

Abstract
During DNA replication, stalled replication forks and DSBs arise when the replication fork encounters ICLs (interstrand crosslinks), covalent protein/DNA intermediates or other discontinuities in the template. Recently, homologous recombination proteins have been shown to function in replication-coupled repair of ICLs in conjunction with the Fanconi anemia (FA) regulatory factors FANCD2-FANCI, and, conversely, the FA gene products have been shown to play roles in stalled replication fork rescue even in the absence of ICLs, suggesting a broader role for the FA network than previously appreciated. Here we show that DNA2 helicase/nuclease participates in resection during replication-coupled repair of ICLs and other replication fork stresses. DNA2 knockdowns are deficient in HDR (homology-directed repair) and the S phase checkpoint and exhibit genome instability and sensitivity to agents that cause replication stress. DNA2 is partially redundant with EXO1 in these roles. DNA2 interacts with FANCD2, and cisplatin induces FANCD2 ubiquitylation even in the absence of DNA2. DNA2 and EXO1 deficiency leads to ICL sensitivity but does not increase ICL sensitivity in the absence of FANCD2. This is the first demonstration of the redundancy of human resection nucleases in the HDR step in replication-coupled repair, and suggests that DNA2 may represent a new mediator of the interplay between HDR and the FA/BRCA pathway.
Keywords: DNA crosslink repair, DNA replication, FANCD2, Fanconi anemia, double-strand break repair, recombination
Introduction
FA (Fanconi anemia) is a disease characterized by developmental abnormalities, bone marrow failure and cancer predisposition. At the cellular level, FA is characterized by extreme sensitivity to ICLs (interstrand crosslinks). When a replication fork encounters an ICL, unwinding cannot occur, and a host of replication proteins, nucleases, translesion polymerases and recombination proteins constitute a multigene repair pathway, the FA/BRCA (breast cancer) pathway, to resolve the block (Fig. S1). The FA/BRCA pathway is fundamental, however, not just to repair of ICLs, but also to the repair of damage elicited by DNA replication fork stalling due to numerous endogenous and exogenous insults.1-7
The recombination step in the FA/BRCA pathway requires resection of duplex ends and is poorly characterized compared with other steps.8-11 Resection of DSBs was first delineated in yeast, Saccharomyces cerevisiae.12 After the induction of a DSB, the MRX (Mre11, Rad50, Xrs2) complex and Sae2 (MRN and CtIP in human) can carry out a limited resection reaction of about 100 bp. A second, long-range resection reaction provides the 3′ ssDNA overhang that binds RPA and RAD51, the DNA strand exchange protein, which are important for HDR (homology-directed repair) and checkpoint signaling. In one long-range resection machine, EXO1 (exonuclease 1) degrades the 5′ strands, but is redundant with a second pathway involving a complex of Sgs1 3′–5′ DNA helicase and the Dna2 nuclease. It is not clear what directs the division of labor between the two resection pathways. Understanding resection in the FA/BRCA pathway, specifically, is important, because end resection enhances the chromatin association of the key regulatory complex, FANCD2/FANCI, at DSBs and directs the choice of pathway in repair, either error-free HDR or NHEJ (nonhomologous end-joining), which can lead to deleterious rearrangements (Fig. S1). MRN (Mre11, Rad50, Nbs1) and CtIP have been implicated in FA/BRCA short-range resection reaction,1,3 but long-range resection may also occur,9 and the long-range apparatus, if any, has not been identified.
DNA2 or EXO1 are good candidates for the predicted long-range resection nucleases. Biochemical reconstitution experiments by our lab and others define at least three DSB long-range resection pathways in human cells.13,14 In one mechanism, BLM helicase and DNA2 nuclease resect DNA; in a second pathway, BLM directly stimulates the 5′–3′ nucleolytic activity of EXO113,14; in the third pathway, DNA2, EXO1 and BLM cooperate in resection.14 In vivo, DNA2 deficiency leads to partial defects in resection,15 and BLM and EXO1 identify two redundant DSB repair pathways.16 Two critical questions are to what extent are the BLM/DNA2 and EXO1 pathways redundant, complementary or cooperative, and how do these proteins protect stressed DNA replication forks.
DNA2 is perhaps of greatest interest with respect to resection of DSBs occurring during replication, because it is an intrinsic replication fork protein.15,17,18 Human DNA2, like yeast DNA2, functions in both mitochondria and nuclei.17,19,20 DNA2 depletion in human cells results in DNA damage, as evidenced by increased chromosome breakage, interchromosomal bridges and micronuclei, induction of γH2AX repair foci and induction of the ATR-dependent S phase checkpoint, resulting in a severe G2 delay of the cell cycle.17,18 The putative role of DNA2 in DNA replication is in processing of Okazaki fragments (or in repair of faulty processing). Breaks, however, did not accumulate during ongoing replication after silencing human DNA2, nor was fork slowing observed.18 Instead, origin firing is suppressed.18 Origin suppression is known to lead to the instability of common DNA fragile sites, long, origin-poor chromosome domains that may experience increased stalling or incomplete or delayed resolution of stalled forks by forks converging from distant origins.21 This suggested to us that DNA2 might also be required both for replication and for a post-replicative DNA repair step in replication completion. We now demonstrate that DNA replication-linked DNA damage accumulates when DNA2 is depleted, at least in part due to defects in repair. We show that DNA2 is involved in resection of DSBs occurring during replication stress, and that EXO1 is redundant with DNA2. DNA2 and EXO1 also redundantly regulate DSB repair pathway choice by inhibiting induction of phosphorylation of the NHEJ-related kinase DNA-PKcs. Most important, in beginning to delineate between the roles of DNA2 and EXO1 in multiple DSB repair pathways, we present evidence that DNA2 plays a prominent role in coordinating resection and checkpoint activation in the FA/BRCA pathway of ICL repair. Given its constitutive role in DNA replication, DNA2 may constitute a direct link between replication fork failure and HDR.
