

Abstract
Chronic inflammation is a risk factor for the development of colon cancer, providing genotoxic insults, growth and pro-angiogenic factors that can promote tumorigenesis and tumor growth. Immunomodulatory agents can interfere with the inflammation that feeds cancer, but their impact on the transformed cell is poorly understood. The calcium/calcineurin signaling pathway, through activation of NFAT, is essential for effective immune responses, and its inhibitors cyclosporin A (CsA) and FK506 are used in the clinics to suppress immunity. Moreover, the kinases GSK3β and mTOR, modulated by PI-3K/Akt, can inhibit NFAT activity, suggesting a cross-talk between the calcium and growth factor signaling pathways. Both NFAT and mTOR activity have been associated with tumorigenesis. We therefore investigated the impact of calcineurin and PI-3K/mTOR inhibition in growth of human colon carcinoma cells. We show that despite the efficient inhibition of NFAT1 activity, FK506 promotes tumor growth, whereas CsA inhibits it due to a delay in cell cycle progression and induction of necroptosis. We found NFκB activation and mTORC1 activity not to be altered by CsA or FK506. Similarly, changes to mitochondrial homeostasis were equivalent upon treatment with these drugs. We further show that, in our model, NFAT1 activation is not modulated by PI3K/mTOR. We conclude that CsA slows cell cycle progression and induces necroptosis of human carcinoma cell lines in a TGFβ-, NFAT-, NFκB- and PI3K/mTOR-independent fashion. Nevertheless, our data suggest that CsA, in addition to its anti-inflammatory capacity, may target transformed colon and esophagus carcinoma cells without affecting non-transformed cells, promoting beneficial tumoristatic effects.
Keywords: NFAT, calcineurin, mTOR, cancer, cyclosporin A, FK506, necroptosis, colon carcinoma
Introduction
Cellular transformation and cancer development are processes that depend upon alterations both of a cell’s physiology and its interaction with the tissue microenvironment.1 Chronic inflammation contributes to both arms of this process.2,3 An initial stage is marked by tissue remodeling and cell death, and the inflammatory mediators produced at the site of inflammation promote cellular recruitment, activation, proliferation and modifications to the blood vessels present at the injured site.4,5 At later stages, when the insult is eliminated, immunoregulatory cells are recruited, and the process of tissue repair ensues until homeostasis is once again reached.5 It is the continuous tissue repair present in sites chronically inflamed that supplies stimuli capable of driving transformation of nearby cells.3
The recurrent injury and tissue repair induced by chronic inflammation leads to cycles of cell recruitment, differentiation/de-differentiation, proliferation and migration as well as angiogenesis fed by immune cells infiltrating the area. Progressively, the quality of the cellular infiltrate present at the site switches from an inflammatory and cytotoxic response toward an immunomodulatory response,6,7 which is then composed mostly by regulatory T cells and type 2 macrophages producing regulatory cytokines, such as IL-6, IL-8 and IL-10, and promoting angiogenesis.3,4,8 The importance of this cycle to the development and maintenance of the tumor is made clear in cases where it is possible to eliminate the insult leading to chronic inflammation, especially in tumors from the gastro-intestinal tract.6,9-11
Based on these observations, it is expected that controlling the inflammatory components that comprise the tumor niche would favor the host toward eliminating transformed cells. Indeed, constant use of anti-inflammatory drugs was shown to reduce the risk of developing certain types of cancer. The chronic administration of small doses of non-steroid anti-inflammatory drugs like aspirin was shown to reduce the incidence of colon cancer12 and slow down the progression of established tumors, prolonging overall patients survival.13,14 Interestingly, the targets of aspirin, cyclooxigenase (Cox) enzymes 1 and 2, are expressed and important not only for the immune cells and the maintenance of the tumor-associated inflammatory milieu, but also for the transformed cells themselves. Cox-2 has been found overexpressed in several different epithelial human tumors,14 and both genetic and pharmacological inactivation of Cox-2 protect from the development of colon cancer.12,15 Therefore, studies that aim at better understanding the impact of immunomodulatory compounds and the signaling pathways they act upon in the direct growth and maintenance of transformed cells may be efficacious against certain types of human cancer.
