chl1/ChlR1 downregulation causes SCC defects, and a mutation in the human gene causes a cohesinopathy-related disease termed Warsaw breakage syndrome.115-119 Compelling evidence suggests that the FPC cooperates with Chl1/ChlR1 to promote SCC. In human cells, Timeless coimmunoprecipitates with ChlR1; Timeless is involved in loading of ChlR1 onto chromatin; and ChlR1 overexpression partially rescues SCC defects due to Timeless or Tipin depletion.13 In fission yeast, Chl1+ overexpression rescues the HU and MMS sensitivity of swi1Δ, while genetic studies in both budding and fission yeast suggest that Swi1 and Chl1 function in the same pathway.110,120
What, then, is the mechanism of FPC-ChlR1-dependent SCC establishment? ChlR1 interacts with cohesin subunits and Fen1.121 Fen1 is a flap endonuclease required for Okazaki fragment processing during lagging-strand DNA synthesis.122 Importantly, ChlR1 enhances Fen1 activity in vitro, suggesting that lagging-strand processing may affect the efficiency of cohesion. Consistently, Fen1 depletion also causes cohesion defects.121 These results support the idea that ChlR1 promotes SCC by facilitating efficient lagging-strand processing. Considering that the level of chromatin-bound ChlR1 is reduced in Timeless depleted cells,13 it is possible that Timeless effectively recruits ChlR1 to promote lagging-strand synthesis (Fig. 1). Consistently, Timeless downregulation leads to ssDNA accumulation in human cells,23 and yeast genetic studies have suggested the role of the Timeless ortholog Swi1 in coordination of leading- and lagging-strand DNA synthesis to prevent accumulation of ssDNA.6,36 Delayed lagging-strand synthesis causes a long stretch of ssDNA, generating a large loop structure at the replication fork, which may hamper replication fork progression through the cohesin ring (Fig. 1).123,124 Therefore, efficient lagging-strand processing may have a critical role in SCC establishment. Interestingly, a recent study showed that Fen1 interacts with Chl1 and the cohesin aceyltransferase Eco1/Ctf7 in budding yeast, suggesting that SCC may be coupled with lagging-strand synthesis.125
It has been suggested that the replication fork pauses at cohesin-loaded sites.126,127 This pausing would generate ssDNA at the lagging strand of the fork, leading to recruitment of RPA. Tipin then binds RPA, tethering Timeless to the lagging strand at the replication fork (Fig. 1). We suggest that a division of labor of Timeless and Tipin occurs once the Timeless-Tipin complex is localized to the replication fork.128 Tipin may transmit the signal of ssDNA accumulation to activate the DNA replication checkpoint. Timeless modulates ChlR1 and Fen1 functions to facilitate lagging-strand synthesis, while Timeless also holds cohesin subunits on chromatin via protein-protein interaction, which, in turn, coordinates lagging-strand synthesis with SCC (Fig. 1). It is also possible that Timeless-dependent stabilization of cohesin subunits creates a favorable environment for Smc3 acetylation by Eco1/Ctf7, although further studies are needed to address this question. Nevertheless, current evidence suggests a critical link between lagging-strand synthesis and cohesion processes.
Concluding Remarks and Future Directions
The pleiotropic phenotypes that are caused by FPC deficiency are all associated with the functions of the FPC at the replication fork. The FPC interacts with a number of proteins, including RPA, Claspin, TRF1/2, Fob1, ChlR1, cohesin and core replisome components. These findings suggest that the FPC acts as a platform for multiple proteins to regulate a variety of transactions at the replication fork. During DNA replication, the replisome encounters numerous impediments, where DNA resident proteins are localized (Fig. 3). Many such DNA resident proteins may contain the Myb DNA-binding domain and regulate stable pausing of the replisome to coordinate DNA replication with other processes that take place on chromatin. However, the replisome must contain factors that interact with the Myb-containing proteins and recognize the problems at various challenging sites throughout the genome. The FPC appears to be the perfect candidate for this job. In this model, the FPC moves with the fork and detects and reconciles problems during DNA replication throughout the genome, stabilizing the replisome while also facilitating proper checkpoint signaling if necessary (Figs. 1 and 3). In the future, it will be interesting to see how genome-wide studies identify DNA damage sites in the absence of the FPC. We anticipate that many of DNA damage sites contain resident proteins that interact with the FPC. From the findings described above, it is also anticipated that many of these resident proteins will contain Myb DNA-binding domains. Future molecular and proteomic studies may identify various interactions between the FPC and other DNA-binding proteins. These interactions can be interrupted genetically or pharmacologically, and loci-specific phenotypes can then be assessed. Such studies would reveal how the FPC locally regulates DNA replication in each case. However, because a vast number of genomic sites require the FPC, we deduce that the FPC controls DNA replication globally.
