

Abstract
We have previously proposed that catabolic fibroblasts generate mitochondrial fuels (such as ketone bodies) to promote the anabolic growth of human cancer cells and their metastasic dissemination. We have termed this new paradigm “two-compartment tumor metabolism.” Here, we further tested this hypothesis by using a genetic approach. For this purpose, we generated hTERT-immortalized fibroblasts overexpressing the rate-limiting enzymes that promote ketone body production, namely BDH1 and HMGCS2. Similarly, we generated MDA-MB-231 human breast cancer cells overexpressing the key enzyme(s) that allow ketone body re-utilization, OXCT1/2 and ACAT1/2. Interestingly, our results directly show that ketogenic fibroblasts are catabolic and undergo autophagy, with a loss of caveolin-1 (Cav-1) protein expression. Moreover, ketogenic fibroblasts increase the mitochondrial mass and growth of adjacent breast cancer cells. However, most importantly, ketogenic fibroblasts also effectively promote tumor growth, without a significant increase in tumor angiogenesis. Finally, MDA-MB-231 cells overexpressing the enzyme(s) required for ketone re-utilization show dramatic increases in tumor growth and metastatic capacity. Our data provide the necessary genetic evidence that ketone body production and re-utilization drive tumor progression and metastasis. As such, ketone inhibitors should be designed as novel therapeutics to effectively treat advanced cancer patients, with tumor recurrence and metastatic disease. In summary, ketone bodies behave as onco-metabolites, and we directly show that the enzymes HMGCS2, ACAT1/2 and OXCT1/2 are bona fide metabolic oncogenes.
Keywords: ketone body, 3-hydroxy-butyrate, cancer metabolism, BDH1, HMGCS2, OXCT isoforms, ACAT isoforms, tumor growth, metastasis
Introduction
Ketone bodies (3-hydroxy-butyrate, aceto-acetate and acetone) are naturally occurring mitochondrial fuels that are normally produced in the liver during periods of starvation.1,2 Then, they are shuttled via the blood stream to the brain, where neuronal cells have the capacity to convert them back into acetyl-CoA, so they can be re-utilized as mitochondrial fuels when nutrients are scarce. Locally, astrocytes also have the capacity to generate ketone bodies, to protect the mitochondrial metabolism of neurons. This biological process is known as “neuon-glia metabolic coupling.”3
We have recently provided evidence that human tumors may also share the same type of metabolic wiring as the brain. Briefly, we proposed that catabolic fibroblasts, with mitochondrial dysfunction, produce ketone bodies in the tumor stroma.4,5 Then, these ketone bodies are re-utilized by adjacent cancer cells, which process these ketone bodies as mitochondrial fuels for oxidative phosphorylation (OXPHOS), to drive anabolic tumor growth. Of course, this would require the expression of certain neuron-specific enzymes in cancer cells, such as ACAT1/2 and OXCT1/2.
Here, to directly test this hypothesis, we have created ketogenic fibroblasts by overexpressing the rate-limiting enzymes required for ketone production (HMGCS2 and BDH1). In addition, we generated MDA-MB-231 human breast cancer cells overexpressing the metabolic enzymes required for effective ketone re-utilization. Our results directly show that ketone body production and re-utilization can drive increased tumor growth and metastasis. Thus, ketone bodies behave as onco-metabolites, and we directly show that the enzymes HMGCS2, ACAT1/2 and OXCT1/2 are bona fide metabolic oncogenes.
As such, the enzymes associated with ketone body production and re-utilization should be considered as new “druggable” targets for anticancer therapy. As ketone bodies are preferentially used only during periods of starvation, we envision that limited side effects would result from anticancer therapy with ketone body inhibitors.
Results
Fibroblasts overexpressing ketogenic enzymes show increased autophagy, resulting in a loss of stromal Cav-1
To investigate the compartment-specific role of ketogenesis in breast cancer, we first overexpressed two key enzymes for ketone generation, HMGCS2 and BDH1, in hTERT-immortalized human fibroblasts. Figure 1 shows that HMGCS2 and BDH1 were highly overexpressed relative to empty vector control cells. Next, we evaluated if HMGCS2 and BDH1 overexpression is sufficient to mimic a starvation-state, inducing autophagy. To this end, HMGCS2, BDH1 and empty-vector control fibroblasts were analyzed by immunoblot with antibodies directed against autophagy markers.