Cyclosporine A (CsA) and tacrolimus (FK506) are immunosuppressive drugs commonly used in transplant recipients and autoimmune patients.16 They inhibit the calcium/calmodulin-dependent phosphatase calcineurin, which, among other substrates, acts upon members of the nuclear factor of activated T cells (NFAT) family of transcription factors, activating them. NFAT function is essential for cytokine production by T cells and normal physiology of B cells, dendritic cell and mast cells and patients treated with these drugs, which are mostly interchangeable, become immunocompromised.17,18 Further work showed expression of NFAT family members also by cells that do not belong to the immune system, and have ascribed both tumor suppressor and oncogenic activity to these transcription factors.18-20 Moreover, another signaling pathway also linked to cellular transformation, the nutrient-sensing PI-3K/mTOR, has been shown to phosphorylate and, in that way, regulate NFAT activity, either through the activity of GSK3,21 which can be inhibited by AKT or by mTOR.22 mTOR is found in complexes containing raptor (mTORC1) of rictor (mTORC2), both of which can be inhibited by Rapamycin in a dose-dependent fashion.23,24 These observations opened the possibility that inhibition of the tumor suppressor activity of NFAT may contribute to tumor growth where a deregulated activation of the PI3K/mTOR pathway is at play. Here we evaluate the dependence of human colon carcinoma cell lines on the NFAT and mTOR signaling pathways, addressing if a potential cross-talk between these two pathways may contribute to the maintenance of the transformed state. We observe that these cell lines do not depend on NFAT activity for survival and growth. However, treatment with cyclosporine A (CsA) is capable of inducing necroptosis and a mild G0/G1 cell cycle arrest. The study of the importance of these pathways to the transformed cell may lead to new treatments that take advantage of well-established immunomodulatory drugs to target the transformed cell directly.
Results
Human colon carcinoma cells are sensitive to CsA but not FK506 in culture
We and others have shown that NFAT family members regulate the homeostasis of cell proliferation and death, playing a role in transformation.20 Specifically relevant to colon cancer, NFAT regulates expression of cox2, which is associated with tumorigenesis and tumor progression.25 Moreover, enhanced activity of PI-3K, or mutations in its counterpart phosphatase PTEN, are often found in colon cancer.26,27 To test if the calcineurin/NFAT and PI-3K/mTOR pathways are important for the growth and maintenance of the transformed phenotype in human colon carcinoma cells, we treated cells of the CACO-2 cell line with the calcineurin inhibitors CsA and FK506 and with the mTORC1 inhibitor rapamycin (Rapa) in vitro. We found CACO-2 cells to be sensitive to both CsA and Rapa treatments, which lead to a reduction in the total numbers and relative growth of these cells (Fig. 1A). These results were corroborated by the differential growth of CACO-2 colonies upon treatment with these drugs (Fig. 1). The effect of CsA and Rapa does not seem due to induced cell death, since despite the reduction in the area of cell colonies (Fig. 1C, left), which suggest impaired growth, there are no differences in the total number of colonies between the different treatments (Fig. 1C, right). Surprisingly, treatment with FK506 did not reduce CACO-2 growth. We observed no differences in the pattern of cell growth as measured by crystal violet staining (Fig. 1A, left), cell counting (Fig. 1A, right), colony formation and colony progression (Fig. 1B and 1C) when compared with cells cultured in control conditions or treated with FK506.