Acknowledgments
We thank Michelle Villasmil and Esteban Martinez for helpful comments and suggestions on this manuscript. This work was funded by the NIH (F31AG035480 to A.R.L and R01GM077604 to E.N.).
Glossary
Abbreviations:
- 2D-gel
two-dimensional gel electrophoresis of chromosomal DNA
- ATR
ataxia-telangiectasia mutated- and Rad3-related
- ATRIP
ATR-interacting protein
- ChIP-on-chip
chromatin immunoprecipitation with microarray analysis
- FPC
fork protection complex
- HU
hydroxyurea
- MCM
mini-chromosome maintenance
- MMS
methyl methanesulfonate
- MPS1
mat1 replication pause site 1
- RFB
replication fork barrier
- RFC
replication factor C
- RPA
replication protein A
- RTS1
replication termination sequence 1
- SAS1
switch-activating site
- SCC
sister chromatid cohesion
- ssDNA
single-stranded DNA
- UV
ultraviolet
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/21989
References
- 1.Unsal-Kaçmaz K, Chastain PD, Qu P-P, Minoo P, Cordeiro-Stone M, Sancar A, et al. The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol Cell Biol. 2007;27:3131–42. doi: 10.1128/MCB.02190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chou DM, Elledge SJ. Tipin and Timeless form a mutually protective complex required for genotoxic stress resistance and checkpoint function. Proc Natl Acad Sci USA. 2006;103:18143–7. doi: 10.1073/pnas.0609251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gotter AL, Suppa C, Emanuel BS. Mammalian TIMELESS and Tipin are evolutionarily conserved replication fork-associated factors. J Mol Biol. 2007;366:36–52. doi: 10.1016/j.jmb.2006.10.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshizawa-Sugata N, Masai H. Human Tim/Timeless-interacting protein, Tipin, is required for efficient progression of S phase and DNA replication checkpoint. J Biol Chem. 2007;282:2729–40. doi: 10.1074/jbc.M605596200. [DOI] [PubMed] [Google Scholar]
- 5.Mayer ML, Pot I, Chang M, Xu H, Aneliunas V, Kwok T, et al. Identification of protein complexes required for efficient sister chromatid cohesion. Mol Biol Cell. 2004;15:1736–45. doi: 10.1091/mbc.E03-08-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noguchi E, Noguchi C, McDonald WH, Yates JR, 3rd, Russell P. Swi1 and Swi3 are components of a replication fork protection complex in fission yeast. Mol Cell Biol. 2004;24:8342–55. doi: 10.1128/MCB.24.19.8342-8355.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calzada A, Hodgson B, Kanemaki M, Bueno A, Labib K. Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev. 2005;19:1905–19. doi: 10.1101/gad.337205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Errico A, Costanzo V, Hunt T. Tipin is required for stalled replication forks to resume DNA replication after removal of aphidicolin in Xenopus egg extracts. Proc Natl Acad Sci USA. 2007;104:14929–34. doi: 10.1073/pnas.0706347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gambus A, Jones RC, Sanchez-Diaz A, Kanemaki M, van Deursen F, Edmondson RD, et al. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat Cell Biol. 2006;8:358–66. doi: 10.1038/ncb1382. [DOI] [PubMed] [Google Scholar]
- 10.Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, et al. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature. 2003;424:1078–83. doi: 10.1038/nature01900. [DOI] [PubMed] [Google Scholar]
- 11.Matsumoto S, Ogino K, Noguchi E, Russell P, Masai H. Hsk1-Dfp1/Him1, the Cdc7-Dbf4 kinase in Schizosaccharomyces pombe, associates with Swi1, a component of the replication fork protection complex. J Biol Chem. 2005;280:42536–42. doi: 10.1074/jbc.M510575200. [DOI] [PubMed] [Google Scholar]
- 12.Tanaka H, Kubota Y, Tsujimura T, Kumano M, Masai H, Takisawa H. Replisome progression complex links DNA replication to sister chromatid cohesion in Xenopus egg extracts. Genes Cells. 2009;14:949–63. doi: 10.1111/j.1365-2443.2009.01322.x. [DOI] [PubMed] [Google Scholar]