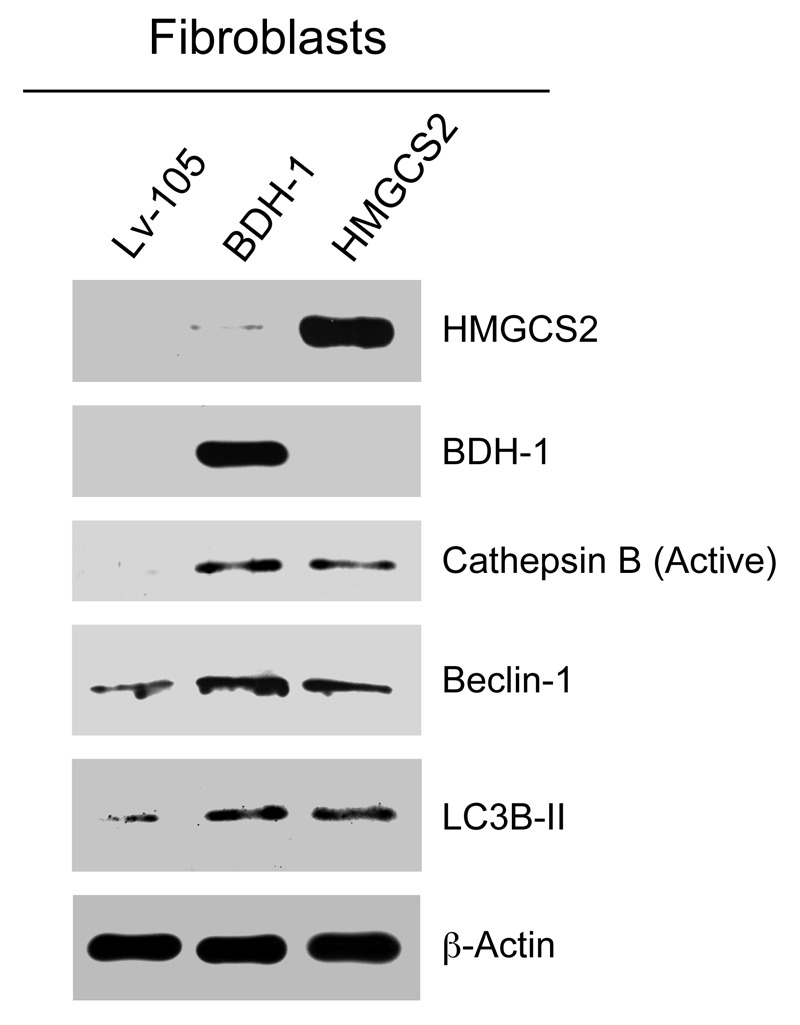
Figure 1. Fibroblasts overexpressing ketogenic enzymes show increased autophagy. hTERT fibroblasts overexpressing the ketogenic enzymes BDH1 and HMGCS2 were generated using a lentiviral approach. Western blot analysis was performed with antibodies against HMGCS2, BDH1, cathepsin B, beclin-1 and LC3B. Note that ketogenic fibroblasts overexpressing BDH1 and HMGCS2 show higher levels of the autophagy markers cathepsin B (active form), beclin-1 and LC3B-II (cleaved form) relative to control fibroblasts. Equal loading was assessed with β-actin.
Figure 1 shows that HMGCS2 and BDH1 overexpression both drive an autophagic program in fibroblasts, leading to the increased expression and activation of Cathepsin B (active form), Beclin-1 and LC3B (cleaved and active form).
Consistent with the idea that HMGCS2 and BDH1 overexpression in fibroblasts sensitizes them to the induction of autophagy, these fibroblasts were also more sensitive to the loss of Cav-1 upon co-culture with human breast cancer cells (MCF7) (Fig. 2). Previous studies have clearly documented that loss of Cav-1 expression under these co-culture conditions is due to its autophagic digestion, which is lysosome-dependent.6,7
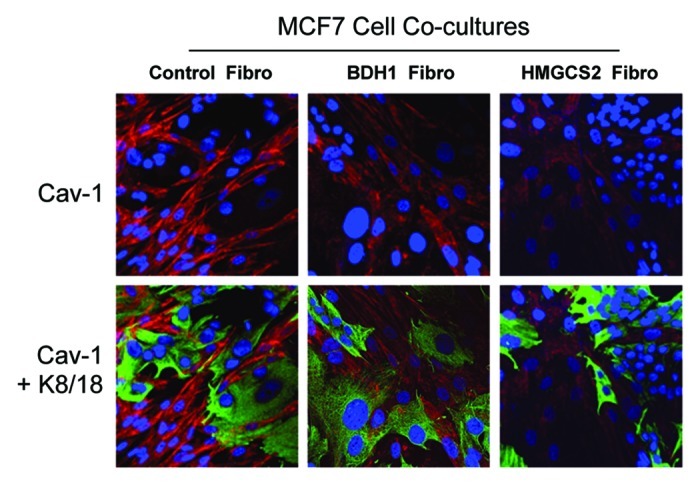
Figure 2. Upon coculture with MCF7 cells, fibroblasts overexpressing BDH1 or HMGCS2 show Cav-1 downregulation. Fibroblasts overexpressing BDH1, HMGCS2 or empty vector control were co-cultured with MCF7 cells for 5 d. Then, cells were fixed and immunostained with anti-Cav-1 (red) and anti-K8–18 (green) antibodies. Nuclei were stained with DAPI (blue). Cav-1 staining is shown in the top panels to better appreciate that upon coculture with MCF7 cells, fibroblasts overexpressing BDH1 or HMGCS2 show downregulation of Cav-1, as compared with MCF7 cells cocultured with control fibroblasts. Original magnification, 40x.
