

Abstract
We have previously suggested that ketone body metabolism is critical for tumor progression and metastasis. Here, using a co-culture system employing human breast cancer cells (MCF7) and hTERT-immortalized fibroblasts, we provide new evidence to directly support this hypothesis. More specifically, we show that the enzymes required for ketone body production are highly upregulated within cancer-associated fibroblasts. This appears to be mechanistically controlled by the stromal expression of caveolin-1 (Cav-1) and/or serum starvation. In addition, treatment with ketone bodies (such as 3-hydroxy-butyrate, and/or butanediol) is sufficient to drive mitochondrial biogenesis in human breast cancer cells. This observation was also validated by unbiased proteomic analysis. Interestingly, an MCT1 inhibitor was sufficient to block the onset of mitochondrial biogenesis in human breast cancer cells, suggesting a possible avenue for anticancer therapy. Finally, using human breast cancer tumor samples, we directly confirmed that the enzymes associated with ketone body production (HMGCS2, HMGCL and BDH1) were preferentially expressed in the tumor stroma. Conversely, enzymes associated with ketone re-utilization (ACAT1) and mitochondrial biogenesis (HSP60) were selectively associated with the epithelial tumor cell compartment. Our current findings are consistent with the “two-compartment tumor metabolism” model. Furthermore, they suggest that we should target ketone body metabolism as a new area for drug discovery, for the prevention and treatment of human cancers.
Keywords: ketone body, 3-hydroxy-butyrate, cancer metabolism, BDH1, HMGCS2, ACAT isoforms, tumor growth, metastasis
Introduction
Ketone bodies are high-energy mitochondrial fuels that burn more efficiently than other mitochondrial fuels.1 Most importantly, they can be utilized under conditions of hypoxia, when oxygen is scarce.2,3 Potentially, this would allow a tumor to grow even in the absence of an optimal blood supply. Thus, ketone body utlization may be important in tumor initiation (before the establishment of a vascular supply) or metastasis (after a tumor has outgrown its blood supply). As such, ketone body utilization could have important implications for both cancer prevention, as well as the effective treatment of advanced metastatic disease.4-6
Little is known about how cancer cells and their surrounding microenvironment, generate and use ketones.7 In fact, until recently, only hepatocytes and astrocytes were thought to generate ketone bodies, which were used during periods of starvation.8 In addition, it was also thought that only neurons9 are equipped with the necessary enzymes for ketone body re-utilization, allowing their conversion to acetyl-CoA and entrance into the mitochondrial TCA cycle, driving oxidative phosphorylation (OXPHOS).
However, here we provide new evidence that cancer-associated fibroblasts express the enzymes required to generate ketone bodies. Conversely, we show that ketone bodies can induce mitochondrial biogenesis in epithelial cancer cells, and that they harbor the necessary enzymes to convert ketone bodies into acetyl-CoA.
Thus, ketone bodies are generated in the tumor stroma, and then they are “fed” to epithelial cancer cells to “fuel” anabolic tumor growth. This tumor-based ketone body shuttle is analogous to the liver-brain and astrocyte-neuron ketone shuttles that have been known for decades. Hence, tumor cells have borrowed a normal physiological process to maintain their anabolic growth, under adverse or hypoxic conditions.
As a consequence, interrupting ketone body production in fibroblasts or preventing ketone body re-utilization in epithelial cancer cells would provide a new strategy for anticancer therapy.
Results
Exploring the relationship between ketogenesis and the tumor stroma
Previously, we have proposed that cancer-associated fibroblasts may be ketogenic.10-12 To further address this issue, we used a co-culture system employing hTERT-immortalized human fibroblasts and MCF7 human breast cancer cells.13
Figure 1 shows that co-culture of MCF7 cells induces the expression of a key enzyme associated with ketone production in cancer-associated fibroblasts, namely HMGCL. Conversely, another enzyme associated with ketone re-utilization, ACAT1, is selectively downregulated in cancer-associated fibroblasts. These results are consistent with the idea that the tumor stroma is highly ketogenic, i.e., associated with ketone body production.
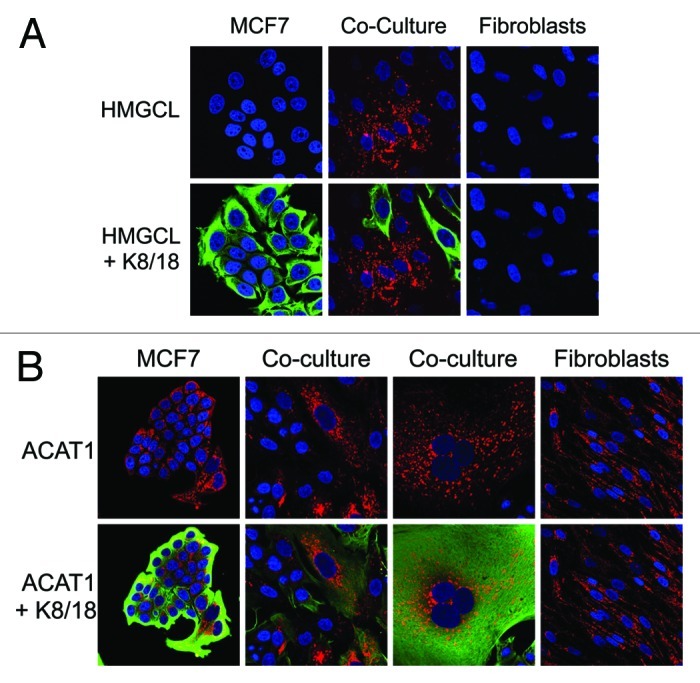
Figure 1. Co-culture with MCF7 cells induces HMGCL expression in fibroblasts and ACAT1 downregulation in fibroblasts. hTERT-fibroblast-MCF7 co-cultures were maintained for 5 d. Then, cells were fixed and immunostained with anti-HMGCL (Fig. 1A) or anti-ACAT1 (Fig. 1B) antibodies. MCF7 cells were identified using anti-K8–18 (green) antibodies. Nuclei were stained with DAPI (blue). (A) HMGCL staining (red) and DAPI (blue) is shown in the top panels to better appreciate the co-culture-induced HMGCL upregulation in fibroblasts. (B) ACAT1 staining (red) and DAPI (blue) is shown in the top panels to better appreciate the co-culture-induced ACAT1 upregulation in MCF7 cells and ACAT1 downregulation in fibroblasts. Original magnification, 40x.