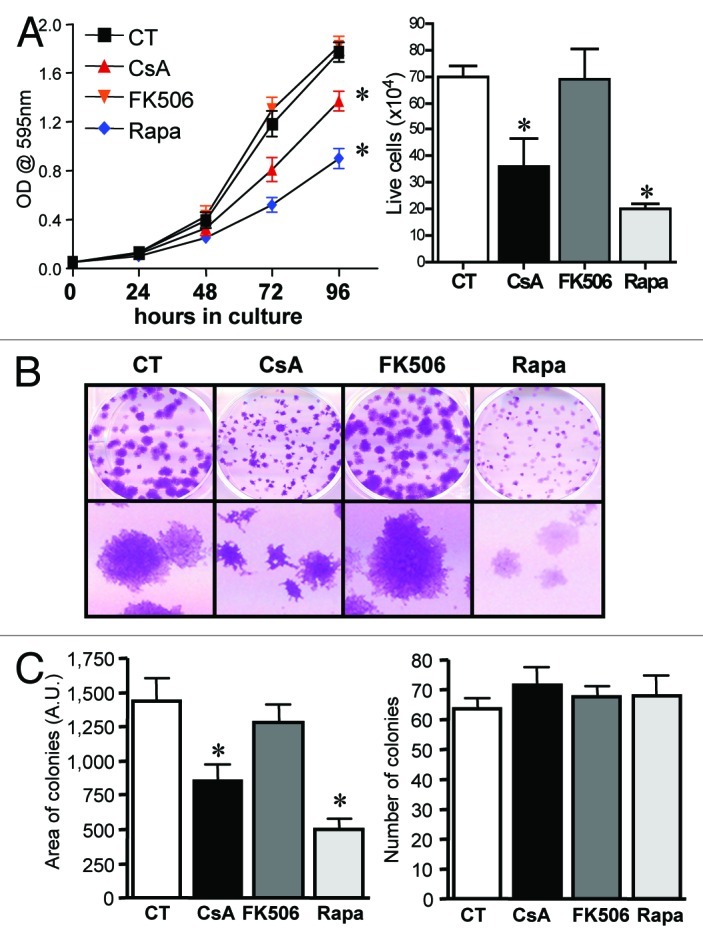
Figure 1. CACO-2 cells are sensitive to CsA but not FK506 in culture. (A) CACO-2 ,cells were cultured in the presence of CsA (2 μM), FK506 (2 μM) or Rapa (20 nM) for 4 d. At each time point samples were fixed and stained with violet crystal (left panel), or at 96 h of culture cell were trypsinized and counted (trypan blue-negative cells; right graph). Results are the mean of three to 17 independent experiments. * marks p < 0.05 when compared with CT group. (B) Clonogenic assay of CACO-2 cells cultured in the presence of specific inhibitors as in panel A. Results are representative of seven independent experiments. (C) Quantification of colony number and area from experiment shown in B. Results are the mean of 12 independent experiments. * marks p < 0.05 when comparing to CT group. (D) Colon carcinoma cell lines were cultured in the presence of indicated inhibitors at concentrations depicted in panel A for 96 h and stained with violet crystal. Results are the mean of three to six independent experiments. * marks p < 0.05 when comparing to CT group. (E) Clonogenic assay of colon carcinoma cell lines cultured in the presence of specific inhibitors as in (B). Results are representative of three independent experiments.
In order to define if the effect these drugs had on CACO-2 cells reflected general characteristics of colon carcinoma cells, HT-29, HCT-116 and LOVO cells, three other human colon carcinoma cells lines were cultured in the presence of CsA, FK506 or Rapa, and their general growth and clonogenic capacity evaluated. Similar to that observed for CACO-2 cells, CsA and Rapa led to decreased cell growth, whereas upon treatment with FK506, a tendency for overgrowth could be observed (Fig. 2A). These data were corroborated by those obtained in clonogenic assays (Fig. 2B), where a reduction in the colony area following treatment with CsA or Rapa were observed, without alteration in colony numbers (data not shown). Taken together, our results suggest that colon carcinoma cells are sensitive to the signaling pathways targeted by CsA and Rapa, but, different than anticipated, the dominant pathways modulated by CsA in these cells differ from those modulated by FK506 since the latter shows no effect.