Fibroblasts overexpressing ketogenic enzymes induce mitochondrial biogenesis in MCF7 cells, as well as cell growth
To examine the paracrine effects of HMGCS2- and BDH1-overexpressing fibroblasts on adjacent cancer cells, they were co-culultured with MCF7 cells under low mitogen conditions.
Figure 3 directly shows that, under co-culture conditions, HMGCS2- and BDH1-overexpressing fibroblasts stimulate an increase in mitochondrial mass and/or biogenesis in MCF7 cells. Note the dramatic increase in the expression of two well-established markers of mitochondrial mass, including a mitochondrial membrane protein and the mitochondrial chaperone, HSP60.
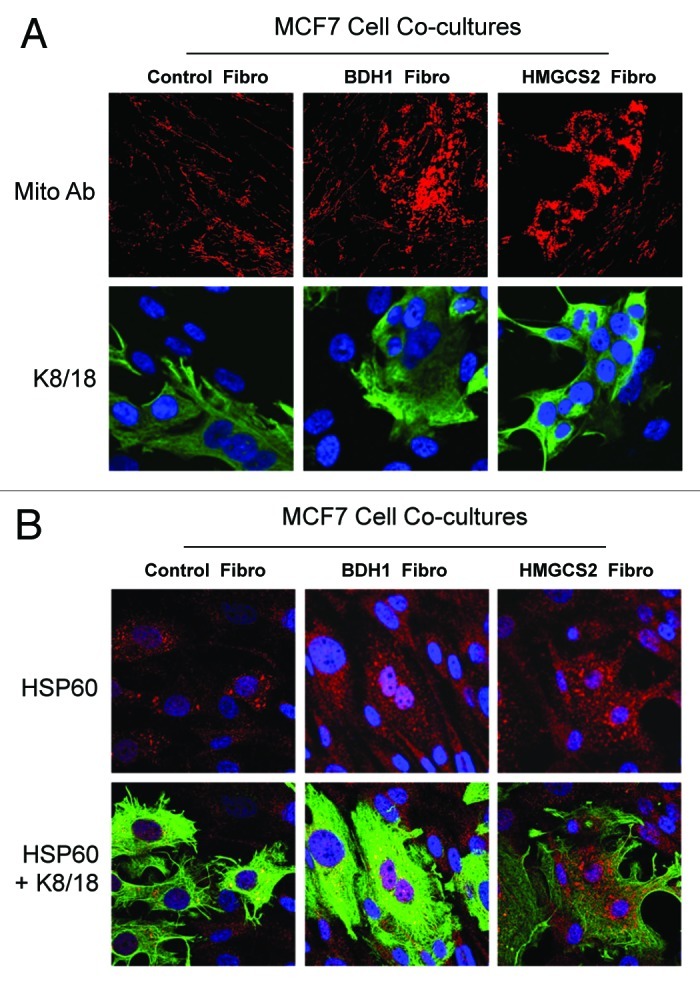
Figure 3. Coculture with fibroblasts overexpressing BDH1 or HMGCS2 induces mitochondrial biogenesis in MCF7 cells. Fibroblasts overexpressing BDH1, HMGCS2 or empty vector control were co-cultured with MCF7 cells for 5 d. Then, cells were fixed and immunostained with anti-mitochondrial membrane (A) or HSP60 (B) antibodies. MCF7 cells were labeled using anti-K8–18 (green) antibodies. (A) Mitochondrial membrane staining (red) is shown in the top panels to better illustrate that fibroblasts overexpressing BDH1 or HMGCS2 induce mitochondrial biogenesis in MCF7 cells. (B) HSP60 staining (red) is shown in the top panels to better appreciate that fibroblasts overexpressing BDH1 or HMGCS2 promote the expression of the mitochondrial chaperon HSP60 in MCF7 cells. Original magnification, 63x.
To examine the paracrine effects of ketogenic fibroblasts on MCF7 cell growth, we used quantitative FACS analysis. For this purpose, we assessed the cellular ratio between GFP(-) fibroblasts and GFP(+) MCF7 cells. Figure 4 shows that co-culture with HMGCS2- and BDH1-overexpressing fibroblasts, increases the proportion of MCF7 cells by > 2-fold, consistent with the idea that increased mitochondrial biogenesis in cancer cells “fuels” their proliferative capacity. FACS tracings are presented in Figure 4A, and bar graphs are shown in Figure 4B.