Serum starvation or Cav-1 downregulation induces the expression of ketogenic enzymes in fibroblasts
Ketones are normally produced under conditions of organismal starvation. Thus, we examined the expression levels of ketogenic enzymes in fibroblasts in response to serum starvation. Interestingly, we observed that two ketognenic enzymes, BDH1 and HMGCS1, were selectively upregulated under conditions of serum starvation (Fig. 2A).
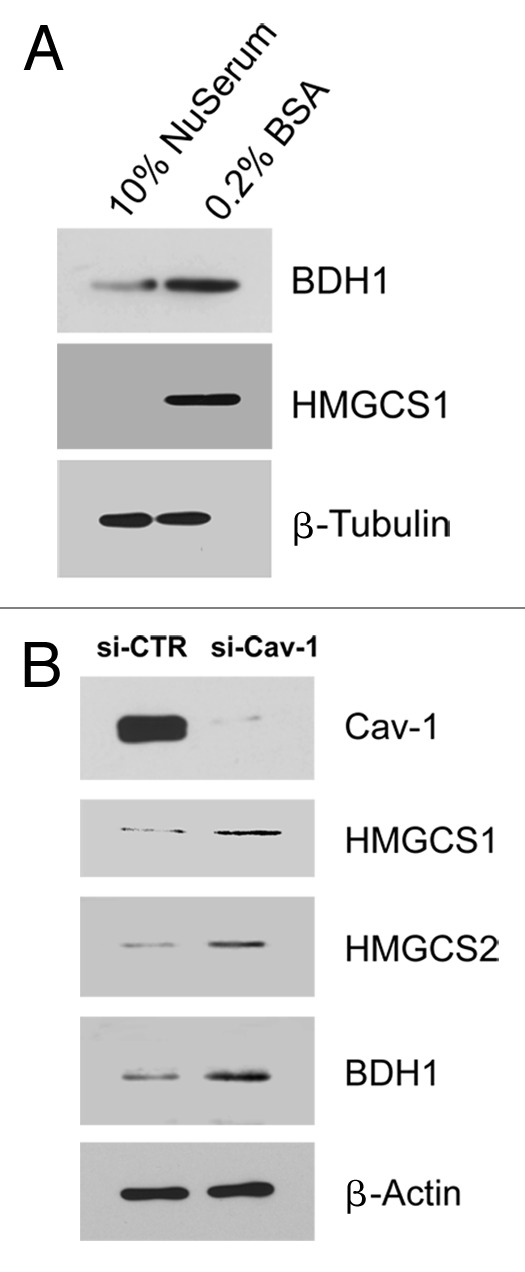
Figure 2. Serum starvation or Cav-1 downregulation induces the expression of ketogenic enzymes in fibroblasts. (A) Serum starvation induces the expression of enzymes for ketone body synthesis. hTERT-fibroblasts were cultured in 10% NuSerum or 0.2% BSA (serum starvation) for 72 h. Cells were then lysed and subjected to western blot analysis with antibodies directed against BDH1 and HMGCS1. Note that when fibroblasts were serum starved, BDH1 and HMGCS1 were upregulated. Equal loading was assessed by β-tubulin immunoblotting. (B) Cav-1 knock-down leads to increased expression of enzymes involved in ketone body synthesis. hTERT-fibroblasts were treated with Cav-1 siRNA or control siRNA for 48 h. Cells were then lysed and subjected to western blot analysis with antibodies directed against Cav-1, HMGCS1, HMGCS2 and BDH1. Note that when Cav-1 levels were knocked-down, HMGCS1, HMGCS2 and BDH1 were upregulated. Equal loading was assessed by β-actin immunoblotting.
Loss of Cav-1 has been shown to result in ketone production in Cav-1 (−/−) deficient mice.10 However, the exact mechanism remains unknown. One hypothesis is that a loss of Cav-1 upregulates the enzymes associated with ketone body production. To test this idea, we transiently knocked-down Cav-1 expression in cultured fibroblasts. Figure 2B shows that an acute loss of Cav-1 expression is indeed sufficient to drive the upregulation of several enzymes required for ketone production (HMGCS1, HMGCS2 and BDH1). It is important to note that a loss of stromal Cav-1 expression in cancer-associated fibroblasts in patients is associated with metastasis and/or overall poor clinical outcome in breast and prostate cancers (reviewed in ref. 6).