Figure 2. Human adenocarcinoma cell lines are sensitive to CsA but not FK506 in culture. (A) Colon carcinoma cell lines were cultured in the presence of indicated inhibitors at concentrations depicted in Figure 1 for 96 h and stained with violet crystal. Results are the mean of three to six independent experiments. * marks p < 0.05 when comparing to CT group. (B) Clonogenic assay of colon carcinoma cell lines cultured in the presence of specific inhibitors as in (B). Results are representative of three independent experiments.
CsA effect is dominant over that of FK506, and does not synergize with Rapa
In order to investigate the dominance of the signaling pathways being affected by the drugs herein tested, we combined the drugs at increasing concentrations, two by two, and evaluated the growth of CACO-2 cells and their clonogenic capacity. When considering the differential effects of CsA and FK506, two hypotheses stood. Since both drugs target calcineurin inhibiting it, it was possible that (1) FK506’s modulation of other pathways promoted cell growth in a dominant fashion to the inhibition of NFAT activation; or (2) CsA promoted growth arrest in a NFAT-independent fashion. To test these hypotheses, we combined CsA and FK506 at increasing concentrations, treating CACO-2 cells in culture with these drug combinations and quantifying cell growth at different times along the culture. As shown in Figure 3A, the effect of FK506 was surmounted by that of CsA, such that when combined, CACO-2 cells showed a reduced growth independently of the FK506 concentration used. Similar results were observed in clonogenic assay, where the highest concentration of each drug was used (Fig. 3D).

Figure 3. Effect of the combination of drugs in the growth of CACO-2 cells. (A, B and C) CACO-2 cells were cultured for 72 h in the presence of indicated inhibitors at concentrations depicted in each panel. Drugs were combined as noted. Samples were stained with violet crystal to quantify cell growth. Results are the mean of two to three independent experiments. (B) * marks p < 0.05 when comparing the group treated with the same Rapa concentration in the absence of FK506. (C) * marks p < 0.05 when comparing the group treated with the same Rapa concentration in the absence of CsA. (D) Clonogenic assay of CACO-2 cells cultured in the presence of CsA (2 μM), FK506 (2 μM) or Rapa (20 nM) alone or in combination, as depicted in the figure. Results are representative of two independent experiments.
We then combined FK506 and Rapa. At low concentrations of FK506 the effect of Rapa was dominant and culminated in a reduced cell growth (Fig. 3B). However, when high enough concentrations of FK506 were used (100- to 1,000-fold more than Rapa), its effect was dominant, and there was no difference in growth when compared with vehicle-treated cells (Fig. 3B). Due to the large excess of FK506 needed for the reversion of the Rapa phenotype, these results are most likely explained by the competition of FK506 and Rapa for their common partner protein, FK506-binding protein 12 (FKBP12), required for both drugs to bind their distinct targets (calcineurin and mTORC1, respectively).28 Data obtained in clonogenic assays corroborate these results (Fig. 3D).
Finally, as observed in the experiments described in Figure 1A and B, treatment with CsA or Rapa led to a reduction in cellular growth. However, the combination of these drugs had no additive effect (Fig. 3C). Analysis of clonogenic experiments carried under similar drug combination conditions, where only the highest concentration of the drugs was tested, provided similar results (Fig. 3D). These data demonstrate that the cytostatic effect of CsA is dominant over that of FK506 and suggest that CsA and Rapa may act on similar pathways, since no synergic effect could be detected in this setting.
Altered growth after treatment with CsA or Rapa is not due to apoptosis
We showed that treatment of colon carcinoma cells with CsA or Rapa led to a reduction in the accumulation of colon carcinoma cells in vitro (Figs. 1 and 2). Despite suggestive data that this phenotype was not caused by induction of cell death (Fig. 1C), we went on to test if the treatments could induce apoptosis. CACO-2 cells were cultured for 24 h, 48 h or 72 h in the presence of CsA, FK506 or Rapa. At each time point, cells and supernatant were collected, stained with propidium iodine (PI) and analyzed by FACS. We did not observe significant cell death induced by any of the treatments in the time points tested (Fig. 4A). Similar results were obtained with HT-29, HCT-116 and LOVO cells assayed in the same conditions (data not shown).