Figure 4. Coculture with fibroblasts overexpressing BDH1 or HMGCS2 increases the MCF7 cell population. GFP (+) MCF7 cells were grown in co-culture with fibroblasts harboring BDH1, HMGCS2 or the empty vector control for 5 d. Then, cells were trypsinized and analyzed by flow cytometry using a 488 nm laser, to identify the GFP (+) MCF7 cell population. Fibroblasts were identified as the GFP (-) cell population. (A) Traces of the relative percentage of MCF7-GFP (+) cells and GFP (-) fibroblasts. (B) Histogram representation of the relative percentage of MCF7 cells and fibroblasts. The presence of fibroblasts overexpressing BDH1 and HMGCS2 increases the proportion of MCF7 cells by > 2-fold. *p < 0.0002.
Fibroblasts overexpressing HMGCS2 promote the tumor growth of MDA-MB-231 breast cancer cells without increased neo-angiogenesis
Since ketogenic fibroblasts appeared to increase the proliferation of human breast cancer cells, we next tested their in vivo activity on tumor growth. Briefly, to evaluate the tumor-promoting properties of HMGCS2 fibroblasts, we used a xenograft model. HMGCS2 fibroblasts were co-injected with MDA-MB-231 human breast cancer cells into the flanks of athymic nude mice and allowed to grow for 4.5 weeks. Figure 5A shows that tumor weight and volume are increased by 2.8-fold in tumors derived from HMGCS2-overexpressing fibroblasts, as compared with empty vector control fibroblasts. However, the tumor-promoting effects of HMGCS2 fibroblasts were not due to increased tumor angiogenesis, as no significant differences in CD31(+) vascular density were noted (Fig. 5B).

Figure 5. Fibroblasts overexpressing HMGCS2 promote tumor growth of MDA-MB-231 breast cancer cells, without an increased vascularity. (A) To evaluate the tumor-promoting properties of HMGCS2 fibroblasts, we used a xenograft model. HMGCS2 or LV-105 control fibroblasts were co-injected with MDA-MB-231 breast cancer cells into the flanks of athymic nude mice. After 4.5 weeks tumors were analyzed. Note that tumor weight and volume are increased by 2.8-fold in tumors derived from HMGCS2 overexpressing fibroblasts, compared with the LV-105 empty vector control. p-values are as indicated. n = 10. (B) Tumor angiogenesis. To evaluate if HMGCS2 fibroblasts promote tumor growth by increasing tumor angiogenesis, frozen tumor sections were analyzed by CD31 immunostaining. Quantification of the number of CD31 (+) vessels per field indicates that vessel density is not increased in tumors overexpressing HMGCS2, relative to control tumors.
MDA-MB-231 human breast cancer cells overexpressing the enzymes necessary for ketone utilization show increased tumor growth and metastatic capacity
MDA-MB-231 breast cancer cells overexpressing ACAT1, ACAT2, OXCT1, OXCT2 or the empty vector were generated using a lentiviral approach. Figure 6 directly shows that we successfully overexpressed ACAT1/2 and OXCT1/2 in MDA-MB-231 cells.
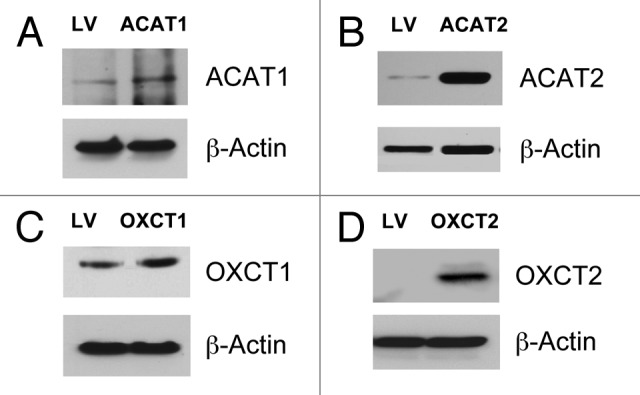
Figure 6. Generation of MDA-MB-231 cells overexpressing enzymes for ketone utilization. MDA-MB-231 breast cancer cells overexpressing ACAT1, ACAT2, OXCT1, OXCT2 or LV-105 empty vector control were generated using a lentiviral approach. Western blot analysis was performed with isoform-specific antibodies to confirm the overexpression of ACAT1 (A), ACAT2 (B), OXCT1 (C), OXCT2 (D), relative to empty vector control cells. Equal loading was assessed with β-actin.