Ketone bodies are sufficient to induce mitochondrial biogenesis in human breast cancer cells
Ketone bodies are known mitochondrial fuels. However, it remains unknown whether treatment with ketone bodies can induce mitochondrial biogenesis. To address this issue, we treated MCF7 cells with a number of mitochondrial fuels. We then monitored the status of mitochondrial mass via immunofluorescence and immunoblotting with a mitochondrial-specific antibody probe.
Figure 3A and B shows that treatment with ketone bodies (3-hydroxy-butyrate, or butanediol) or L-lactate was indeed sufficient to increase mitochondrial mass. Similarly, ketone bodies were especially effective at upregulating the expression of multiple functional subunits of the PDH complex (Fig. 3C). Thus, if the necessary mitochondrial fuels are abundant in their microenvironment, cancer cells will adapt and generate the necessary “power plants” to burn the available mitochondrial fuel source.

Figure 3. Lactate or ketone bodies induces mitochondrial mass and metabolism in MCF7 cells. MCF7 cells were cultured in the presence of 10 mM L-lactate, 10 mM β-hydroxybutyrate, 10 mM butanediol, or vehicle alone for 2 d. (A) MCF7-treated cells were fixed and immunostained with anti-intact mitochondrial membrane antibody. Mitochondrial staining (red) is shown in the top panels to better appreciate that lactate and ketone body increase mitochondrial mass. Mitochondrial staining (red) and DAPI (blue) is shown in the bottom panels. Original magnification, 40×. (B) MCF7-treated cells were lysed and subjected to western blot analysis with antibodies directed against the intact mitochondrial membrane. Equal loading was assessed by β-actin immunoblotting. (C) MCF7-treated cells were lysed and subjected to western blot analysis with antibodies directed against multiple functional subunits of PDH complex. Equal loading was assessed by β-actin immunoblotting. Note that lactate, β-hydroxy-butyrate and butanediol increase the expression of subunits of the PDH enzymatic complex.
This concept was also validated by unbiased proteomic analysis of MCF7 that were treated with ketone bodies (3-hydroxy-butyrate) or L-lactate. These results are summarized in Tables S1 and S2. Note that both mitochondrial fuels induce the protein expression of mitochondrial proteins and chaperones. The latter would be required for increased protein folding, a further indication of the onset of anabolic growth.
The MCT1 inhibitor CHC inhibits mitochondrial biogenesis in MCF7 cells under co-culture conditions
We have previously shown that the co-culture of fibroblasts with human breast cancer cells is also sufficient to promote mitochondrial biogenesis in adjacent breast cancer cells. We suggested that this may be due to the secretion of L-lactate and ketone bodies by cancer-associated fibroblasts during co-culture. Then, L-lactate and ketone bodies would presumably be taken up by the MCT1 mono-carboxylate transporter in cancer cells.
To further address this issue, here we examined the activity of an MCT1 inhibitor, known as CHC,14 under conditions of co-culture. Such an inhibitor would block the uptake of endogenous L-lactate and ketone bodies generated by cancer-associated fibroblasts during co-culture. Figure 4 shows that CHC-treatment prevents mitochondrial biogenesis in MCF7-fibroblasts co-cultures, as predicted. Thus, the uptake of endogenous L-lactate and ketone bodies is also required to drive mitochondrial biogenesis under co-culture conditions.
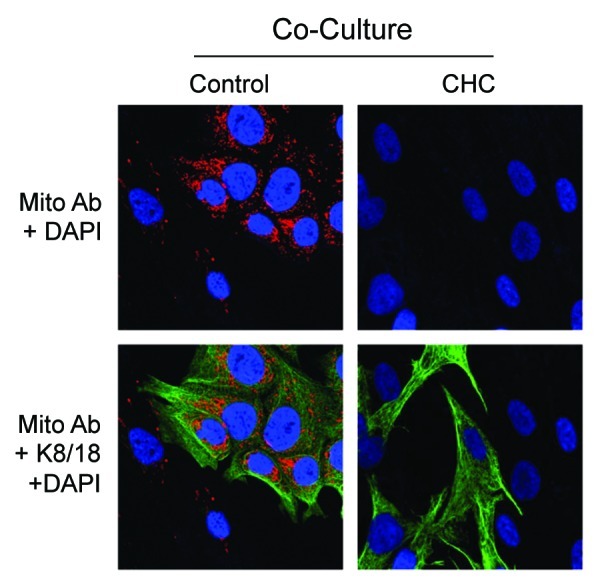
Figure 4. The MCT1 inhibitor CHC inhibits mitochondrial biogenesis in MCF7 cells. Co-cultures of MCF7 and fibroblasts were maintained for 5 d. Forty-eight h prior to fixation, cells were incubated with 10 mM CHC or vehicle alone. Then, cells were fixed and immunostained with antibodies against the intact mitochondrial membrane (red). and K8–18 (green). Nuclei were stained with DAPI (blue). Mitochondrial membrane staining is shown in the top panels to better appreciate that the MCT1 inhibitor CHC blocks the co-culture-induced increase in mitochondrial mass. Original magnification, 40×.