- 13.Leman AR, Noguchi C, Lee CY, Noguchi E. Human Timeless and Tipin stabilize replication forks and facilitate sister-chromatid cohesion. J Cell Sci. 2010;123:660–70. doi: 10.1242/jcs.057984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egel R, Beach DH, Klar AJ. Genes required for initiation and resolution steps of mating-type switching in fission yeast. Proc Natl Acad Sci USA. 1984;81:3481–5. doi: 10.1073/pnas.81.11.3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park H, Sternglanz R. Identification and characterization of the genes for two topoisomerase I-interacting proteins from Saccharomyces cerevisiae. Yeast. 1999;15:35–41. doi: 10.1002/(SICI)1097-0061(19990115)15:1<35::AID-YEA340>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 16.Foss EJ. Tof1p regulates DNA damage responses during S phase in Saccharomyces cerevisiae. Genetics. 2001;157:567–77. doi: 10.1093/genetics/157.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B, Elledge SJ. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science. 1996;271:357–60. doi: 10.1126/science.271.5247.357. [DOI] [PubMed] [Google Scholar]
- 18.Murakami H, Okayama H. A kinase from fission yeast responsible for blocking mitosis in S phase. Nature. 1995;374:817–9. doi: 10.1038/374817a0. [DOI] [PubMed] [Google Scholar]
- 19.Noguchi E, Noguchi C, Du LL, Russell P. Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Mol Cell Biol. 2003;23:7861–74. doi: 10.1128/MCB.23.21.7861-7874.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee BS, Grewal SI, Klar AJ. Biochemical interactions between proteins and mat1 cis-acting sequences required for imprinting in fission yeast. Mol Cell Biol. 2004;24:9813–22. doi: 10.1128/MCB.24.22.9813-9822.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paulsen RD, Cimprich KA. The ATR pathway: fine-tuning the fork. DNA Repair (Amst) 2007;6:953–66. doi: 10.1016/j.dnarep.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 22.Unsal-Kaçmaz K, Mullen TE, Kaufmann WK, Sancar A. Coupling of human circadian and cell cycles by the timeless protein. Mol Cell Biol. 2005;25:3109–16. doi: 10.1128/MCB.25.8.3109-3116.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith KD, Fu MA, Brown EJ. Tim-Tipin dysfunction creates an indispensible reliance on the ATR-Chk1 pathway for continued DNA synthesis. J Cell Biol. 2009;187:15–23. doi: 10.1083/jcb.200905006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–8. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 25.Chini CC, Chen J. Human claspin is required for replication checkpoint control. J Biol Chem. 2003;278:30057–62. doi: 10.1074/jbc.M301136200. [DOI] [PubMed] [Google Scholar]
- 26.Kumagai A, Dunphy WG. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol Cell. 2000;6:839–49. doi: 10.1016/S1097-2765(05)00092-4. [DOI] [PubMed] [Google Scholar]
- 27.Nakaya R, Takaya J, Onuki T, Moritani M, Nozaki N, Ishimi Y. Identification of proteins that may directly interact with human RPA. J Biochem. 2010;148:539–47. doi: 10.1093/jb/mvq085. [DOI] [PubMed] [Google Scholar]
- 28.Kemp MG, Akan Z, Yilmaz S, Grillo M, Smith-Roe SL, Kang TH, et al. Tipin-replication protein A interaction mediates Chk1 phosphorylation by ATR in response to genotoxic stress. J Biol Chem. 2010;285:16562–71. doi: 10.1074/jbc.M110.110304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lopez-Mosqueda J, Maas NL, Jonsson ZO, Defazio-Eli LG, Wohlschlegel J, Toczyski DP. Damage-induced phosphorylation of Sld3 is important to block late origin firing. Nature. 2010;467:479–83. doi: 10.1038/nature09377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zegerman P, Diffley JF. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature. 2010;467:474–8. doi: 10.1038/nature09373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Masai H, Matsumoto S, You Z, Yoshizawa-Sugata N, Oda M. Eukaryotic chromosome DNA replication: where, when, and how? Annu Rev Biochem. 2010;79:89–130. doi: 10.1146/annurev.biochem.052308.103205. [DOI] [PubMed] [Google Scholar]
- 32.Zou L, Stillman B. Assembly of a complex containing Cdc45p, replication protein A, and Mcm2p at replication origins controlled by S-phase cyclin-dependent kinases and Cdc7p-Dbf4p kinase. Mol Cell Biol. 2000;20:3086–96. doi: 10.1128/MCB.20.9.3086-3096.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Masai H, Miyake T, Arai K. hsk1+, a Schizosaccharomyces pombe gene related to Saccharomyces cerevisiae CDC7, is required for chromosomal replication. EMBO J. 1995;14:3094–104. doi: 10.1002/j.1460-2075.1995.tb07312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown GW, Kelly TJ. Purification of Hsk1, a minichromosome maintenance protein kinase from fission yeast. J Biol Chem. 1998;273:22083–90. doi: 10.1074/jbc.273.34.22083. [DOI] [PubMed] [Google Scholar]