Next, we assessed the tumorigenic effects of the overexpression expression ACAT isoforms or OXCT isoforms, using a xenograft model. As quantitatively similar results were obtained with both ACAT and OXCT isoforms, results are presented as ACAT1/2 and OXCT1/2, for simplicity.
Figure 7A shows that overexpression of ACAT1/2 and OXCT1/2 in human breast cancer cells directly promotes tumor growth. Note MDA-MB-231 cells overexpressing ACAT1/2 and OXCT1/2 show large increases in tumor growth, with up to a > 3-fold increase in tumor volume. However, little or no increases were observed in tumor angiogenesis (Fig. 7B). Thus, increased tumor vascularity could not account for the observed increases in tumor growth.

Figure 7. Overexpression of ACAT1/2 and OXCT1/2 in breast cancer cells promotes tumor growth. (A) Tumor growth. A xenograft model employing MDA-MB-231 breast cancer cells overexpressing an empty vector (Lv-105), ACAT1/2 or OXCT1/2 were injected into the flanks of athymic nude mice. Tumor weights and volumes were measured 4 weeks post-injection. Note that MDA-MB-231 cells overexpressing ACAT1/2 and OXCT1/2 greatly promote tumor growth, resulting in a 2-fold increase in tumor weight and a 3-fold increase in tumor volume, respectively, relative to control cells. (B) Tumor angiogenesis. Tumor frozen sections were cut and immunostained with anti-CD31 antibodies and vascular density (number of vessels per field) was quantified. Note that ACAT1/2 overexpression leads to a 20% increase in vessel density, while no significant change was seen in xenografts with MDA-MB-231 overexpressing OXCT1/2.
Finally, we used a lung colonization assay to assess possible effects on metastatic capacity. MDA-MB-231 cells overexpressing ACAT1/2 or OXCT1/2 were injected into the tail veins of athymic nude mice. After 8 weeks, the lungs were insuflated, and lung metastasis were scored. Figure 8 shows that MDA-MB-231 cells overexpressing ACAT1/2 greatly promote lung metastasis, as compared with control cells and OXCT1/2-overexpressing cells.
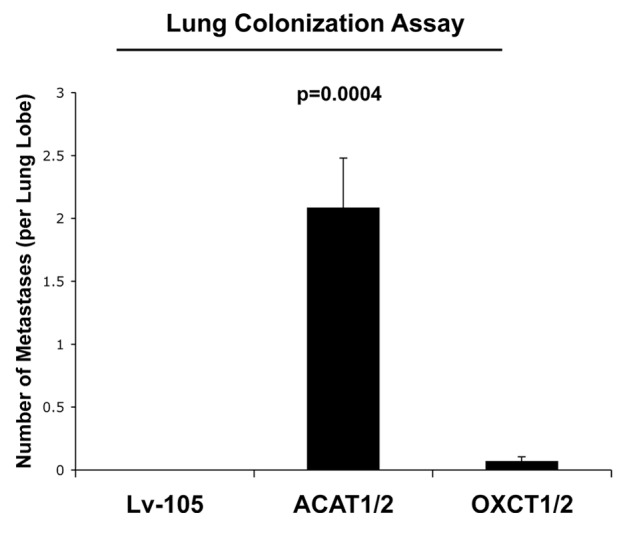
Figure 8. Overexpression of ACAT1/2 in breast cancer cells promotes lung metastasis. We used a lung metastasis assay model of MDA-MB-231 cells overexpressing empty vector (Lv-105), ACAT1/2 or OXCT1/2 injected into the tail vein of athymic nude mice. After 8 weeks, lungs were insuflated and lung metastases were scored. Note that MDA-MB-231 cells overexpressing ACAT1/2 greatly promote lung metastasis as compared with control cells and OXCT1/2-overexpressing cells.
Proteomic profile of MDA-MB-231 cells treated with ketone bodies
To assess the possible phenotypic effects of ketone body utilization on parental MDA-MD-231, they were treated with 3-hydroxy-butyrate. The results of this analysis are summarized in Table S1.
Note that treatment with 3-hydroxy-butyrate quantitatively increases the expression of protein markers associated with (1) protein synthesis, (2) an epithelial-mesenchymal transition (EMT), (3) protein folding and calcium homeostasis, as well as certain (4) neuronal proteins. Interestingly, neurons are known to be one of the few cell types that express the rare enzymes associated with ketone utilization (ACAT1/2 and OXCT1/2).1,3
Discussion
Previously, we showed that i.p. injection of 3-hydroxy-butyrate in an MDA-MB-231 xenograft model significantly increased tumor growth by ~2.5-fold, without any increases in tumor angiogenesis.8 Similarly, treatment of MCF7 breast cancer cells with 3-hydroxy-butyrate was combined with genome-wide transcriptional profiling to generate a novel gene signature that effectively predicts tumor recurrence, metastasis and poor clinical outcome in human breast cancer patients.9 However, these results do not establish a direct cause-effect relationship between ketone body production, re-utilization and tumor progression.