Results
DNA2 and EXO1 nucleases play redundant roles in resection of replication-associated DSBs
To study the respective roles of DNA2 and EXO1 in replication-linked DSB repair in vivo, we either singly or simultaneously depleted DNA2 and EXO1 using shDNA2 and siEXO1, respectively, in U2OS cells. We achieved a depletion efficiency of at least 80% for each of the targeted mRNAs (Fig. 1A). (Neither DNA2 nor EXO1 protein, very low abundance proteins, are detectable on western with available antibodies). The cell cycle distribution of such deletions shows an increased S and G2 population, the portion of the cell cycle we wish to monitor.18 We then investigated resection of CPT (camptothecin)-induced damage in the depleted cells. CPT inhibits DNA replication by inhibition of topoisomerase events critical for maintaining normal topological tension during elongation. Such inhibition has been proposed to underlie formation of aberrant intermediates with double-stranded ends (reversed forks).22 In addition, CPT causes DNA damage by stabilizing topoisomerase I-DNA adducts and preventing DNA ligation after cleavage, leading to DSBs upon subsequent DNA replication. Key proteins involved in HDR, such as the MRN/CtIP complex, are important in the repair of the latter type of CPT-induced damage.23-25 The MRN complex and RPA colocalize at sites of DNA damage after treatment with CPT,24 and cells defective in resection are sensitive to CPT.16 It is not possible, as was done for studying synchronously induced, site-specific DSBs in yeast, to directly measure resection in human cells using hybridization to specific probes. We therefore used several indirect methods from which the 3′ ssDNA produced by resection can be inferred.26-28 We first examined the phosphorylation status of RPA, a heterotrimeric ssDNA-binding complex comprised of RPA1, RPA2 and RPA3, which binds the resected single-stranded DNA. ssDNA-bound RPA2 is phosphorylated by the ATR checkpoint kinase at serine 3329 and then by the PI3 kinase DNA-PKcs at serine 4 and 830 in response to DNA DSB damage. Therefore, impaired hyperphosphorylation of RPA2 can be used as a surrogate for reduced production of ssDNA by resection.16 Cells with DNA2 and/or EXO1 depletion were treated with CPT and extracts prepared. Immunoblotting experiments using antibodies that recognize RPA2-pS4/S8 and RPA2-pS33 revealed that in the presence of DNA damage, RPA2 was robustly phosphorylated in SCR (scrambled) controls. The DNA2/EXO1 doubly depleted cells had markedly diminished levels of RPA2-pS4/S8 and pS33 compared to the SCR control and to singly depleted DNA2 and EXO1 (Fig. 1B). Notably, in the DNA2-depleted cells, there was a modest but reproducible increase in RPA phosphorylation even in the absence of CPT (Fig. 1B, lanes 2 and 4, and see also Fig. 2), which we have shown elsewhere is likely due to endogenous DNA damage occurring in the absence of DNA2.18

Figure 1. DNA2 and EXO1 are required for resection at CPT-induced damage. (A) Targeting of DNA2 and/or EXO1 with shDNA2 and EXO1 siRNA resulted in at least 80% reduction of the respective mRNA levels. (B) After exposure to CPT (1 μM, 1 h), phosphorylation of RPA2 S4/S8, S33 and CHK1 S345 is reduced in DNA2/EXO1-depleted U2OS cells. (C) A second shRNA targeting DNA2 (pRESQ shDNA2’) used together with EXO1 siRNA for double depletion, recapitulated results shown in (B). (D) Retrovirally expressed RNAi-resistant DNA2 restored RPA phosphorylation in cells depleted using shDNA2’ and EXO1 siRNA. Chk1 is also shown in addition to RPA (Figure presents an image with intervening lanes removed for clarity.). Experiments were performed at least twice and representative results are shown. See Experimental Procedures for details.

Figure 2. DNA2 and EXO1 are required for resection at ICL-induced damage. (A) shDNA2/EXO1 siRNA-treated cells exposed to cisplatin (15 μM, 24 h) show reduced phosphorylation of RPA2 S4/S8, S33 and Chk1 S345 compared with shSCR-treated cells . (B) shDNA2’/EXO1 siRNA-treated cells exposed to cisplatin (15 μM, 24 h) show reduced phosphorylation of RPA2 S4/S8, S33 and Chk1 S345 compared with shSCR-treated cells and ectopically expressed, RNAi-resistant DNA2 restored RPA and Chk1 phosphorylation in depleted U2OS cells (C). CDDP,cisplatin. Experiments were performed at least twice. See also Figure S2. See Experimental Procedures for details.
We confirmed that the diminished levels of RPA2-pS4/S8 phosphorylation after CPT were not due to off-target shRNA effects in two ways. First, we repeated the experiments with another shRNA, shDNA2′, described previously.17,18 Interestingly, shDNA2′ significantly reduced RPA phosphorylation even in the presence of EXO1 (Fig. 1C; Fig. S2), confirming our previous in vitro work and another recent report.14,15 This appeared to be due to a greater reduction in levels of DNA2 by shDNA2′ than shDNA2 (Fig. S2 and not shown). Second, we rescued RPA phosphorylation by ectopically expressing RNAi-resistant DNA2 (compare Fig. 1C and D), also as we described previously.17,18Figure S2 verifies the efficiency of expression of RNAi-resistant DNA2 cDNA.
Role of DNA2 resection in repair of interstrand crosslinks
It is likely that DNA2 and EXO1 function in conjunction with other proteins, such as components of the FA/BRCA network. Cisplatin causes ICLs between two opposite guanines, which physically block converging replication forks. This lesion is thought to be converted to a DSB during replication-associated ICL removal by the FA/BRCA pathway (Fig. S1). Mechanistic studies have demonstrated a role for at least 700 bp of resection and RAD51-dependent strand invasion into the sister chromatid in replication-coupled repair of a single, site-specific cisplatin ICL in Xenopus extracts.9-11 However, the long-range nuclease(s), if any, involved in resection have not been identified.
When DNA2- or DNA2 and EXO1-depleted cells were treated with cisplatin, RPA2-pS33 was drastically reduced in the double depletion compared with controls (Fig. 2A), as with CPT (Fig. 1). Interestingly, when exposed to cisplatin, DNA2-depleted cells, but not EXO1-depletes, had a reduction in RPA2-pS33 equivalent to that seen in the DNA2/EXO1 co-depleted cells (compare Fig. 2A lane 6 and lane 8). Similar results were obtained with a second shRNA, shDNA2′ (Fig. 2B and C), and expressing wild-type, RNAi-resistant DNA2 rescued the DNA2 depletion defects in cisplatin-treated cells. (The results for single knockdowns show some variability from experiment to experiment, but the double knockdowns are consistently defective). RPA phosphorylation was also diminished when we used a second shRNA, shDNA2’ (Fig. 2B and C; Fig. S2), ruling out off-target effects. [The reduced RPA-pS33 did not seem to significantly affect RPA2-pS4/S8 (Fig. 2A)]. This raises the intriguing question of whether there is a threshold phosphorylation at S33 that can “prime” the hyperphosphorylation of pS4/S8.31 Alternatively, DNA2 may have another role in repair of ICL damage that more directly affects the phosphorylation of RPA2-S33. The ICL-induced reduction in RPA2-pS33 observed in DNA2 singly-depleted cells, but not in EXO1-depleted cells, correlates with increased sensitivity of DNA2 depletions to chronic or acute treatment with cisplatin and physical interaction between DNA2 and FANCD2 (see below). Thus, we propose that DNA2 and EXO1 also participate in ICL repair during DNA replication, even though the initiating lesion is somewhat different from that encountered by CPT (Fig. 1).
Failure to hyperactivate the ATR and Chk1 DNA damage response in DNA2- and/or EXO1-depleted cells treated with cisplatin and other replication-coupled DNA damaging agents
The RPA-coated ssDNA overhangs generated by resection are crucial for ATR checkpoint kinase recruitment and full activation.27,32-34 Once activated, ATR phosphorylates the effector checkpoint kinase, Chk1, on S345 to promote cell cycle delay and repair. Thus, the appearance of Chk1-pS345 provides both an additional readout for resection and an indirect measure of ATR activation. DNA2 and/or EXO1 were depleted from U2OS cells, and cells were exposed to cisplatin or CPT, as above. With cisplatin, consistent Chk1-S345 reduction was seen only with the double depletion (Fig. 2A and B). Phosphorylation could be rescued by expression of RNAi-resistant DNA2 (Fig. 2C, lane 4; Fig. S2B).