Lactate and/or ketone bodies induce Smad2/3 activation in MCF7 cells
It remains unknown what signaling pathways may be activated by ketone bodies and L-lactate in order to drive mitochondrial biogenesis in human breast cancer cells. Figure 5 shows that L-lactate and 3-hydroxy-butyrate both induce Smad2/3 hyper-phosphorylation as early as 5 to 15 min after treatment, which appears to be sustained up to 24 h. These results are consistent with the idea that L-lactate and ketone bodies can induce the onset of an EMT and/or “stemness” in human breast cancer cells.12
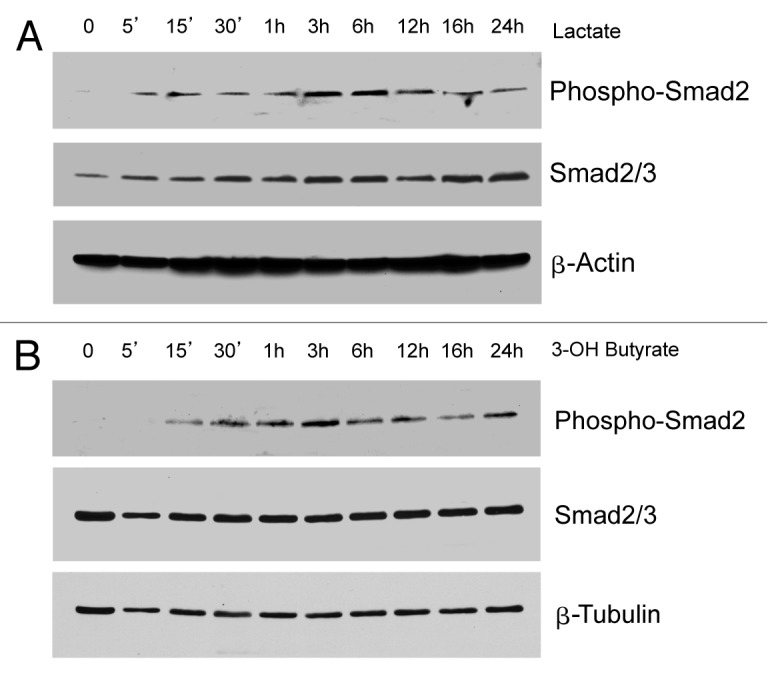
Figure 5. Lactate or ketone bodies induces Smad2 activation in MCF7 cells. MCF7 cells were treated with 10 mM L-lactate (Fig. 5A) or 10 mM β-hydroxybutyrate (Fig. 5B) for the indicated times. Then, cells were lysed and subjected to western blot analysis with antibodies directed against phospho-Smad2 and total Smad2/3. Equal loading was assessed by β-actin immunoblotting. Note that treatment with lactate or β-hydroxybutyrate is sufficient to induce the activation of the Smad2 pathway, suggesting an EMT induction.
Cancer-associated fibroblasts and ketone bodies induce the mitochondrial anti-stress response in MCF7 cells
Here, we also examined the onset of the mitochondrial anti-stress response, using specific molecular protein markers, namely HSP60, TRAP1 and SCO1. Figure 6A shows that co-culture of MCF7 cells with fibroblasts is sufficient to induce the expression of HSP60 in MCF7 cancer cells. Similarly, Figure 6B shows that when MCF7 cells were treated with 3-hydroxy-butyrate, this was sufficient to induce the upregulation of TRAP1 and SCO1. These results are consistent with the onset of mitochondrial biogenesis in the tumor cell compartment.
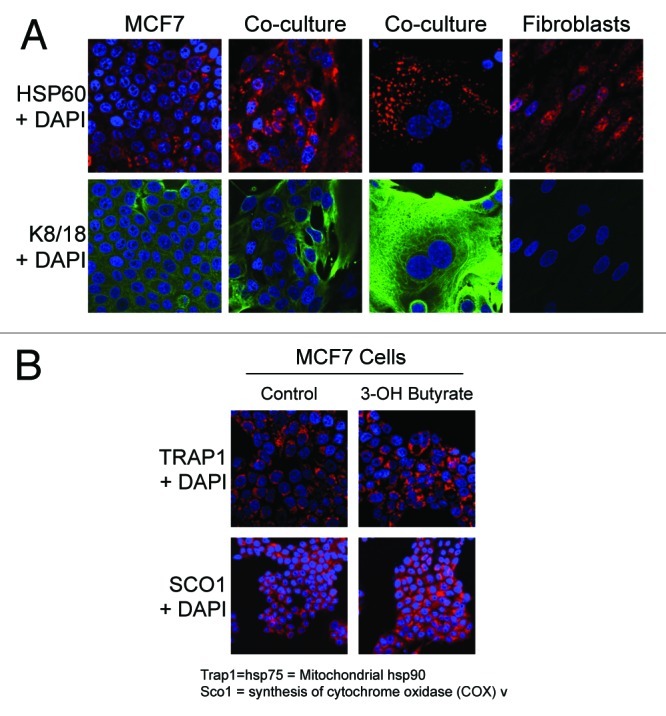
Figure 6. Co-culture with fibroblasts and β-hydroxybutyrate treatment induce mitochondrial anti-stress responses in MCF7 cells. (A) hTERT-fibroblast-MCF7 co-cultures were maintained for 5 d. Then, cells were fixed and immunostained with anti-HSP60 (red) and anti-K8–18 (green) antibodies. Nuclei were stained with DAPI (blue). HSP60 staining is shown in the top panels to better appreciate the co-culture-induced HSP60 upregulation in MCF7 cells. (B) MCF7 cells were cultured in the presence of 10 mM β-hydroxybutyrate or vehicle alone for 2 d. Then, cells were fixed and immunostained with anti-TRAP1 or SCO1 antibodies. Nuclei were stained with DAPI (blue). Note that β-hydroxybutyrate treatment induces the expression of TRAP1 and SCO1 in MCF7 cells, indicating an increased mitochondrial anti-stress responses. Original magnification, 40×.