To address this issue, here we have provided new genetic evidence that ketone bodies are critical for promoting tumor growth and metastasis. First, we generated ketogenic fibroblasts, by overexpressing the rate-limiting enzymes required for ketogenesis, namely HMGCS2 and BHD1. As a complementary approach, we also generated human breast cancer cells that were equipped with the necessary enzymes to drive ketone body re-utilization. Importantly, our results directly show that ketogenic fibroblasts promote the growth of adjacent breast cancer cells, by driving increased mitochondrial biogenesis. Thus, the tumor stroma may serve as a reservoir for ketone body production, while cancer cells upregulate the enzymes required for ketone body re-utilization, driving oxidative mitochondrial metabolism (OXPHOS) in epithelial cancer cells (Fig. 9).
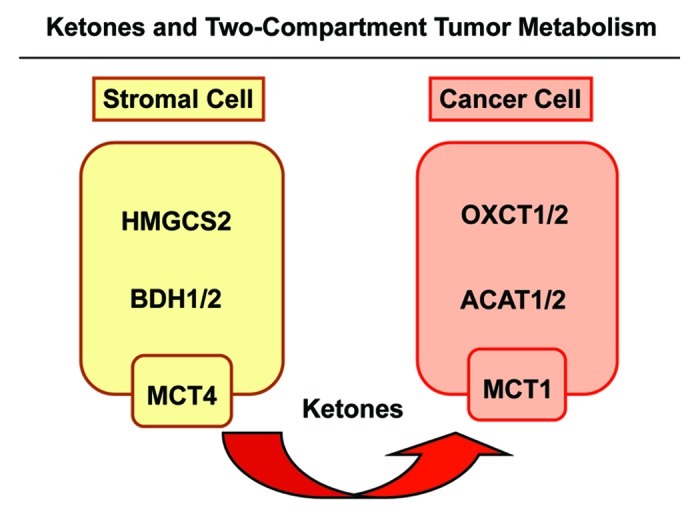
Figure 9. Ketones and two-compartment tumor metabolism. Cancer-associated fibroblasts are catabolic and undergo autophagy/mitophagy. This results in ketone body production, via HGMCS2 and BDH isotorms, driving the production of 3-hydroxy-butyrate in the tumor stroma. These ketone bodies are then transferred from stromal fibroblasts to adjacent cancer cells, via mono-carboxylate transporters (MCT1/4). Then, in cancer cells, stromally derived ketone bodies are converted to acetyl-CoA, using the metabolic enzymes OXCT1/2 and ACAT1/2. After conversion to acetyl-CoA, energy is generated via the TCA cycle and oxidative mitochondrial metabolism (OXPHOS).
To prevent this form of “two-compartment tumor metabolism,” ketone inhibitors should be designed to halt ketone body production in cancer-associated fibroblasts and ketone body re-utilization in epithelial cancer cells. This simple strategy could effectively starve cancer cells to death by “cutting off their fuel supply.”
Finally, it is worth noting that ketogenic fibroblasts were more prone to a loss of stromal Cav-1 expression. In breast cancer patients, a loss of stromal Cav-1 expression is associated with increased tumor recurrence, metastasis, drug resistance and overall poor clinical outcome.10-13 Thus, stromal Cav-1 could be used as a biomarker to select patients that would be more likely to benefit from therapy with ketone inhibitors, allowing biomarker-based treatment stratification and personalized cancer therapy.
Materials and Methods
Materials
Antibodies were as follows: HMGCS2 (AV41562, Sigma); ACAT1 (HPA007569, Sigma); ACAT2 (HPA025736, Sigma); BDH1 (ab6834, Abcam); HMGCL (WH03155, Sigma); cathepsin B (sc13985, Santa Cruz Biotech); Beclin-1 (NBPI-00085, Novus Biologicals); LC3B (ab48394, Abcam); Cav-1 (sc-894, Santa Cruz Biotech); HSP60 (ab46798, Abcam); cytokeratin 8/18 (20R-CP004, Fitzgerald Industries International); surface of intact mitochondria (MAB1273, Millipore); β-tubulin and β-actin (Sigma); secondary antibodies for immunofluorescence were Alexa-Green 488 nm and Alexa Orange-Red 546 nm (Invitrogen). Other reagents were as follows: 4,6-diamidino-2- phenylindole (DAPI) (D3571), Prolong Gold Antifade mounting reagent (P36930) (Invitrogen).