With CPT, we consistently observed a greater reduction in Chk1-pS345 phosphorylation in doubly DNA2/EXO1-depleted cells than in either single depletion (Fig. 1B). The reduction in doubly depleted cells was equivalent to that observed with CtIP depletion, further verifying that this readout correlates with a resection defect.16 In the presence of RNAi-resistant DNA2, we observed no reduction in Chk1 phosphorylation in response to CPT (Fig. 1D). We also note that in the absence of exogenous damage, there is a modest increase in RPA2-pS4/S8 and Chk1-pS345 (Figs. 1 and 2), again consistent with our recent demonstration that there is endogenous DNA replication damage due to DNA2 deficiency and activation of the DNA damage response.18 We conclude, that DNA2 and EXO1 participate in partially redundant pathways of resection in response to cisplatin or CPT, though DNA2 appears to have a greater role than EXO1 or may coordinate with EXO1.
To more directly demonstrate a role for DNA2 and EXO1 in ssDNA generation, we monitored ssDNA accumulation using BrdU (5-bromodeoxyuridine) antibody. BrdU is incorporated into double-stranded DNA but is only detectable with anti-BrdU antibody in regions of single-stranded DNA. Cells with single or double depletion of DNA2 and EXO1 were cultured in BrdU for several generations to uniformly label chromosomes, be exposed to CPT and be stained for BrdU foci without denaturation. CtIP depletion was used as a positive control.25,35 We observed a significant reduction in BrdU focus formation in cells having simultaneous DNA2 and EXO1 depletion (6%) compared with DNA2 or EXO1 depletion alone (19% and 29%, respectively), similar to the reduction seen in CtIP-depleted cells (Fig. 3A).25,35 The single DNA2- or EXO1-depleted cells were also slightly impaired compared with the SCR-treated controls, which showed 36% of cells with BrdU foci, suggesting partially independent repair pathways that cannot compensate for each other’s absence.

Figure 3. Formation of DNA damage foci is compromised after the depletion of DNA2/EXO1. Left panels are representative immunofluorescence images of BrdU, RPA and RAD51 foci and right panels are quantification of the cells shown on the left panel. (A) After exposure to CPT, DNA2/EXO1 U2OS-depleted cells have significantly fewer single-stranded BrdU foci compared with DNA2 or EXO1 single depletion. Cells with single or double depletion of DNA2 and/or EXO1 were cultured in BrdU, exposed to CPT (1 μM, 1 h) and stained with α-BrdU antibodies without denaturation. (B) After exposure to CPT (1 μM, 1 h), phospho-RPA2 S4/S8 foci are significantly less in DNA2/EXO1-depleted cells compared with DNA2 or EXO1 depletion. (C) RAD51 foci are significantly diminished in DNA2/EXO1-depleted cells compared with DNA2 or EXO1 depletion after exposure to CPT (1 μM, 1 h). (D) After exposure to cisplatin (15 μM, 24 h), DNA2-depleted cells have reduced RAD51 foci formation. Error bars indicate mean ± SEM for n = 3 independent experiments and *p < 0.05, **p < 0.01. Comparisons were also made with CtIP, as a control, but were not shown on the graph for clarity. Statistical analyses were done using an unpaired two-tailed t-test. At least 100 cells were counted for each independent experiment. See Experimental Procedures for details.
We next monitored RPA2-pS4/S8 foci.16,35 DNA2- and/or EXO1-depleted cells were exposed to CPT and immunostained for RPA2-pS4/S8 foci and for γH2AX, a marker of chromatin at DSBs, to identify cells that had undergone damage. The number of cells with RPA2-pS4/S8 foci in γH2AX-positive cells was then determined. DNA2- or EXO1-depleted cells had reduced levels of RPA focus formation (26% and 23%, respectively) compared with the SCR cells (37%). DNA2 and EXO1 double-depleted cells, had significantly fewer RPA foci (10%) (Fig. 3B). Control cells with CtIP depletion had levels of RPA foci (6%) similar to those of the double depletion.
The RPA complex that binds to ssDNA during resection is replaced by RAD51, which forms a dynamic nucleoprotein filament involved in strand invasion. A reduction in RAD51 foci would also suggest impaired resection. We examined RAD51 focus formation after exposure to cisplatin or CPT in DNA2 and/or EXO1-depleted cells. In response to CPT, DNA2/EXO1 doubly depleted cells had 33% fewer foci than the SCR control and 25% and 29% fewer foci than EXO1 or DNA2 single depletions, respectively (Fig. 3D). We also saw a marked reduction in RAD51 foci in cisplatin-treated, DNA2-depleted cells compared with the SCR control (Fig. 3D). Instructively, RPA2-pS4/S8 is essential for recruitment of RAD51 to ssDNA after exposure to hydroxyurea, which stalls replication forks.36 The smaller reduction in RAD51 foci than in BrdU or RPA foci (Fig. 3A–C) most likely reflects that, in the absence of DNA2, some RAD51 continues to be recruited to the stalled forks to protect the replication fork from collapse before any break and need for resection occurs. We have demonstrated that DNA2 deficiency in itself induces replication fork stress.17,18 The role of RAD51 in fork protection rather than fork repair has been reported recently in Xenopus and human.37-39 In fact, RAD51 is recruited to ICLs before the initial incisions, that is, before its role in repair of the broken sister chromatid.9 Interpretation of the RAD51 data illustrates the unique challenges imposed by working with a gene involved both in constitutive DNA replication and in repair of stressed replication forks.
DNA2- and EXO1-depleted cells accumulate unrepaired cisplatin and CPT-induced breaks
To verify a defect in DSB repair, we examined whether the cells accumulated broken DNA in two ways. First, we monitored accumulation of CPT-induced DSBs using PFGE (pulse field gel electrophoresis) after DNA2 and/or EXO1 depletion. Cells were treated with DNA-PK inhibitor NU7441 to reduce potentially compensating repair by NHEJ and exposed to CPT for 2 h. Even cells singly depleted of DNA2 or EXO1 showed accumulation of DSBs compared with the scrambled control. The DNA2 and EXO1 doubly depleted cells, however, had even higher levels of unrepaired DSBs than either of the single depletions (Fig. 4A, typical gel representative of four biological replicates). This again suggests synergy between DNA2 and EXO1 depletions. As a second demonstration of increased DSBs, we monitored the frequency of gaps in sister chromatids using a chromosomal aberration assay and found an increase in gaps/DSBs in the double depletes and in DNA2-depleted cells treated with cisplatin or CPT compared with untreated cells (Fig. 4B). (We attribute the apparent difference in level of CPT-induced breaks in the EXO1-depletes monitored by PFGE in Fig. 4A compared with Fig. 4B to the absence of NHEJ inhibitor in the metaphase spread experiments.)