Compartmentalization of ketone production and ketone re-utilization in human breast cancers
Based on our model of “two-compartment tumor metabolism,”4,5,15,16 we would predict that the enzymes required for ketone body production would be compartmentalized within the tumor stroma. Conversely, we would hypothesize that the enzymes required for ketone re-utilization would be selectively localized within epithelial cancer cells.
To test this hypothesis, we used a series of human breast cancer samples, selected based on a loss of stromal Cav-1, which is a marker of a lethal tumor microenvironment.17,18 In accordance with our hypothesis, ACAT1, a key enzyme required for ketone re-utilization, was preferentially localized to epithelial cancer cells (Fig. 7). In contrast, Figure 8 shows that as series of enzymes required for ketone body production (HMGCS2, HMGCL and BDH1) were all mainly localized to the tumor stroma.
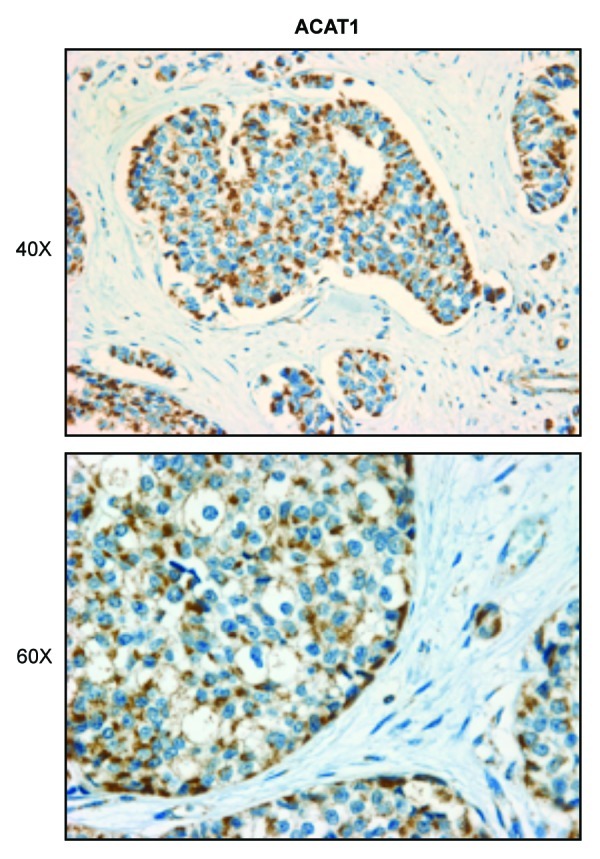
Figure 7. Compartmentalization of ketone metabolism: Human breast cancers show increased expression of a key enzyme for ketone utilization in the epithelial tumor cells. Paraffin-embedded tissue sections from human breast cancer samples were immunostained with antibodies directed against ACAT1, a key enzyme for ketone utilization. Slides were counterstained with hematoxylin. Note that ACAT1 is highly overexpressed in the epithelial tumor cell compartment. Original magnification, 40× and 60×.
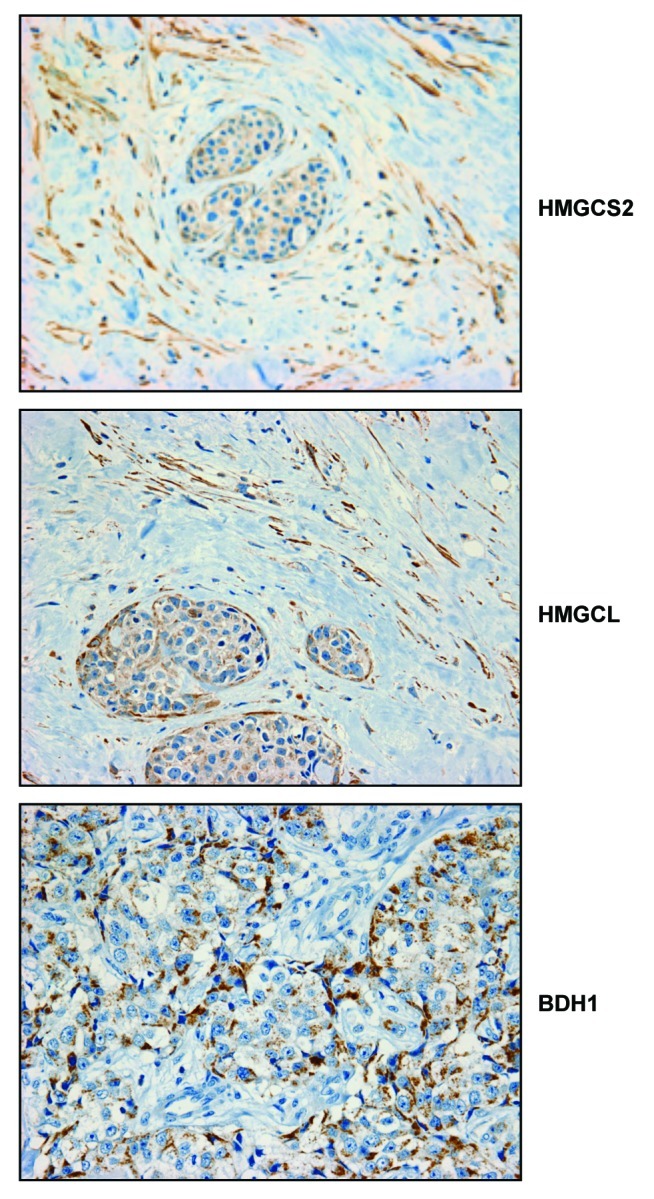
Figure 8. Compartmentalization of ketone metabolism: Human breast cancers show increased expression of key enzymes for ketone generation in the stromal compartment. Paraffin-embedded tissue sections from human breast cancer samples were immunostained with antibodies directed against HMGCS2, HMGCL and BDH1, key enzymes for ketone generation. Slides were counterstained with hematoxylin. Note that HMGCS2, HMGCL and BDH1 are highly overexpressed in the stromal compartment of human breast cancers. Original magnification, 40×.