Cell cultures
Human skin fibroblasts immortalized with telomerase reverse transcriptase protein (hTERT-BJ-1) were originally purchased from Clontech, Inc. The breast cancer cell lines MCF7 and MDA-MB-231 were purchased from ATCC. All cells were maintained in DMEM with 10% Fetal Bovine Serum (FBS) and Penicillin 100 units/mL-Streptomycin 100 µg/mL.
Lentivirus production and cell line derivation
For lentivirus production, 293Ta-packaging cells (Genecopoeia) were transfected with different lentiviral plasmids (all from Genecopoeia). The following plasmids were used: EX-neg-Lv105 (the empty vector control), EX-V0811-Lv105 (encoding BDH1), EX-I0546-Lv105 (encoding HMGCS2), EX-C0185-Lv105 (encoding ACAT1), EX-C0676-Lv105 (encoding ACAT2), EX-M0222-Lv105 (encoding OXCT1) and EX-T1301-Lv105 (encoding OXCT2). All plasmids contained a puromycin resistance marker. The resulting lentiviral particles were then used to infect fibroblasts and MDA-MB-231 cells to obtain stable cell lines overexpressing the target gene of interest.
Immunoblotting
Cells were harvested in lysis buffer (10 mM TRIS-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 60 mM octylglucoside) containing protease inhibitors (Roche Applied Science) and phosphatase inhibitors (Roche Applied Science) and centrifuged at 13,000 × g for 15 min at 4°C to remove insoluble debris. Protein concentrations were analyzed using the BCA reagent (Pierce). Thirty μg of proteins were loaded and separated by SDS-PAGE and transferred to a 0.2 μm nitrocellulose membrane (Fisher Scientific). After blocking for 30 min in TBST (10 mM TRIS-HCl pH 8.0, 150 mM NaCl, 0.05% Tween-20) with 5% nonfat dry milk, membranes were incubated with the primary antibody for 1 h, washed and incubated for 30 min with horseradish peroxidase-conjugated secondary antibodies. The membranes were washed and incubated with an enhanced chemiluminescence substrate (ECL; Thermo Scientific).
Co-cultures of MCF7 cells and fibroblasts
hTERTfibroblasts and MCF7 cells were plated on glass coverslips in 12-well plates in 1 ml of complete media. MCF7 cells were plated within 2 h of fibroblast plating. The total number of cells per well was 1 ×105, at a 5:1 fibroblast-to-epithelial cell ratio. The day after plating, media was changed to DMEM with 10% NuSerum (a low-protein alternative to FBS; BD Biosciences) and Pen-Strep. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2 for 5 d.
Immunocytochemistry
Immunocytochemistry was performed as previously described.14,15 All steps were performed at room temperature. Briefly, after 30 min fixation in 2% paraformaldehyde, cells were permeabilized for 10 min with immunofluorescence (IF) buffer (PBS, 0.2% BSA, 0.1% TritonX-100). Then, cells were incubated for 10 min with NH4Cl in PBS to quench free aldehyde groups. Primary antibodies were incubated in IF buffer for 1 h. After washing with IF buffer (3×, 10 min each), cells were incubated for 30 min with fluorochrome-conjugated secondary antibodies diluted in IF buffer. Finally, slides were washed with IF buffer (3×, 10 min each), incubated with the DAPI nuclear stain and mounted.
Confocal microscopy
Images were collected with a Zeiss LSM510 meta confocal system using a 405 nm Diode excitation laser with a band pass filter of 420–480 nm, a 488 nm Argon excitation laser with a band pass filter of 505–550 nm, and a 543 nm HeNe excitation laser with a 561–604 nm filter. Images were acquired with 40× or 63× objectives, as stated in the figure legends.
Flow cytometric analysis
GFP (+) MCF7 cells were grown in co-culture with hTERTfibroblasts for 5 d. Then, cells were trypsinized and analyzed by flow cytometry. The GFP (+) MCF7 cell population was identified using the 488 nm laser. Fibroblasts were identified as the GFP (-) cell population. The percentage of GFP (+) cells over the total cell population was calculated using FlowJo 8.8 software.
Animal studies
Animals were housed and maintained in a pathogen-free environment/barrier facility at the Kimmel Cancer Center at Thomas Jefferson University under National Institutes of Health (NIH) guidelines. Mice were kept on a 12-h light/dark cycle with ad libitum access to chow and water. Approval for all animal protocols used for this study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC). Briefly, MDA-MB-231 tumor cells (1 × 106 cells) alone or mixed with fibroblasts (3 × 105 cells) were resuspended in 100 μl of sterile PBS and were co-injected into the flanks of athymic NCr nude mice (NCRNU; Taconic Farms; 6–8 weeks of age). After 4–5 weeks, mice were sacrificed; tumors were excised, measured and frozen in liquid nitrogen cooled isopentane.