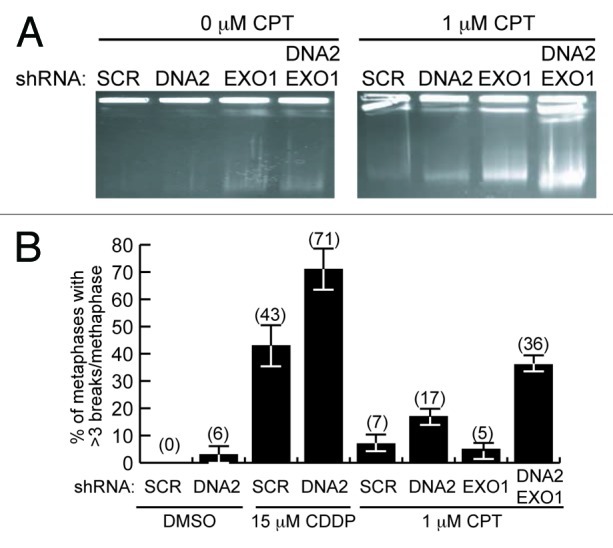
Figure 4. DNA2/EXO1-depleted U2OS cells accumulate DSBs and broken chromosomes after CPT or cisplatin replication inhibition. (A) Pulse-field gel electrophoresis shows markedly higher amounts of unrepaired DNA in DNA2/EXO1-depleted cells than in the DNA2 or EXO1-depleted cells. (B) After the simultaneous depletion of DNA2 and EXO1, the number of breaks per metaphase spread observed in the absence of DNA damage (DMSO) increases upon exposure to cisplatin or to CPT. At least 100 cells were counted in each independent experiment. See Experimental Procedures and Supplemental Material for details. CDDP, cisplatin.
DNA2 repair pathways intersect with the FA/BRCA pathway: DNA2 interaction with FANCD2
In yeast, one member of the FA/BRCA family that is conserved is FANCM/Mph1. High-copy expression of Mph1 suppresses the lethality of a yeast dna2 helicase-dead mutant, and purified Mph1 stimulates DNA2 nuclease.40 This suggested human DNA2 might function in the FA/BRCA pathway, and we next addressed whether DNA2 and EXO1 act in conjunction with any of the human FA pathway factors. FANCD2 is a central component of the FA/BRCA pathway, and after monoubiquitylation is essential, in complex with FANCI, to induce the initial incisions and for the sequential TLS, NER and HDR steps in ICL removal (Fig. S1). Also, like DNA2, it is constitutively associated with replication forks.7 We first asked if DNA2 interacts with FANCD2. We find that DNA2 and FANCD2 show surprisingly robust interaction, given the low levels of DNA2 present in nuclei. FANCD2 co-immunoprecipitates with endogenous DNA2 in extracts incubated with α-DNA2. In the reverse experiment, we failed to find DNA2 in FANCD2 immunoprecipitates (Fig. 5A). However, DNA2 is expressed at very low levels, and a large fraction is mitochondrial rather than nuclear. Therefore, we overexpressed DNA2 in HEK293T cells. In this case, FANCD2 was able to co-immunoprecipitate DNA2 (Fig. 5A, lower panel). We further confirmed the DNA2/FANCD2 interaction by adding purified FLAG-DNA2 to extracts expressing endogenous levels of FANCD2 and demonstrating that the purified DNA2 protein co-immunoprecipitated with FANCD2 using α-FANCD2 antibody (Fig. 5A). Note that Figure 5A, top right, serves as the negative control for the coimmunoprecipitation in the lower panels. Since we have shown that DNA2 coimmunoprecipitates with And1, a replication fork protein, this DNA2/FANCD2 interaction may imply that they interact at replication forks.17,18

Figure 5. DNA2 physically interacts with and functions downstream of FANCD2 and participates in SSA in vivo. (A) In U2OS cells, antibody against endogenous DNA2 coimmunoprecipitates FANCD2, but antibody against FANCD2 does not coimmunoprecipitate detectable amounts of DNA2 (top panel). When DNA2 is overexpressed or recombinant DNA2 added to the whole-cell extract, however, DNA2 is detectable in the FANCD2 immunoprecipitate (bottom panel). Whole-cell extracts were prepared from asynchronous HEK293T cells transfected with pcDNA3.1-DNA2–6His-4Myc. (B) DNA2 was overexpressed in HEK293T cells with or without cisplatin treatment. Lysates were incubated in the presence of DNaseI,59 and immunoprecipitation performed with FANCD2 antibody. (C) FANCD2 monoubiquitylation was unaffected in DNA2-depleted cells exposed to cisplatin or MMC (mitomycin C). (D) Top panel is a diagram of the U2OS DR-GFP reporter used to examine HDR after I-SceI expression. DNA2 depletion caused an increase in GFP+ cells. (E) Top panel is a diagram of the U2OS SA-GFP reporter used to examine SSA after I-SceI expression. DNA2-depleted cells had a significant decrease in SSA. (F) Depletion of DNA2 in FANCD2−/− does not affect the level of HDR. HDR was measured in FANCD2−/− alone and in FANCD2−/− after shDNA2 knockdown, i.e., cells doubly deficient in FANCD2 and DNA2 using the DR-GFP assay.
We next determined whether the interaction was likely due to protein/protein interaction by repeating the IPs in extracts treated with DNaseI. We also monitored interaction in the presence of ICL damage. As shown in Figure 5C, DNA2 and FANCD2 interact in the absence of DNA, suggesting protein/protein interaction. Interestingly, when cells were treated with cisplatin, we saw interaction of DNA2 with both ubiquitylated and unmodified FANCD2. In addition, we noticed that a faint band always detected in the DNA2 IPs with DNA2 antibody was significantly increased after ICL damage, suggesting that DNA2 is modified upon ICL induction, as it is in yeast during HU treatment.41
To determine if DNA2 was required to induce FANCD2 ubiquitylation, we determined whether FANCD2 was ubiquitylated in DNA2-depleted cells with ICL damage. DNA2 depletion did not reduce FANCD2 monoubiquitylation after treatment of cells with either cisplatin or with mitomycin C, another ICL-inducing agent. This implies that DNA2 is not needed for FANCD2 ubiquitylation and, given that the two proteins interact, that DNA2 likely functions downstream of (or in parallel with) FANCD2 (Fig. 5C)
Depletion of DNA2 leads to a reduction in SSA (single-strand annealing), a specific subpathway of HDR
The FA pathway components have been implicated in replication-coupled HDR repair of preformed DSBs previously.42 To determine if DNA2, like other FA proteins, was involved in HDR at preformed DSBs, we used a reporter, DR-GFP, composed of direct repeats of two mutated GFP genes.43 One repeat is inactivated by insertion of an I-Scel recognition site and the other repeat is a GFP fragment (Fig. 5D, insert from ref. 44). Upon induction of I-SceI endonuclease, repair of the I-SceI DSB by HDR (gene conversion) leads to reconstitution of GFP, allowing the efficiency of HDR to be quantified by flow cytometry. Surprisingly, GFP+ cells increased, rather than decreased, in cells treated with shDNA2 (Fig. 5D). Since DNA2 is involved in long-range resection, we reasoned that the increase in GFP-positive cells could be explained if DNA2 depletion reduced a subpathway of HDR that competes with gene conversion, called SSA (single-strand annealing).44 In SSA, DSBs are resected until regions of microhomology flanking the DSB are exposed and anneal to each other, creating a deletion of intervening DNA when joining occurs. SSA would result in a deletion of GFP and thus oppose gene conversion in the DR-GFP reporter assay. If DNA2 participated in SSA resection, then silencing of DNA2 might increase HDR. When we used a reporter that measures reconstitution of the GFP gene by SSA (Fig. 5E), we found a significant decrease in SSA in the shDNA2-treated cells. Instructively, other FA/BRCA mutants, notably FANCA- and FANCD2-deficient cells, similarly have reduced SSA (Nakanishi, 2005). The stimulation of SSA by DNA2 also suggests a role for DNA2 in resection upstream of BRCA2, important for loading of RAD51, since BRCA2 inhibits SSA.45 We conclude that DNA2 is required for HDR, supporting recent studies by others.15
We also analyzed HDR in cells lacking both DNA2 and FANCD2 using a GFP assay similar to that described above. As reported previously, FANCD2-deficient PD20 cells are mildly defective in HDR.45 Depletion of DNA2 in PD20 cells does not affect the level of HDR (Fig. 5F). Since the phenotype of the double deplete is similar to FANCD2 deficiency and not to DNA2 deficiency (in U2OS cells), this suggests that DNA2 functions downstream of FANCD2.