- 35.Shimmoto M, Matsumoto S, Odagiri Y, Noguchi E, Russell P, Masai H. Interactions between Swi1-Swi3, Mrc1 and S phase kinase, Hsk1 may regulate cellular responses to stalled replication forks in fission yeast. Genes Cells. 2009;14:669–82. doi: 10.1111/j.1365-2443.2009.01300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sommariva E, Pellny TK, Karahan N, Kumar S, Huberman JA, Dalgaard JZ. Schizosaccharomyces pombe Swi1, Swi3, and Hsk1 are components of a novel S-phase response pathway to alkylation damage. Mol Cell Biol. 2005;25:2770–84. doi: 10.1128/MCB.25.7.2770-2784.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bensimon A, Simon A, Chiffaudel A, Croquette V, Heslot F, Bensimon D. Alignment and sensitive detection of DNA by a moving interface. Science. 1994;265:2096–8. doi: 10.1126/science.7522347. [DOI] [PubMed] [Google Scholar]
- 38.Tourrière H, Versini G, Cordón-Preciado V, Alabert C, Pasero P. Mrc1 and Tof1 promote replication fork progression and recovery independently of Rad53. Mol Cell. 2005;19:699–706. doi: 10.1016/j.molcel.2005.07.028. [DOI] [PubMed] [Google Scholar]
- 39.Mirkin EV, Mirkin SM. Replication fork stalling at natural impediments. Microbiol Mol Biol Rev. 2007;71:13–35. doi: 10.1128/MMBR.00030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lisby M, Rothstein R, Mortensen UH. Rad52 forms DNA repair and recombination centers during S phase. Proc Natl Acad Sci USA. 2001;98:8276–82. doi: 10.1073/pnas.121006298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lisby M, Mortensen UH, Rothstein R. Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat Cell Biol. 2003;5:572–7. doi: 10.1038/ncb997. [DOI] [PubMed] [Google Scholar]
- 42.Urtishak KA, Smith KD, Chanoux RA, Greenberg RA, Johnson FB, Brown EJ. Timeless Maintains Genomic Stability and Suppresses Sister Chromatid Exchange during Unperturbed DNA Replication. J Biol Chem. 2009;284:8777–85. doi: 10.1074/jbc.M806103200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aguilera A, Gómez-González B. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet. 2008;9:204–17. doi: 10.1038/nrg2268. [DOI] [PubMed] [Google Scholar]
- 44.Jones RM, Petermann E. Replication fork dynamics and the DNA damage response. Biochem J. 2012;443:13–26. doi: 10.1042/BJ20112100. [DOI] [PubMed] [Google Scholar]
- 45.Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nat Rev Genet. 2005;6:729–42. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- 46.Bastia D, Mohanty BK. Termination of DNA replication. In: DePamphilis ML, ed. DNA replication and human disease. Cold Spring Harbor, N. Y.: Cold Spring Harbor Laboratory Press, 2006:155-174. [Google Scholar]
- 47.Labib K, Hodgson B. Replication fork barriers: pausing for a break or stalling for time? EMBO Rep. 2007;8:346–53. doi: 10.1038/sj.embor.7400940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu G, Leffak M. Instability of (CTG)n•(CAG)n trinucleotide repeats and DNA synthesis. Cell Biosci. 2012;2:7. doi: 10.1186/2045-3701-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–40. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 50.Dalgaard JZ, Klar AJ. swi1 and swi3 perform imprinting, pausing, and termination of DNA replication in S. pombe. Cell. 2000;102:745–51. doi: 10.1016/S0092-8674(00)00063-5. [DOI] [PubMed] [Google Scholar]
- 51.Kaykov A, Holmes AM, Arcangioli B. Formation, maintenance and consequences of the imprint at the mating-type locus in fission yeast. EMBO J. 2004;23:930–8. doi: 10.1038/sj.emboj.7600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vengrova S, Dalgaard JZ. RNase-sensitive DNA modification(s) initiates S. pombe mating-type switching. Genes Dev. 2004;18:794–804. doi: 10.1101/gad.289404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eydmann T, Sommariva E, Inagawa T, Mian S, Klar AJS, Dalgaard JZ. Rtf1-mediated eukaryotic site-specific replication termination. Genetics. 2008;180:27–39. doi: 10.1534/genetics.108.089243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mohanty BK, Bairwa NK, Bastia D. The Tof1p-Csm3p protein complex counteracts the Rrm3p helicase to control replication termination of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2006;103:897–902. doi: 10.1073/pnas.0506540103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kobayashi T. The replication fork barrier site forms a unique structure with Fob1p and inhibits the replication fork. Mol Cell Biol. 2003;23:9178–88. doi: 10.1128/MCB.23.24.9178-9188.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kobayashi T, Horiuchi T. A yeast gene product, Fob1 protein, required for both replication fork blocking and recombinational hotspot activities. Genes Cells. 1996;1:465–74. doi: 10.1046/j.1365-2443.1996.d01-256.x. [DOI] [PubMed] [Google Scholar]