Quantification of tumor angiogenesis
Six-μm tumor frozen sections were fixed with 4% paraformaldehyde in PBS for 10 min at 4°C and washed 3× with PBS. A three-step biotin-streptavidin-horseradish peroxidase method was used for antibody detection. After fixation, the sections were blocked with 10% rabbit serum and incubated overnight at 4°C with rat monoclonal CD31 antibody (550274, BD Biosciences). The sections were then incubated with biotinylated rabbit anti-rat IgG (Vector Labs) and streptavidin-HRP (Dako). Immunoreactivity was revealed with 3, 3′ diaminobenzidine. For quantitation of vessels, CD31-positive vessels were counted in 4–6 fields of each tumor using a 20× objective lens and an ocular grid (0.25 mm2 per field). The total numbers of vessel per unit area was calculated, and the data was represented graphically.
Proteomic analysis
2D DIGE (two-dimensional difference gel electrophoresis) and mass spectrometry protein identification were run by Applied Biomics. Image scans were performed immediately following the SDS-PAGE using Typhoon TRIO (Amersham BioSciences) following the protocols provided. The scanned images were then analyzed by Image QuantTL software (GE-Healthcare) and then subjected to in-gel analysis and cross-gel analysis using DeCyder software version 6.5 (GE-Healthcare). The ratio of protein differential expression was obtained from in-gel DeCyder software analysis. The selected spots were picked by an Ettan Spot Picker (GE-Healthcare), following the DeCyder software analysis and spot picking design. The selected protein spots were subjected to in-gel trypsin digestion, peptides extraction, desalting and followed by MALDI-TOF/TOF (Applied Biosystems) analysis to determine the protein identity.
Experimental metastasis assay
To analyze metastatic potentials, a lung colonization assay was performed. Briefly, MDA-MB-231 cancer cells (1 × 106 cells) were re-suspended in 100 μl of sterile PBS and injected in the tail vein of athymic NCr nude mice (NCRNU; Taconic Farms; at 6–8 weeks of age). After 8 weeks, mice were sacrificed, and the lungs were insuflated with 15% India ink dye, washed in water and bleached in Fekete’s solution (70% ethanol, 3.7% paraformaldehyde, 0.75 M glacial acetic acid). A low-power stereo microscope was then used to determine the number of negatively stained metastatic colonies per lung lobe.
Supplementary Material
Acknowledgments
U.E.M. was supported by a Young Investigator Award from the Margaret Q. Landenberger Research Foundation. Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental material may be found here: www.landesbioscience.com/journals/cc/article/22137
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22137
References
- 1.Henderson ST. Ketone bodies as a therapeutic for Alzheimer’s disease. Neurotherapeutics. 2008;5:470–80. doi: 10.1016/j.nurt.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laffel L. Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab Res Rev. 1999;15:412–26. doi: 10.1002/(SICI)1520-7560(199911/12)15:6<412::AID-DMRR72>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 3.Guzmán M, Blázquez C. Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol Metab. 2001;12:169–73. doi: 10.1016/S1043-2760(00)00370-2. [DOI] [PubMed] [Google Scholar]
- 4.Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Power surge: supporting cells “fuel” cancer cell mitochondria. Cell Metab. 2012;15:4–5. doi: 10.1016/j.cmet.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 5.Martinez-Outschoorn UE, Pestell RG, Howell A, Tykocinski ML, Nagajyothi F, Machado FS, et al. Energy transfer in “parasitic” cancer metabolism: mitochondria are the powerhouse and Achilles’ heel of tumor cells. Cell Cycle. 2011;10:4208–16. doi: 10.4161/cc.10.24.18487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010;9:2423–33. doi: 10.4161/cc.9.12.12048. [DOI] [PubMed] [Google Scholar]
- 7.Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, et al. Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol. 2011;43:1045–51. doi: 10.1016/j.biocel.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010;9:3506–14. doi: 10.4161/cc.9.17.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle. 2011;10:1271–86. doi: 10.4161/cc.10.8.15330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol. 2012;7:423–67. doi: 10.1146/annurev-pathol-011811-120856. [DOI] [PubMed] [Google Scholar]
- 11.Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, et al. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol. 2009;174:2023–34. doi: 10.2353/ajpath.2009.080873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, Philp NJ, Lin Z, Gandara R, et al. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle. 2012;11:1108–17. doi: 10.4161/cc.11.6.19530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu KN, Queenan M, Brody JR, Potoczek M, Sotgia F, Lisanti MP, et al. Loss of stromal caveolin-1 expression in malignant melanoma metastases predicts poor survival. Cell Cycle. 2011;10:4250–5. doi: 10.4161/cc.10.24.18551. [DOI] [PubMed] [Google Scholar]
- 14.Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle. 2011;10:4047–64. doi: 10.4161/cc.10.23.18151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle. 2011;10:1772–83. doi: 10.4161/cc.10.11.15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.