Resection represses induction of the NHEJ pathway
When the HDR pathway is compromised, the NHEJ pathway is capable of DNA repair during S/G2-phase.46 DNA-PKcs is the protein kinase responsible for signaling during NHEJ. We hypothesized that NHEJ and, therefore, DNA-PKcs autophosphorylation would also be increased in DNA2/EXO1-depleted cells under replication stress. We used phospho-specific antibodies to examine DNA-PKcs autophosphorylation at S2056 and found a markedly increased signal in the double DNA2/EXO1-depleted compared with the singly depleted cells (Fig. 6A). We conclude that long-range resection is needed to block activation of NHEJ and short range is not sufficient. Interestingly, in cisplatin-treated cells, we did not observe an increase in DNA-PKcs phosphorylation (Fig. 6B). This could be explained by the previous demonstration that FANCD2 inhibits the appearance of DNA-PKcs pS2056 foci in mitomycin C-treated cells.47 In keeping with this, we demonstrated DNA-PKcs phosphorylation in cisplatin-treated PD20 cells and inhibition of phosphorylation in PD20 cells complemented by FANCD2 (Fig. 6C).

Figure 6. The depletion of DNA2/EXO1 increases phosphorylation of DNA-PKcs after CPT but does not after Cisplatin treatment. (A) Cells treated with CPT (1 mM, 1 h) showed extensive phosphorylation of DNA-PKcs. (B) Cells treated with cisplatin (15 μM, 24 h) showed no hyperphosphorylation of DNA-PKcs S2056. (C) In PD20 cells lacking FANCD2, DNA-PKcs S2056 is hyperphosphorylated after exposure to cisplatin (left) and expression of wild-type FANCD2 in PD20 cells inhibits the hyperphosphorylation (right), as proposed in the text. See Experimental Procedures for details. CDDP, cisplatin.
DNA2 and EXO1 suppress genome instability due to cisplatin, CPT or MMS treatment
The results of the cisplatin experiments and interaction of DNA2 with FANCD2 suggested that DNA2 might mediate interaction between HDR and the FA/BRCA pathway. Genetic interaction with components of the FA pathway during a DNA damage response is another approach to verify this. We were interested in whether silencing of human DNA2 and/or EXO1 led to hypersensitivity to ICLs, if there was synergy between DNA2/EXO1 depletion and mutants in the FA/BRCA pathway, and if cells with silenced DNA2/EXO1 were sensitive to additional agents that cause replication stress.
First, we studied cisplatin sensitivity. SnmB1/Apollo, like its yeast Pso2 ortholog, is a 5′ nuclease implicated in formation (or resection) of DSBs occurring during ICL repair.3 Although yeast pso2 mutants are only mildly sensitive to cisplatin, we observed that a pso2 dna2–1 double mutant was more sensitive to cisplatin than pso2 or dna2–1 single mutants, consistent with a role for DNA2 in the repair of ICL in yeast (Fig. S3).
In human cells, the single DNA2 or EXO1 depletions were not significantly more sensitive to acute cisplatin treatment than the controls, but the doubly depleted cells showed marked hypersensitivity to cisplatin at 0.6 μM, a low concentration, chosen to emphasize S phase repair (Fig. 7A). This level of sensitivity is significant and is similar, for instance, to the sensitivity to CPT demonstrated for CtIP depletion.48 The synergy between the DNA2 and EXO1 depletions argues that they participate in alternative pathways for repair of cisplatin damage. In a second experiment, cells were exposed to chronic, low-level cisplatin treatment (1, 2, 4 μM for 6 d). DNA2 knockdowns showed hypersensitivity even in the presence of EXO1 (Fig. 7B). This result may also be suggested by the results shown in Figure 7A, compare shDNA2 to shSCR-depleted cells, and is consistent with RPA and Chk1 phosphorylation deficiency results in Figure 2A, lanes 6 and 8.

Figure 7. DNA2 and EXO1 are required for survival after exposure to cisplatin but do not increase cisplatin sensitivity of FANCD2-deficient cells. (A) Clonogenic assay of survival of DNA2- and/or EXO1-depleted cells exposed to cisplatin for 24 h. (B) Survival of DNA2-depleted cells exposed to cisplatin for 6 d. (C) PD20 cells with DNA2 and/or EXO1-depletion or PD20 cells complemented with FANCD2 were exposed to cisplatin (24 h) and assayed for survival. (D and E) Survival of DNA2 and/or EXO1-depleted cells exposed to CPT (1 h) or MMS (1 h) and assayed for survival. Error bars indicate mean ± SEM for n ≥ 4 independent experiments. *p < 0.05. See Experimental Procedures for details. CDDP, cisplatin.
We then examined if DNA2 and or EXO1 appeared to affect the FA/BRCA repair pathway. We depleted DNA2 and/or EXO1 in FANCD2-deficient PD20 cells and performed cisplatin sensitivity studies. As shown in Figure 7C, neither doubly (FANCD2/DNA2 or FANCD2/EXO1) nor triply (FANCD2/DNA2/EXO1) depleted cells were significantly more sensitive to acute exposure than FANCD2-deficient cells, suggesting that DNA2/EXO1 and FANCD2 are in the same ICL repair pathway. In fact, there was an increase (p < 0.05) in resistance to cisplatin in the DNA2/FANCD2 doubly deficient cells. This could be explained by an increase in NHEJ or some other repair pathway in the absence of DNA2 resection or to the accumulation of toxic recombination intermediates, if DNA2-mediated repair is active in the absence of FANCD2. Since DNA2/EXO1 depletion causes significant sensitivity to cisplatin but doesn’t increase ICL sensitivity in FANCD2-deficient PD20 cells, and because, as shown in Figure 5, DNA2 depletion does not further affect HDR in FANCD2-deficient PD20 cells, it is possible that DNA2/EXO1 and FANCD2 act in the same pathway, act coordinately, or that FANCD2 is upstream of DNA2/EXO1.
Since endogenous replication-associated damage is likely not due to crosslinks but to general replication fork failure, we also examined hypersensitivity to CPT and MMS. We observed that resistance to CPT was significantly diminished in cells with DNA2/EXO1 double depletion compared with the respective single depletions (Fig. 7D). This is consistent with the observation that BLM/EXO1 doubly depleted cells are significantly more sensitive to CPT than BLM or EXO1 single depletions.16
MMS is a DNA methylating agent that predominantly forms N7-methylguanine adducts postulated to be converted to DSBs during replication49,50 and are repaired by HDR.50,51 When DNA2 and EXO1 were simultaneously depleted, we saw a synergistic response in MMS sensitivity, with the double depletions being more sensitive than the single depletions. A distinct hierarchy of sensitivity was apparent. DNA2-depleted cells were more sensitive than EXO1-depleted cells or the scrambled control, meaning that DNA2 can compensate for the loss of EXO1, but EXO1 cannot compensate for the loss of DNA2 (Fig. 7E). We conclude that, while DNA2 and EXO1 may function in independent pathways, these pathways may be partially redundant for repair of MMS-induced DNA damage.