Consistent with the observed localization of ACAT1 within epithelial cancer cells (Fig. 7), HSP60 (a protein involved in the folding and assembly of mitochondrial proteins) was also specifically localized to the epithelial cancer cell compartment (Fig. 9).

Figure 9. HSP60 expression is increased in the epithelial tumor compartment of human breast cancers. Paraffin-embedded tissue sections from human breast cancer samples were immunostained with antibodies directed against HSP60, a protein involved in the folding and assembly of mitochondrial proteins. Slides were counterstained with hematoxylin. Note that HSP60 is highly overexpressed in the epithelial tumor compartment. Original magnification, 40× and 60×.
Discussion
Recently, we proposed a new paradigm to understand how energy is transferred from the host to tumor cells.19 In this model, fibroblasts are catabolic and undergo mitochondrial dysfunction. One consequence of mitochondrial dysfunction is the onset of ketone production.20-22 In fact, elevated serum ketone levels in newborns are used to diagnose inborn errors in mitochondrial metabolism and, especially, complex I deficiency.20-22
Once ketones are produced in the tumor stroma, they become available for use as mitochondrial fuels in cells that have the necessary machinery to convert ketones back into acetyl-CoA. To utilize ketones, tumor cells upregulate the same metabolic enzymes found in neurons, such as ACAT1. Thus, the enzymes required for ketone body production and/or utilization represent new potential biomarkers for cancer prognosis and new “druggable” targets for anticancer therapy.
In direct support of this notion, treatment of MCF7 cells with ketones was coupled with genome-wide transcriptional profiling, to generate a prognostic gene signature.12 This ketone-induced gene signature was specifically associated with increased tumor recurrence, metastasis and poor clinical outcome in human breast cancer patients.12
As such, ketone-associated biomarkers could be used for treatment stratification of the patient population for identifying cancer patients that might benefit from ketone inhibitors to be administered as anticancer therapy. One such biomarker is a loss of stromal Cav-1, which is a marker of a lethal tumor microenvironment in breast and prostate cancer as well as melanoma.6 In fact, analysis of Cav-1 (−/−) deficient mice shows elevated ketone production in the mammary fat pad as well as lung tissue.10 Tumors grown in a Cav-1-deficient mammary fat pad are ~5 times larger, consistent with the idea that ketones can promote tumor growth.23
Obviously, further work will be required to translate these findings into clinical practice, allowing the implementation of a personalized cancer medicine approach to patient therapy. In this regard, the development of new modalities for the non-invasive imaging of ketone production and/or re-utilization24 in human tumors may also be warranted, especially for diagnostic purposes and for monitoring the efficacy of related anticancer treatment(s).
Materials and Methods
Materials
Antibodies were as follows: HMGCL (WH03155, Sigma); HMGCS1 (ab67242, Abcam); HMGCS2 (AV41562, Sigma); ACAT1 (HPA007569, Sigma); BDH1 (ab6834, Abcam); Cav-1 (610407, BD Biosciences); cytokeratin 8/18 (20R-CP004, Fitzgerald Industries International); surface of intact mitochondria (MAB1273, Millipore); Mitoprofile PDH cocktail (MSP02, Mitosciences); phospho-SMAD2 (3101, Cell Signaling); SMAD2/3 (BD610842, BD Biosciences); HSP60 (ab46798, Abcam); TRAP1(ab62721, Abcam); SCO1 (ab88658, Abcam); secondary antibodies for immunofluorescence were Alexa green 488 nm and Alexa Orange-Red 546 nm (Invitrogen). Other reagents were as follows: L-lactate, 3-hydroxy-butyrate, butanediol, N-acetyl cysteine (NAC), BSA, alpha-cyano-4-hydroxycinnamate (CHC) were from Sigma. 4,6-diamidino-2- phenylindole (DAPI) (D3571) and Prolong Gold Antifade mounting reagent (P36930) were from Invitrogen.
Cell Cultures
Human skin fibroblasts immortalized with telomerase reverse transcriptase protein (hTERT-BJ-1) were originally purchased from Clontech, Inc. The breast cancer cell lines MCF7 and MDA-MB 231 were purchased from ATCC. All cells were maintained in DMEM with 10% Fetal Bovine Serum (FBS) and Penicillin 100 units/mL-Streptomycin 100 µg/mL.