- 57.Krings G, Bastia D. Sap1p binds to Ter1 at the ribosomal DNA of Schizosaccharomyces pombe and causes polar replication fork arrest. J Biol Chem. 2005;280:39135–42. doi: 10.1074/jbc.M508996200. [DOI] [PubMed] [Google Scholar]
- 58.Mejía-Ramírez E, Sánchez-Gorostiaga A, Krimer DB, Schvartzman JB, Hernández P. The mating type switch-activating protein Sap1 Is required for replication fork arrest at the rRNA genes of fission yeast. Mol Cell Biol. 2005;25:8755–61. doi: 10.1128/MCB.25.19.8755-8761.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sánchez-Gorostiaga A, López-Estraño C, Krimer DB, Schvartzman JB, Hernández P. Transcription termination factor reb1p causes two replication fork barriers at its cognate sites in fission yeast ribosomal DNA in vivo. Mol Cell Biol. 2004;24:398–406. doi: 10.1128/MCB.24.1.398-406.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krings G, Bastia D. swi1- and swi3-dependent and independent replication fork arrest at the ribosomal DNA of Schizosaccharomyces pombe. Proc Natl Acad Sci USA. 2004;101:14085–90. doi: 10.1073/pnas.0406037101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gerber JK, Gögel E, Berger C, Wallisch M, Müller F, Grummt I, et al. Termination of mammalian rDNA replication: polar arrest of replication fork movement by transcription termination factor TTF-I. Cell. 1997;90:559–67. doi: 10.1016/S0092-8674(00)80515-2. [DOI] [PubMed] [Google Scholar]
- 62.Brewer BJ, Lockshon D, Fangman WL. The arrest of replication forks in the rDNA of yeast occurs independently of transcription. Cell. 1992;71:267–76. doi: 10.1016/0092-8674(92)90355-G. [DOI] [PubMed] [Google Scholar]
- 63.Grummt I, Maier U, Ohrlein A, Hassouna N, Bachellerie JP. Transcription of mouse rDNA terminates downstream of the 3′ end of 28S RNA and involves interaction of factors with repeated sequences in the 3′ spacer. Cell. 1985;43:801–10. doi: 10.1016/0092-8674(85)90253-3. [DOI] [PubMed] [Google Scholar]
- 64.Kuhn A, Bartsch I, Grummt I. Specific interaction of the murine transcription termination factor TTF I with class-I RNA polymerases. Nature. 1990;344:559–62. doi: 10.1038/344559a0. [DOI] [PubMed] [Google Scholar]
- 65.López-estraño C, Schvartzman JB, Krimer DB, Hernández P. Co-localization of polar replication fork barriers and rRNA transcription terminators in mouse rDNA. J Mol Biol. 1998;277:249–56. doi: 10.1006/jmbi.1997.1607. [DOI] [PubMed] [Google Scholar]
- 66.Ward TR, Hoang ML, Prusty R, Lau CK, Keil RL, Fangman WL, et al. Ribosomal DNA replication fork barrier and HOT1 recombination hot spot: shared sequences but independent activities. Mol Cell Biol. 2000;20:4948–57. doi: 10.1128/MCB.20.13.4948-4957.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao A, Guo A, Liu Z, Pape L. Molecular cloning and analysis of Schizosaccharomyces pombe Reb1p: sequence-specific recognition of two sites in the far upstream rDNA intergenic spacer. Nucleic Acids Res. 1997;25:904–10. doi: 10.1093/nar/25.4.904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Azvolinsky A, Giresi PG, Lieb JD, Zakian VA. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol Cell. 2009;34:722–34. doi: 10.1016/j.molcel.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deshpande AM, Newlon CS. DNA replication fork pause sites dependent on transcription. Science. 1996;272:1030–3. doi: 10.1126/science.272.5264.1030. [DOI] [PubMed] [Google Scholar]
- 70.Hodgson B, Calzada A, Labib K. Mrc1 and Tof1 regulate DNA replication forks in different ways during normal S phase. Mol Biol Cell. 2007;18:3894–902. doi: 10.1091/mbc.E07-05-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sekedat MD, Fenyö D, Rogers RS, Tackett AJ, Aitchison JD, Chait BT. GINS motion reveals replication fork progression is remarkably uniform throughout the yeast genome. Mol Syst Biol. 2010;6:353. doi: 10.1038/msb.2010.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sabouri N, McDonald KR, Webb CJ, Cristea IM, Zakian VA. DNA replication through hard-to-replicate sites, including both highly transcribed RNA Pol II and Pol III genes, requires the S. pombe Pfh1 helicase. Genes Dev. 2012;26:581–93. doi: 10.1101/gad.184697.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Voineagu I, Freudenreich CH, Mirkin SM. Checkpoint responses to unusual structures formed by DNA repeats. Mol Carcinog. 