Discussion
In our study, we established a role for human DNA2 in resection of DSBs, especially those occurring during repair of replication forks stalled by ICLs. This work provides in vivo confirmation that resection pathways are conserved from yeast to man, as previously suggested, but not proved, by our in vitro reconstitution studies as52 and extends a report by others.15 We also discovered that DNA2 functions in checkpoint activation (see model in Fig. S1E). This extends genetic studies implicating HDR proteins in ICL repair and recent elegant mechanistic studies showing ICL to be RAD51-dependent and to involve sister chromatid exchange, which presumably requires resection of a double-strand end for strand invasion.9 Importantly, we demonstrated interaction between DNA2 and the central regulatory protein in the FA/BRCA pathway, FANCD2, physically linking DNA2 to ICL repair, and showed that FANCD2-DNA2-EXO1 triply deficient cells are not more sensitive to ICLs than FANCD2 mutants, suggesting function in the same pathway and action on the same substrates. DNA2 likely acts downstream of FANCD2, consistent with a role in resection of the DSB that occurs after unhooking of the crosslink.9 DNA2 is likely to act upstream of BRCA2, since DNA2 stimulates SSA, an HDR pathway inhibited by BRCA2. Although FA deficiency is most prominently linked to ICL repair defects, the FA pathway clearly deals with a wide range of endogenous and exogenous template blocks that cause replication fork stalling. In viewing the FA pathway as a general response to replication fork stalling, our study answers a question left open by our recent studies of the role of human DNA2 in DNA replication. We did not find a defect in Okazaki fragment processing in DNA2-deficient cells, as we had in yeast. Yet we found accumulation of DSBs, as shown in the untreated (as well as CPT-treated cells in Fig. 4) DNA2-depleted cells, and an increase in γH2AX and RAD51 foci as well as Chk1 activation.18 In addition, we found that forks moved at normal rates, but the cell cycle was delayed in G2 by the checkpoint, and that this is due to depressed replication origin firing, which is known to lead to fork collapse, for instance, in already origin-depleted regions such as common fragile sites.18,21 Linking DNA2 to the FA/BRCA pathway supports a role for DNA2 during S and G2 in repair of stalled and/or collapsed forks, even in the absence of exogenous DNA damage. DNA2 may have evolved a more important role in post-replication repair in human, or our assays for ssDNA breaks during S phase in DNA2 knockdowns, i.e., alkaline comet assays during BrdU pulse-labeling protocols, may not have been sensitive enough to detect damage only in a subset of Okazaki fragments.18
A second important implication of our work is that it is likely consistent with our in vitro reconstitution studies,14 that DNA2 is the nuclease that functions with BLM helicase in parallel to the EXO1 pathway in DSB repair.16 Presumably, depletion of BLM incapacitates DNA2 function by depriving it of its substrate, BLM-helicase unwound 5′ssDNA, just as occurs in vitro.14,16
A third aspect of our work went beyond DNA2 and investigated the full complement of proteins involved in resection by studying not just DNA2 but also EXO1. To a large extent, we find that the DNA2 pathway is redundant with and parallel to resection by EXO1, as evidenced by the synergistic drug sensitivity and defects in resection in response to double depletions. However, several of our results also suggest more prominent functions for DNA2 than EXO1, as in yeast, such as in removal of ICLs (Figs. 2 and 7). The single DNA2 depletion showed reduced RPA and Chk1 phosphorylation, while EXO1 silencing did not, and double depletion showed only a marginal, if any, increase in RPA and Chk1 phosphorylation. DNA2 depletion also had a greater effect than EXO1 depletion when accumulation of unrepaired breaks was studied (Fig. 4A). This further suggests that DNA2 and EXO1 cannot completely compensate for the absence of the other, and that they therefore do not define completely redundant pathways. It may suggest that DNA2 and EXO1 actually act together in one resection pathway on the same molecules, rather than acting in compensating ways, or that they act in independent pathways for different types of damage. This is consistent with findings in the in vitro reconstituted reaction, where there is clearly a pathway involving both DNA2 and EXO1 acting on the same molecule.14 It is also consistent with results reported in yeast.53
A fourth important finding relates to the roles of DNA2 and EXO1 in mediating HDR. DNA2 depletion decreased SSA, leading to an increase in gene conversion (Fig. 5). DNA2 associates with a DSB and at least one role of DNA2 is downstream of (or in parallel with) FANCD2’s role in HDR (Fig. 5).
DNA2 and EXO1 also influence the frequency of HDR vs. NHEJ. The HDR pathway preferentially repairs DSBs that occur during S- and G2-phase of the cell cycle. This optimizes precise repair of the breaks and error-free restart of replication forks. It has been proposed that interfering with the HDR resection pathway facilitates genome instability, and that this is mediated by the activation of NHEJ pathway during S-phase. Supporting this, deletion of CtIP results in an increase in NHEJ.25 Double depletion of CtIP and EXO1 in CPT-treated cells results in an increase in DNA-PKcs autophosphorylation at serine 2056 and increase in genome rearrangements.54 The identification of the redundant DNA2 and EXO1 pathways allowed us to determine if inhibiting the long-range resection pathway alone also led to activation of NHEJ, i.e., even in the presence of CtIP and active short-range resection. Phosphorylation of DNA-PKcs in the doubly depleted cells treated with CPT suggests that this is the case. Importantly, in the case of cisplatin-treated cells we did not observe any significant increase in DNA-PKcs hyperphosphorylation. We attribute this observation to FANCD2 preventing aberrant NHEJ during ICL repair by inhibiting DNA-PKcs phosphorylation.47
DNA2 has been linked to protecting replication forks from collapse at replication fork barriers, such as the ribosomal replication fork barrier in S. cerevisiae,55,56 and recently in Schizosaccharomyces pombe.41 Our results could also be explained by a role for DNA2 in protecting forks in human cells. New roles of BRCA2 and stabilization of RAD51 filaments in protection of stalled replication forks, rather than repair, already provide evidence for such a pathway.37-39,57 Interestingly, RAD51 also associates with forks stalled at ICLs before the initial repair incisions.9 The linkage of DNA2 to the FA/BRCA pathway is also particularly interesting in view of recent results suggesting that DNA2 is overproduced in response to a specific type of replicative stress caused by induction of oncogenes such as RAS.15 DNA2 overproduction was also observed in cells undergoing the early stages of transformation of normal epithelial cells into breast tumor cells.15 A similar pattern was found for ovarian and pancreatic cells undergoing evolution to full transformation. Finally, an inverse correlation between breast tumor grade and level of DNA2 was established. It will be interesting to investigate whether DNA2 can be used to identify more genes specifically interacting with the FA/BRCA pathway that may serve as markers for these diseases in earlier stages than is now possible.