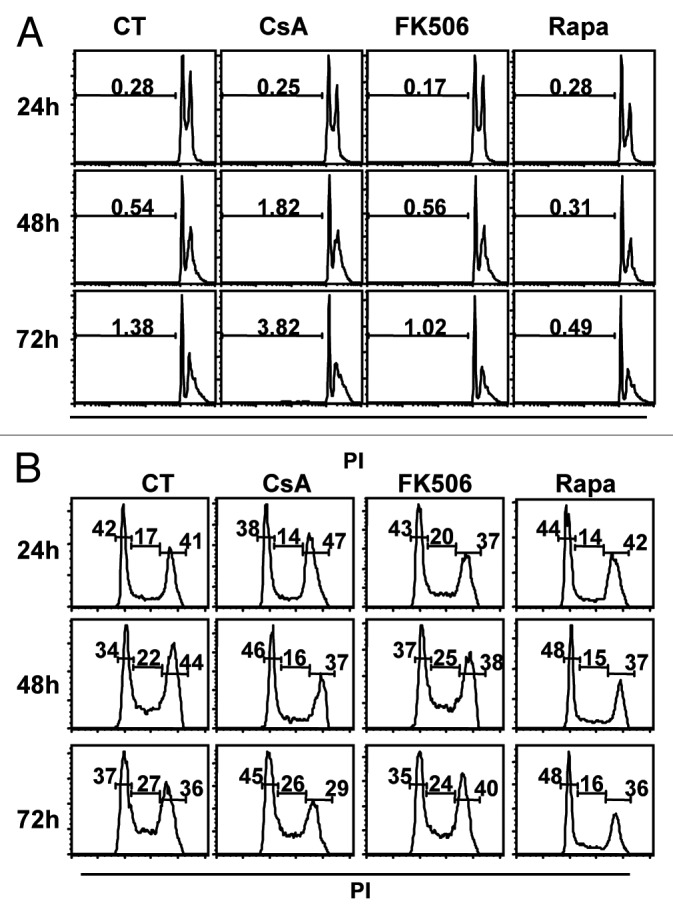
Figure 4. Treatment with CsA and Rapa does not induce cell death but leads to a small delay in cell cycle progression. Analysis of total DNA content of CACO-2 cells cultured in control conditions or treated with CsA (2 μM), FK506 (2 μM) or Rapa (20 nM). At indicated time-points cells were collected and their nuclei DNA content analyzed by propidium iodine (PI) staining and FACS in logarithmic scale to favor SubG0 (A) or in linear scale to favor 2–4 n DNA content analysis (B). Data are representative of six independent experiments.
Treatment with CsA and Rapa leads to a G0/G1 cell cycle arrest
Cellular expansion depends on a balance between cell death and proliferation. Since we observe an important reduction in total cells upon treatment with these drugs despite the absence of apoptosis, we evaluated cell cycle progression. CACO-2 cells were cultured as described above, and the distribution of cells along the phases of the cell cycle assayed by PI staining and FACS analysis. We detected an accumulation of cells at the G0/G1 stage upon treatment with CsA or Rapa, which is not seen in the control condition or when cells are treated with FK506 (Fig. 4B). Similar results were obtained when the other carcinoma cell lines were analyzed in similar conditions (data not shown).