2009;48:309–18. doi: 10.1002/mc.20512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Voineagu I, Narayanan V, Lobachev KS, Mirkin SM. Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc Natl Acad Sci USA. 2008;105:9936–41. doi: 10.1073/pnas.0804510105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Darlow JM, Leach DR. The effects of trinucleotide repeats found in human inherited disorders on palindrome inviability in Escherichia coli suggest hairpin folding preferences in vivo. Genetics. 1995;141:825–32. doi: 10.1093/genetics/141.3.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–58. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 77.Voineagu I, Surka CF, Shishkin AA, Krasilnikova MM, Mirkin SM. Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat Struct Mol Biol. 2009;16:226–8. doi: 10.1038/nsmb.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shishkin AA, Voineagu I, Matera R, Cherng N, Chernet BT, Krasilnikova MM, et al. Large-scale expansions of Friedreich’s ataxia GAA repeats in yeast. Mol Cell. 2009;35:82–92. doi: 10.1016/j.molcel.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu G, Chen X, Gao Y, Lewis T, Barthelemy J, Leffak M. Altered replication in human cells promotes DMPK (CTG)(n) · (CAG)(n) repeat instability. Mol Cell Biol. 2012;32:1618–32. doi: 10.1128/MCB.06727-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Makovets S, Herskowitz I, Blackburn EH. Anatomy and dynamics of DNA replication fork movement in yeast telomeric regions. Mol Cell Biol. 2004;24:4019–31. doi: 10.1128/MCB.24.9.4019-4031.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miller KM, Rog O, Cooper JP. Semi-conservative DNA replication through telomeres requires Taz1. Nature. 2006;440:824–8. doi: 10.1038/nature04638. [DOI] [PubMed] [Google Scholar]
- 82.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–10. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 83.Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, et al. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ohki R, Ishikawa F. Telomere-bound TRF1 and TRF2 stall the replication fork at telomeric repeats. Nucleic Acids Res. 2004;32:1627–37. doi: 10.1093/nar/gkh309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grandin N, Charbonneau M. Mrc1, a non-essential DNA replication protein, is required for telomere end protection following loss of capping by Cdc13, Yku or telomerase. Mol Genet Genomics. 2007;277:685–99. doi: 10.1007/s00438-007-0218-0. [DOI] [PubMed] [Google Scholar]
- 86.Leman AR, Dheekollu J, Deng Z, Lee SW, Das MM, Lieberman PM, et al. Timeless preserves telomere length by promoting efficient DNA replication through human telomeres. Cell Cycle. 2012;11:2337–47. doi: 10.4161/cc.20810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xhemalce B, Riising EM, Baumann P, Dejean A, Arcangioli B, Seeler J-S. Role of SUMO in the dynamics of telomere maintenance in fission yeast. Proc Natl Acad Sci USA. 2007;104:893–8. doi: 10.1073/pnas.0605442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moser BA, Nakamura TM. Protection and replication of telomeres in fission yeast. Biochem Cell Biol. 2009;87:747–58. doi: 10.1139/O09-037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Uhlmann F, Nasmyth K. Cohesion between sister chromatids must be established during DNA replication. Curr Biol. 1998;8:1095–101. doi: 10.1016/S0960-9822(98)70463-4. [DOI] [PubMed] [Google Scholar]
- 90.Gerlich D, Koch B, Dupeux F, Peters J-M, Ellenberg J. Live-cell imaging reveals a stable cohesin-chromatin interaction after but not before DNA replication. Curr Biol. 2006;16:1571–8. doi: 10.1016/j.cub.2006.06.068. [DOI] [PubMed] [Google Scholar]
- 91.Strunnikov AV, Larionov VL, Koshland D. SMC1: an essential yeast gene encoding a putative head-rod-tail protein is required for nuclear division and defines a new ubiquitous protein family. J Cell Biol. 1993;123:1635–48. doi: 10.1083/jcb.123.6.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Michaelis C, Ciosk R, Nasmyth K. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell. 1997;91:35–45. doi: 10.1016/S0092-8674(01)80007-6. [DOI] [PubMed] [Google Scholar]