Experimental Procedures
Cell culture and transfections
U2OS, U2OS-DR-GFP (Pierce, 1999) and U2OS-SA-GFP (Gunn, 2011) cells were gifts from Dr. W. Dunphy, California Institute of Technology; Dr. M. Jasin, Memorial Sloan-Kettering Cancer Center and Dr. J. Stark, City of Hope. PD20 fancD2−/− lines were from the Fanconi Anemia Research Fund. Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37°C in 5% CO2.
Virus production and infection
Virus was produced in HEK293T cells as described.17,58 Briefly, cells were transfected with pLKO.1 shSCR, pLKO.1 shDna2 or pRESQ shDna2’ and pCMVΔR8.2 and pCMV-VSV-G (the latter two at 8:1 ratio) using BioT (Bioland Scientific). Virus was recovered 48 h post-transfection, and infections were performed on U2OS cells overnight in the presence of 10 μg/ml of protamine sulfate. Transduced cells were selected with 2 μg/ml of puromycin for 48 h. The following sequences were used for the hDna2 short hairpins, 5′-CATAGCCAGTAGTATTCGATG-3′ for shDna2, 5′-GCAGTATCTCCTCTAGCTAGT for pRESQ shDna2’ and 5′-AAGGTTAAGTCGCCCTCGCTC-3′ for shSCR.17,18
For depletion of EXO1, siRNA duplexes, purchased from Dharmacon, were transfected into cells using oligofectamine (Life Technologies). siRNA sequences are as follows, siCNTL (luciferase 5′-CGUACGCGGAAUACUUCGATT-3′) and siEXO1 (5′-CAAGCCUAUUCUCGUAUUUTT-3′) (Gravel, 2008). Depletion of DNA2 and EXO1 was determined by Taqman qualitative PCR (Qiagen). Experiments were performed 48 h post-siRNA transfection.
Knockdown rescue experiment
For complementation studies, virus was produced as described above using plasmids, pBabeHygro-3xFlag-DNA2, pRESQ shSCR and pRESQ shDna2′17,18. U2OS cells overexpressing FLAG-tagged DNA2 were selected in 200 μg/ml hygromycin for 5 d and then transduced with the virus-containing pRESQ shSCR or pRESQ shDna2′, which targets the 3′UTR of endogenous DNA2 but not the exogenous DNA2, and selected with 2 μg/ml of puromycin for 48 h.18 Cells were then treated with control siCNTL or siEXO1 for 48 h, exposed to CPT (1 μM, 1 h) or cisplatin (15 μM, 24 h), or no damaging agent and harvested and assayed for RPA2 pS4/S8, pS33 and Chk1 pS345 by western blotting.
Clonogenic and cytotoxicity assays
Survival data for methyl methane sulfonate (MMS) and PD20 cells treated with cisplatin (CDDP) was generated from a cytotoxicity assay and for camptothecin (CPT) or cisplatin from a clonogenic assay. Briefly, 48 h after depletion of DNA2 and/or EXO1, cells were serial diluted in media and inoculated into 12 well plates, left to attach overnight and treated with MMS (0, 0.02, 0.03, 0.04, 0.05%) or CPT (0, 0.001, 0.01, 0.1 μM) for 1 h and cisplatin (0, 0.625, 1.25, 2.5 μM) for 24 h, washed and cultured. For chronic exposure, cells were treated for 6 d with 0, 1, 2, 4 μM cisplatin. For susceptibility to MMS, and cisplatin (for PD20 cells) 48 h post-treatment, cells were fixed and stained with 1% crystal violet in methanol and viability count determined by imaging cell density using near infrared imaging (Li-Cor). For CPT and cisplatin, colonies were allowed to form for 14 d prior to fixation, staining with crystal violet and enumeration of visible colonies.
Immunofluoresence microscopy
This was performed essentially as described (Duxin, 2009). Details can be found in Supplemental Material.
Recombination assay
DNA2 was depleted from DR-GFP or SA-GFP cells. Cells were transfected with an empty plasmid or a plasmid-expressing I-SceI endonuclease for 48 h. GFP+ cells were determined by flow cytometry.
Statistical analysis
Student’s t-tests were performed at p < 0.05.
Supplementary Material
Acknowledgments
We thank Joon Lee and William Dunphy from the Dunphy laboratory for comments on the manuscript. We thank Shelly Diamond for expert performance and analysis of GFP by flow cytometry. We thank the Fanconi Anemia Research Fund for the FANCD2 antibodies and PD20 cell lines. This work was supported by a Breast Cancer grant from Congressionally Directed Medical Research Programs (J.L.C.), GM100186, the Ellison Foundation and the Ross Fellowship from the Biology Division, California Institute of Technology (K.K.K).
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22215
References
- 1.Constantinou A. Rescue of replication failure by Fanconi anaemia proteins. Chromosoma. 2012;121:21–36. doi: 10.1007/s00412-011-0349-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D’Andrea AD. S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood. 2002;100:2414–20. doi: 10.1182/blood-2002-01-0278. [DOI] [PubMed] [Google Scholar]
- 3.Moldovan GL, D’Andrea AD. How the fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–49. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howlett NG, Taniguchi T, Durkin SG, D’Andrea AD, Glover TW. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genet. 2005;14:693–701. doi: 10.1093/hmg/ddi065. [DOI] [PubMed] [Google Scholar]
- 5.Zhu W, Dutta A. Activation of fanconi anemia pathway in cells with re-replicated DNA. Cell Cycle. 2006;5:2306–9. doi: 10.4161/cc.5.20.3364. [DOI] [PubMed] [Google Scholar]
- 6.Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, Hurov KE, Luo J, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song IY, Barkley LR, Day TA, Weiss RS, Vaziri C. A novel role for Fanconi anemia (FA) pathway effector protein FANCD2 in cell cycle progression of untransformed primary human cells. Cell Cycle. 2010;9:2375–88. doi: 10.4161/cc.9.12.11900. [DOI] [PubMed] [Google Scholar]
- 8.Ben-Yehoyada M, Wang LC, Kozekov ID, Rizzo CJ, Gottesman ME, Gautier J. Checkpoint signaling from a single DNA interstrand crosslink. Mol Cell. 2009;35:704–15. doi: 10.1016/j.molcel.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long DT, Räschle M, Joukov V, Walter JC. Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science. 2011;333:84–7. doi: 10.1126/science.1204258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knipscheer P, Räschle M, Smogorzewska A, Enoiu M, Ho TV, Schärer OD, et al. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Räschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, et al. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008;134:969–80. doi: 10.1016/j.cell.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–71. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 13.Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci USA. 2008;105:16906–11. doi: 10.1073/pnas.0809380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011;25:350–62. doi: 10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peng G, Dai H, Zhang W, Hsieh HJ, Pan MR, Park YY, et al. Human nuclease/helicase DNA2 alleviates replication stress by promoting DNA end resection. Cancer Res. 2012;72:2802–13. doi: 10.1158/0008-5472.CAN-11-3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–72. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duxin JP, Dao B, Martinsson P, Rajala N, Guittat L, Campbell JL, et al. Human Dna2 is a nuclear and mitochondrial DNA maintenance protein. Mol Cell Biol. 2009;29:4274–82. doi: 10.1128/MCB.01834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duxin JP, Moore HR, Sidorova J, Karanja K, Honaker Y, Dao B, et al. Okazaki fragment processing-independent role for human Dna2 enzyme during DNA replication. J Biol Chem. 2012;287:21980–91. doi: 10.1074/jbc.M112.359018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Budd ME, Reis CC, Smith S, Myung K, Campbell JL. Evidence suggesting that Pif1 helicase functions in DNA replication with the Dna2 helicase/nuclease and DNA polymerase delta. Mol Cell Biol. 2006;26:2490–500. doi: 10.1128/MCB.26.7.2490-2500.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng L, Zhou M, Guo Z, Lu H, Qian L, Dai H, et al. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol Cell. 2008;32:325–36. doi: 10.1016/j.molcel.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Debatisse M, Le Tallec B, Letessier A, Dutrillaux B, Brison O. Common fragile sites: mechanisms of instability revisited. Trends Genet. 2012;28:22–32. doi: 10.1016/j.tig.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Bermejo R, Lai MS, Foiani M. Preventing replication stress to maintain genome stability: resolving conflicts between replication and transcription. Mol Cell. 2012;45:710–8. doi: 10.1016/j.molcel.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 23.O’Connell BC, Adamson B, Lydeard JR, Sowa ME, Ciccia A, Bredemeyer AL, et al. A genome-wide camptothecin sensitivity screen identifies a mammalian MMS22L-NFKBIL2 complex required for genomic stability. Mol Cell. 2010;40:645–57. doi: 10.1016/j.molcel.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robison JG, Lu L, Dixon K, Bissler JJ. DNA lesion-specific co-localization of the Mre11/Rad50/Nbs1 (MRN) complex and replication protein A (RPA) to repair foci. J Biol Chem. 2005;280:12927–34. doi: 10.1074/jbc.M414391200. [DOI] [PubMed] [Google Scholar]
- 25.Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459:460–3. doi: 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X, Zhao R, Glick GG, Cortez D. Function of the ATR N-terminal domain revealed by an ATM/ATR chimera. Exp Cell Res. 2007;313:1667–74. doi: 10.1016/j.yexcr.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou L, Liu D, Elledge SJ. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci USA. 2003;100:13827–32. doi: 10.1073/pnas.2336100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–11. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- 29.Vassin VM, Anantha RW, Sokolova E, Kanner S, Borowiec JA. Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA-replication stress. J Cell Sci. 2009;122:4070–80. doi: 10.1242/jcs.053702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liaw H, Lee D, Myung K. DNA-PK-dependent RPA2 hyperphosphorylation facilitates DNA repair and suppresses sister chromatid exchange. PLoS One. 2011;6:e21424. doi: 10.1371/journal.pone.0021424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anantha RW, Vassin VM, Borowiec JA. Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. J Biol Chem. 2007;282:35910–23. doi: 10.1074/jbc.M704645200. [DOI] [PubMed] [Google Scholar]
- 32.Shiotani B, Zou L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol Cell. 2009;33:547–58. doi: 10.1016/j.molcel.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zou L. Single- and double-stranded DNA: building a trigger of ATR-mediated DNA damage response. Genes Dev. 2007;21:879–85. doi: 10.1101/gad.1550307. [DOI] [PubMed] [Google Scholar]
- 34.MacDougall CA, Byun TS, Van C, Yee MC, Cimprich KA. The structural determinants of checkpoint activation. Genes Dev. 2007;21:898–903. doi: 10.1101/gad.1522607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, et al. Human CtIP promotes DNA end resection. Nature. 2007;450:509–14. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi W, Feng Z, Zhang J, Gonzalez-Suarez I, Vanderwaal RP, Wu X, et al. The role of RPA2 phosphorylation in homologous recombination in response to replication arrest. Carcinogenesis. 2010;31:994–1002. doi: 10.1093/carcin/bgq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol. 2010;17:1305–11. doi: 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–42. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22:106–16. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kang YH, Kang MJ, Kim JH, Lee CH, Cho IT, Hurwitz J, et al. The MPH1 gene of Saccharomyces cerevisiae functions in Okazaki fragment processing. J Biol Chem. 2009;284:10376–86. doi: 10.1074/jbc.M808894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu J, Sun L, Shen F, Chen Y, Hua Y, Liu Y, et al. The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell. 2012;149:1221–32. doi: 10.1016/j.cell.2012.04.030. [DOI] [PubMed] [Google Scholar]
- 42.Nakanishi K, Cavallo F, Brunet E, Jasin M. Homologous recombination assay for interstrand cross-link repair. Methods Mol Biol. 2011;745:283–91. doi: 10.1007/978-1-61779-129-1_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–8. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gunn A, Bennardo N, Cheng A, Stark JM. Correct end use during end joining of multiple chromosomal double strand breaks is influenced by repair protein RAD50, DNA-dependent protein kinase DNA-PKcs, and transcription context. J Biol Chem. 2011;286:42470–82. doi: 10.1074/jbc.M111.309252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D’Andrea AD, et al. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc Natl Acad Sci USA. 2005;102:1110–5. doi: 10.1073/pnas.0407796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pierce AJ, Hu P, Han M, Ellis N, Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001;15:3237–42. doi: 10.1101/gad.946401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, et al. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell. 2010;39:25–35. doi: 10.1016/j.molcel.2010.06.026. [DOI] [PubMed] [Google Scholar]
- 48.Polo SE, Blackford AN, Chapman JR, Baskcomb L, Gravel S, Rusch A, et al. Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol Cell. 2012;45:505–16. doi: 10.1016/j.molcel.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beranek DT. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat Res. 1990;231:11–30. doi: 10.1016/0027-5107(90)90173-2. [DOI] [PubMed] [Google Scholar]
- 50.Nikolova T, Ensminger M, Löbrich M, Kaina B. Homologous recombination protects mammalian cells from replication-associated DNA double-strand breaks arising in response to methyl methanesulfonate. DNA Repair (Amst) 2010;9:1050–63. doi: 10.1016/j.dnarep.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 51.Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23:81–5. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 52.Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011;25:350–62. doi: 10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu Z, Chung W-H, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–94. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eid W, Steger M, El-Shemerly M, Ferretti LP, Peña-Diaz J, König C, et al. DNA end resection by CtIP and exonuclease 1 prevents genomic instability. EMBO Rep. 2010;11:962–8. doi: 10.1038/embor.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weitao T, Budd M, Campbell JL. Evidence that yeast SGS1, DNA2, SRS2, and FOB1 interact to maintain rDNA stability. Mutat Res. 2003;532:157–72. doi: 10.1016/j.mrfmmm.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 56.Weitao T, Budd M, Hoopes LL, Campbell JL. Dna2 helicase/nuclease causes replicative fork stalling and double-strand breaks in the ribosomal DNA of Saccharomyces cerevisiae. J Biol Chem. 2003;278:22513–22. doi: 10.1074/jbc.M301610200. [DOI] [PubMed] [Google Scholar]
- 57.Hashimoto Y, Puddu F, Costanzo V. RAD51- and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat Struct Mol Biol. 2012;19:17–24. doi: 10.1038/nsmb.2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saharia A, Guittat L, Crocker S, Lim A, Steffen M, Kulkarni S, et al. Flap endonuclease 1 contributes to telomere stability. Curr Biol. 2008;18:496–500. doi: 10.1016/j.cub.2008.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang X, Andreassen PR, D’Andrea AD. Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol Cell Biol. 2004;24:5850–62. doi: 10.1128/MCB.24.13.5850-5862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.