CsA-induced cell cycle arrest is not due to production of TGFβ
Whereas TGFβ treatment may lead to the activation of calcineurin and consequently of NFAT proteins,29-31 in some model systems, treatment with CsA leads to the production of TGFβ.32-34 TGFβ exposure induces an arrest in cell cycle progression of primary endothelial cells while it promotes growth of transformed cells.35 Since we have observed an arrest at the G0/G1 stage of cellular proliferation upon treatment with CsA, it is possible that CsA exposure leads to TGFβ production and, consequently, cell cycle arrest of colon adenocarcinoma cells. Even though FK506 is more potent than CsA in inducing TGFβ expression and signaling in rat renal mesangial cells,36 an important variation among cell lines has been observed,32-34 making it important to test if in our model the cell cycle arrest induced by CsA and not FK506 is mediated by TGFβ. The production of TGFβ can be inferred based on the amount of latency-associated peptide (LAP) present in culture supernatants, since this peptide is part of the small latent complex of TGFβ, and its release is necessary for the formation and release of mature TGFβ. We have therefore measured hLAP in the supernatant of CACO-2 cell cultures following treatment with CsA or FK506. Despite finding a tendency for overproduction upon exposure to CsA, there was no significant difference between the amounts of hLAP detected in control-conditioned medium and that following any of the treatments (Fig. 5A). Indeed, inactivation of TGFβ1 in CsA-treated cultures by the use of blocking TGFβ1 antibody did not restore CACO-2 growth (Fig. 5B). TGFβ is therefore not the mechanism behind the cell cycle arrest of CsA-treated CACO-2 cells.
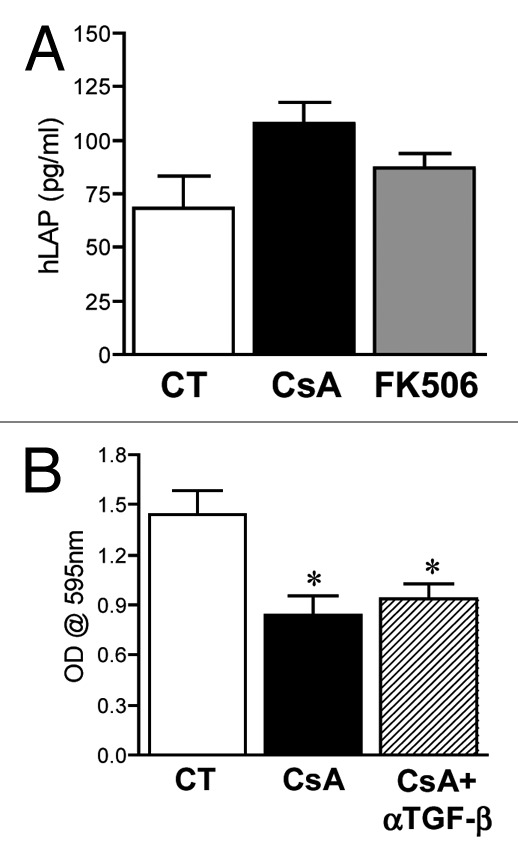
Figure 5. Arrest in cell proliferation following CsA treatment is not due to TGFβ production. (A) CACO-2 cells were cultured in control conditions or treated with CsA (2 μM) or FK506 (2 μM) for 96 h in culture, the supernatant was collected and hLAP quantified by ELISA. Results are the mean of seven independent experiments. (B) CACO-2 cells were cultured in control conditions, treated with CsA (2 μM) alone or in the presence of 10 μg/ml of αTGFβ for 72 h in culture, fixed and stained with violet crystal. Results are the mean of three independent experiments. * marks p < 0.05 when compared with CT group.
CsA induces necroptosis in CACO-2 cells
Given that an altered but modest progression through the cell cycle was observed when cells were treated with CsA or Rapa, we investigated if the cells could be dying by a programmed cell death other than apoptosis. We therefore evaluated the viability of CACO-2 cells upon the different treatments by looking at PI incorporation in a normo-osmotic buffer. Indeed, CACO-2 cells treated with CsA had a higher percentage of PI+ cells than those receiving any other treatment (Fig. 6A). Cells cultured in the presence of FK506 behave like control cells. Surprisingly, treatment with Rapa increased the viability of CACO-2 cells, reducing the percentage of PI+ cells after 48 h in culture (Fig. 6A).