- 93.Schmiesing JA, Ball AR, Jr., Gregson HC, Alderton JM, Zhou S, Yokomori K. Identification of two distinct human SMC protein complexes involved in mitotic chromosome dynamics. Proc Natl Acad Sci USA. 1998;95:12906–11. doi: 10.1073/pnas.95.22.12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Haering CH, Löwe J, Hochwagen A, Nasmyth K. Molecular architecture of SMC proteins and the yeast cohesin complex. Mol Cell. 2002;9:773–88. doi: 10.1016/S1097-2765(02)00515-4. [DOI] [PubMed] [Google Scholar]
- 95.Losada A, Yokochi T, Kobayashi R, Hirano T. Identification and characterization of SA/Scc3p subunits in the Xenopus and human cohesin complexes. J Cell Biol. 2000;150:405–16. doi: 10.1083/jcb.150.3.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sumara I, Vorlaufer E, Gieffers C, Peters BH, Peters JM. Characterization of vertebrate cohesin complexes and their regulation in prophase. J Cell Biol. 2000;151:749–62. doi: 10.1083/jcb.151.4.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Skibbens RV, Corson LB, Koshland D, Hieter P. Ctf7p is essential for sister chromatid cohesion and links mitotic chromosome structure to the DNA replication machinery. Genes Dev. 1999;13:307–19. doi: 10.1101/gad.13.3.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tóth A, Ciosk R, Uhlmann F, Galova M, Schleiffer A, Nasmyth K. Yeast cohesin complex requires a conserved protein, Eco1p(Ctf7), to establish cohesion between sister chromatids during DNA replication. Genes Dev. 1999;13:320–33. doi: 10.1101/gad.13.3.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang J, Shi X, Li Y, Kim BJ, Jia J, Huang Z, et al. Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol Cell. 2008;31:143–51. doi: 10.1016/j.molcel.2008.06.006. [DOI] [PubMed] [Google Scholar]
- 100.Liu J, Krantz ID. Cornelia de Lange syndrome, cohesin, and beyond. Clin Genet. 2009;76:303–14. doi: 10.1111/j.1399-0004.2009.01271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McNairn AJ, Gerton JL. Cohesinopathies: One ring, many obligations. Mutat Res. 2008;647:103–11. doi: 10.1016/j.mrfmmm.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 102.Vega H, Waisfisz Q, Gordillo M, Sakai N, Yanagihara I, Yamada M, et al. Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat Genet. 2005;37:468–70. doi: 10.1038/ng1548. [DOI] [PubMed] [Google Scholar]
- 103.Rabitsch KP, Tóth A, Gálová M, Schleiffer A, Schaffner G, Aigner E, et al. A screen for genes required for meiosis and spore formation based on whole-genome expression. Curr Biol. 2001;11:1001–9. doi: 10.1016/S0960-9822(01)00274-3. [DOI] [PubMed] [Google Scholar]
- 104.Chan RC, Chan A, Jeon M, Wu TF, Pasqualone D, Rougvie AE, et al. Chromosome cohesion is regulated by a clock gene paralogue TIM-1. Nature. 2003;423:1002–9. doi: 10.1038/nature01697. [DOI] [PubMed] [Google Scholar]
- 105.Formosa T, Nittis T. Dna2 mutants reveal interactions with Dna polymerase alpha and Ctf4, a Pol alpha accessory factor, and show that full Dna2 helicase activity is not essential for growth. Genetics. 1999;151:1459–70. doi: 10.1093/genetics/151.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hanna JS, Kroll ES, Lundblad V, Spencer FA. Saccharomyces cerevisiae CTF18 and CTF4 are required for sister chromatid cohesion. Mol Cell Biol. 2001;21:3144–58. doi: 10.1128/MCB.21.9.3144-3158.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mayer ML, Gygi SP, Aebersold R, Hieter P. Identification of RFC(Ctf18p, Ctf8p, Dcc1p): an alternative RFC complex required for sister chromatid cohesion in S. cerevisiae. Mol Cell. 2001;7:959–70. doi: 10.1016/S1097-2765(01)00254-4. [DOI] [PubMed] [Google Scholar]
- 108.Naiki T, Kondo T, Nakada D, Matsumoto K, Sugimoto K. Chl12 (Ctf18) forms a novel replication factor C-related complex and functions redundantly with Rad24 in the DNA replication checkpoint pathway. Mol Cell Biol. 2001;21:5838–45. doi: 10.1128/MCB.21.17.5838-5845.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Warren CD, Eckley DM, Lee MS, Hanna JS, Hughes A, Peyser B, et al. S-phase checkpoint genes safeguard high-fidelity sister chromatid cohesion. Mol Biol Cell. 2004;15:1724–35. doi: 10.1091/mbc.E03-09-0637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ansbach AB, Noguchi C, Klansek IW, Heidlebaugh M, Nakamura TM, Noguchi E. RFCCtf18 and the Swi1-Swi3 complex function in separate and redundant pathways required for the stabilization of replication forks to facilitate sister chromatid cohesion in Schizosaccharomyces pombe. Mol Biol Cell. 2008;19:595–607. doi: 10.1091/mbc.E07-06-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Errico A, Cosentino C, Rivera T, Losada A, Schwob E, Hunt T, et al. Tipin/Tim1/And1 protein complex promotes Pol alpha chromatin binding and sister chromatid cohesion. EMBO J. 2009;28:3681–92. doi: 10.1038/emboj.2009.