Co-cultures of MCF7 Cells and Fibroblasts
hTERT-fibroblasts and MCF7 cells were plated on glass coverslips in 12-well plates in 1 ml of complete media. MCF7 cells were plated within 2 h of fibroblast plating. The total number of cells per well was 1 × 105. Experiments were performed at a 5:1 fibroblast-to-epithelial cell ratio. As controls, monocultures of fibroblasts and MCF7 cells were seeded using the same number of cells as the corresponding co-cultures. The day after plating, media was changed to DMEM with 10% NuSerum (a low protein alternative to FBS; BD Biosciences) and Pen-Strep. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2. 10mM CHC was added to fresh media 48 h prior to fixation on day 5 of co-culture of MCF7 cells and BJ-1 fibroblasts.
Immunocytochemistry
Immunocytochemistry was performed as previously described ADD REF. All steps were performed at room temperature. Briefly, after 30 min fixation in 2% paraformaldehyde, cells were permeabilized for 10 min with immunofluorescence (IF) buffer (PBS, 0.2% BSA, 0.1% TritonX-100). Then, cells were incubated for 10 min with NH4Cl in PBS to quench free aldehyde groups. Primary antibodies were incubated in IF buffer for 1 h. After washing with IF buffer (3×, 10 min each), cells were incubated for 30 min with fluorochrome-conjugated secondary antibodies diluted in IF buffer. Finally, slides were washed with IF buffer (3×, 10 min each), incubated with the nuclear stain and mounted.
Confocal microscopy
Images were collected with a Zeiss LSM510 meta confocal system using a 405 nm Diode excitation laser with a band pass filter of 420–480 nm, a 488 nm Argon excitation laser with a band pass filter of 505–550 nm and a 543 nm HeNe excitation laser with a 561–604 nm filter. Images were acquired with 20×, 40× or 63× objectives, as stated in the figure legends.
Cav-1 knockdown
siRNA-mediated Cav-1 knockdown was achieved using the HiPerFect transfection reagents (Qiagen) as per manufacturer instructions, with minor modifications. Briefly, cells were seeded in complete media and were transfected within 30 min of plating. Transfections were performed by mixing Cav-1 siRNA (SI00299635, Qiagen) or control siRNA (1022076, Qiagen) with serum-free media and HiPerFect reagent. The mixture was vortexed for 30 sec and left for 15 min at room temperature before adding it drop-wise onto freshly seeded cells. Three hours after transfection, complete media was added to the cells. Cav-1 knockdown was monitored 36–48 h after transfection.
Mitochondrial detection
An antibody against the intact surface of mitochondria (MAB 1273, Millipore) was used to visualize mitochondria by immunocytochemistry. Western blot analysis was performed using cocktails of anti-mitochondrial antibodies detecting PDH subunits.
Immunoblotting
hTERT-fibroblasts and MCF7 cells were harvested in lysis buffer (10 mM TRIS-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 60 mM octylglucoside) containing protease inhibitors (Roche Applied Science) and phosphatase inhibitors (Roche Applied Science) and centrifuged at 13,000 × g for 15 min at 4°C to remove insoluble debris. Protein concentrations were analyzed using the BCA reagent (Pierce). Thirty μg of proteins were loaded and separated by SDS-PAGE and transferred to a 0.2 μm nitrocellulose membrane (Fisher Scientific). After blocking for 30 min in TBST (10 mM TRIS-HCl pH 8.0, 150 mM NaCl, 0.05% Tween-20) with 5% nonfat dry milk, membranes were incubated with the primary antibody for 1 h, washed and incubated for 30 min with horseradish peroxidase-conjugated secondary antibodies. The membranes were washed and incubated with an enhanced chemi-luminescence substrate (ECL; Thermo Scientific).
Proteomic analysis
2D DIGE (two-dimensional difference gel electrophoresis) and mass spectrometry protein identification were run by Applied Biomics. Image scans were performed immediately following the SDS-PAGE using Typhoon TRIO (Amersham BioSciences) following the protocols provided. The scanned images were then analyzed by Image QuantTL software (GE-Healthcare) and then subjected to in-gel analysis and cross-gel analysis using DeCyder software version 6.5 (GE-Healthcare). The ratio of protein differential expression was obtained from in-gel DeCyder software analysis. The selected spots were picked by an Ettan Spot Picker (GE-Healthcare) following the DeCyder software analysis and spot picking design. The selected protein spots were subjected to in-gel trypsin digestion, peptides extraction, desalting and followed by MALDI-TOF/TOF (Applied Biosystems) analysis to determine the protein identity.
Immunohistochemistry
Immunohistochemical staining was performed essentially as we previously described.13,26 Briefly, 5-µm sections from paraffin-embedded breast cancer tissue were de-paraffinized, and then rehydrated by passage through a graded series of ethanol. Antigen retrieval was performed by microwaving the slides in 100 mM sodium citrate buffer for 15 min. Endogenous peroxidase activity was quenched with 3% H2O2 for 20 to 30 min. Then, slides were washed with phosphate-buffered saline (PBS) and blocked with 10% goat serum in PBS for 1 h at room temperature. Samples were incubated with the primary antibodies diluted in 10% goat serum/PBS overnight at 4°C. The following antibodies were used: ACAT1 (HPA00428, Sigma), BDH1 (ab93931, Abcam), HMGCL (HPA004727, Sigma), HMGCS2 (AV41562, Sigma), HSP60 (ab46798, Abcam). After washing in PBS (three times, 5 min each), slides were stained with the LSAB2 system kit (Dako Cytomation), according to the manufacturer’s recommendations. Briefly, samples were incubated with biotinylated linker antibodies for 30 min, washed in PBS (three times, 5 min each) and then incubated with a streptavidin-horseradish peroxidase-conjugated solution for 30 min. After washing, samples were incubated with the diaminobenzidine reagent, until color production developed. Finally, the slides were washed in PBS and counterstained with hematoxylin, dehydrated and mounted with coverslips. Importantly, critical negative controls were performed in parallel for all of the immunohistochemical studies.