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dheekollu J, Wiedmer A, Hayden J, Speicher D, Gotter AL, Yen T, et al. Timeless links replication termination to mitotic kinase activation. PLoS ONE. 2011;6:e19596. doi: 10.1371/journal.pone.0019596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Smith-Roe SL, Patel SS, Simpson DA, Zhou YC, Rao S, Ibrahim JG, et al. Timeless functions independently of the Tim-Tipin complex to promote sister chromatid cohesion in normal human fibroblasts. Cell Cycle. 2011;10:1618–24. doi: 10.4161/cc.10.10.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wu Y, Suhasini AN, Brosh RM., Jr. Welcome the family of FANCJ-like helicases to the block of genome stability maintenance proteins. Cell Mol Life Sci. 2009;66:1209–22. doi: 10.1007/s00018-008-8580-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.L Holloway S. CHL1 is a nuclear protein with an essential ATP binding site that exhibits a size-dependent effect on chromosome segregation. Nucleic Acids Res. 2000;28:3056–64. doi: 10.1093/nar/28.16.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Petronczki M, Chwalla B, Siomos MF, Yokobayashi S, Helmhart W, Deutschbauer AM, et al. Sister-chromatid cohesion mediated by the alternative RF-CCtf18/Dcc1/Ctf8, the helicase Chl1 and the polymerase-alpha-associated protein Ctf4 is essential for chromatid disjunction during meiosis II. J Cell Sci. 2004;117:3547–59. doi: 10.1242/jcs.01231. [DOI] [PubMed] [Google Scholar]
- 117.Skibbens RV. Chl1p, a DNA helicase-like protein in budding yeast, functions in sister-chromatid cohesion. Genetics. 2004;166:33–42. doi: 10.1534/genetics.166.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.van der Lelij P, Chrzanowska KH, Godthelp BC, Rooimans MA, Oostra AB, Stumm M, et al. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am J Hum Genet. 2010;86:262–6. doi: 10.1016/j.ajhg.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Parish JL, Rosa J, Wang X, Lahti JM, Doxsey SJ, Androphy EJ. The DNA helicase ChlR1 is required for sister chromatid cohesion in mammalian cells. J Cell Sci. 2006;119:4857–65. doi: 10.1242/jcs.03262. [DOI] [PubMed] [Google Scholar]
- 120.Xu H, Boone C, Brown GW. Genetic dissection of parallel sister-chromatid cohesion pathways. Genetics. 2007;176:1417–29. doi: 10.1534/genetics.107.072876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Farina A, Shin JH, Kim DH, Bermudez VP, Kelman Z, Seo YS, et al. Studies with the human cohesin establishment factor, ChlR1. Association of ChlR1 with Ctf18-RFC and Fen1. J Biol Chem. 2008;283:20925–36. doi: 10.1074/jbc.M802696200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liu Y, Kao HI, Bambara RA. Flap endonuclease 1: a central component of DNA metabolism. Annu Rev Biochem. 2004;73:589–615. doi: 10.1146/annurev.biochem.73.012803.092453. [DOI] [PubMed] [Google Scholar]
- 123.Uhlmann F. A matter of choice: the establishment of sister chromatid cohesion. EMBO Rep. 2009;10:1095–102. doi: 10.1038/embor.2009.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lengronne A, McIntyre J, Katou Y, Kanoh Y, Hopfner KP, Shirahige K, et al. Establishment of sister chromatid cohesion at the S. cerevisiae replication fork. Mol Cell. 2006;23:787–99. doi: 10.1016/j.molcel.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 125.Rudra S, Skibbens RV. Sister chromatid cohesion establishment occurs in concert with lagging strand synthesis. Cell Cycle. 2012;11:2114–21. doi: 10.4161/cc.20547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sherwood R, Takahashi TS, Jallepalli PV. Sister acts: coordinating DNA replication and cohesion establishment. Genes Dev. 2010;24:2723–31. doi: 10.1101/gad.1976710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Onn I, Heidinger-Pauli JM, Guacci V, Unal E, Koshland DE. Sister chromatid cohesion: a simple concept with a complex reality. Annu Rev Cell Dev Biol. 2008;24:105–29. doi: 10.1146/annurev.cellbio.24.110707.175350. [DOI] [PubMed] [Google Scholar]
- 128.Noguchi E. Division of labor of the replication fork protection complex subunits in sister chromatid cohesion and Chk1 activation. Cell Cycle. 2011;10:2055–6. doi: 10.4161/cc.10.13.15805. [DOI] [PMC free article] [PubMed] [Google Scholar]