Supplementary Material
Acknowledgments
U.E.M. was supported by a Young Investigator Award from the Margaret Q. Landenberger Research Foundation. Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.). We thank Drs. Ruth Birbe and Agnieszka Witkiewicz (TJU; Department of Pathology) for providing human breast cancer samples.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental material may be found here: www.landesbioscience.com/journals/cc/article/22136
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22136
References
- 1.Henderson ST. Ketone bodies as a therapeutic for Alzheimer’s disease. Neurotherapeutics. 2008;5:470–80. doi: 10.1016/j.nurt.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zou Z, Sasaguri S, Rajesh KG, Suzuki R. dl-3-Hydroxybutyrate administration prevents myocardial damage after coronary occlusion in rat hearts. Am J Physiol Heart Circ Physiol. 2002;283:H1968–74. doi: 10.1152/ajpheart.00250.2002. [DOI] [PubMed] [Google Scholar]
- 3.Suzuki M, Suzuki M, Kitamura Y, Mori S, Sato K, Dohi S, et al. Beta-hydroxybutyrate, a cerebral function improving agent, protects rat brain against ischemic damage caused by permanent and transient focal cerebral ischemia. Jpn J Pharmacol. 2002;89:36–43. doi: 10.1254/jjp.89.36. [DOI] [PubMed] [Google Scholar]
- 4.Martinez-Outschoorn UE, Pestell RG, Howell A, Tykocinski ML, Nagajyothi F, Machado FS, et al. Energy transfer in “parasitic” cancer metabolism: mitochondria are the powerhouse and Achilles’ heel of tumor cells. Cell Cycle. 2011;10:4208–16. doi: 10.4161/cc.10.24.18487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Power surge: supporting cells “fuel” cancer cell mitochondria. Cell Metab. 2012;15:4–5. doi: 10.1016/j.cmet.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 6.Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol. 2012;7:423–67. doi: 10.1146/annurev-pathol-011811-120856. [DOI] [PubMed] [Google Scholar]
- 7.Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, et al. Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol. 2011;43:1045–51. doi: 10.1016/j.biocel.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laffel L. Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab Res Rev. 1999;15:412–26. doi: 10.1002/(SICI)1520-7560(199911/12)15:6<412::AID-DMRR72>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 9.Guzmán M, Blázquez C. Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol Metab. 2001;12:169–73. doi: 10.1016/S1043-2760(00)00370-2. [DOI] [PubMed] [Google Scholar]
- 10.Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle. 2010;9:3485–505. doi: 10.4161/cc.9.17.12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010;9:3506–14. doi: 10.4161/cc.9.17.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle. 2011;10:1271–86. doi: 10.4161/cc.10.8.15330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010;9:2423–33. doi: 10.4161/cc.9.12.12048. [DOI] [PubMed] [Google Scholar]
- 14.Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–42. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I, et al. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal. 2012;16:1264–84. doi: 10.1089/ars.2011.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 17.Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, et al. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol. 2009;174:2023–34. doi: 10.2353/ajpath.2009.080873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, Philp NJ, Lin Z, Gandara R, et al. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle. 2012;11:1108–17. doi: 10.4161/cc.11.6.19530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A, et al. Understanding the “lethal” drivers of tumor-stroma co-evolution: emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor microenvironment. Cancer Biol Ther. 2010;10:537–42. doi: 10.4161/cbt.10.6.13370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirano M, Davidson M, DiMauro S. Mitochondria and the heart. Curr Opin Cardiol. 2001;16:201–10. doi: 10.1097/00001573-200105000-00008. [DOI] [PubMed] [Google Scholar]
- 21.DiMauro S, Bonilla E, Davidson M, Hirano M, Schon EA. Mitochondria in neuromuscular disorders. Biochim Biophys Acta. 1998;1366:199–210. doi: 10.1016/S0005-2728(98)00113-3. [DOI] [PubMed] [Google Scholar]
- 22.DiMauro S, Hirano M. Mitochondria and heart disease. Curr Opin Cardiol. 1998;13:190–7. [PubMed] [Google Scholar]
- 23.Mercier I, Camacho J, Titchen K, Gonzales DM, Quann K, Bryant KG, et al. Caveolin-1 and accelerated host aging in the breast tumor microenvironment: chemoprevention with rapamycin, an mTOR inhibitor and anti-aging drug. Am J Pathol. 2012;181:278–93. doi: 10.1016/j.ajpath.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Authier S, Tremblay S, Dumulon V, Dubuc C, Ouellet R, Lecomte R, et al. [11C] acetoacetate utilization by breast and prostate tumors: a PET and biodistribution study in mice. Mol Imaging Biol. 2008;10:217–23. doi: 10.1007/s11307-008-0143-6. [DOI] [PubMed] [Google Scholar]
- 25.Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle. 2011;10:4047–64. doi: 10.4161/cc.10.23.18151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle. 2011;10:1772–83. doi: 10.4161/cc.10.11.15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.