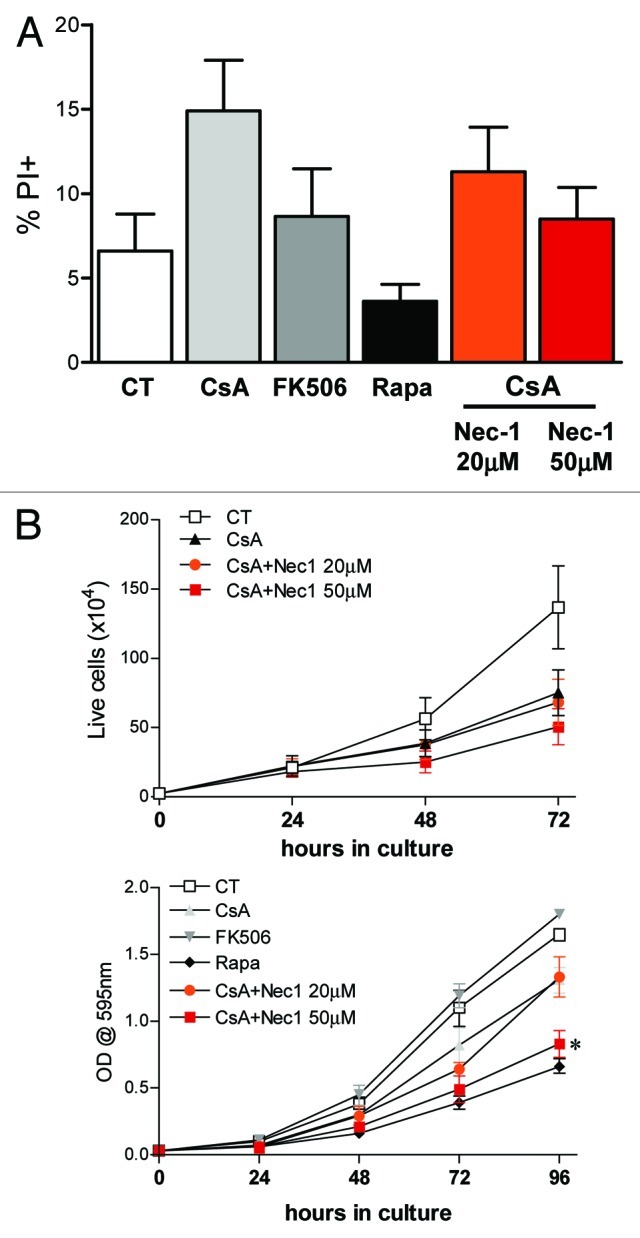
Figure 6. Treatment with CsA induces necroptosis. (A) CACO-2 cells were treated with vehicle, CsA, FK506, Rapa or a combination of CsA and Nec-1 as depicted in the figure for 48h. Data are the average of four independent experiments. (B) CACO-2 cells were cultured in the presence of drugs as described in A for 4 d. At each time point samples were trypsinized and counted (upper panel) or fixed and stained with violet crystal (lower panel). Results are the mean of three independent experiments. * marks p < 0.05 when comparing to group treated only with CsA, in the absence of Nec-1.
CsA has been shown to interfere with mitochondrial homeostasis, an effect not attributed to FK506.37 Therefore, a mitochondria-mediated cell death could be at play. To test if the reduced viability was a consequence of necroptosis, we concomitantly treated CACO-2 cells with CsA and increasing concentrations of necrostatin 1 (Nec-1).38 Nec-1 was able to revert the increase in PI+ cells in a dose-dependent manner, taking it to the levels of control cultures (Fig. 6A). Since treatment with Nec-1 reverted the decrease in viability induced by CsA, we analyzed if it reverted the growth arrest pattern of these cells as well. Surprisingly, despite the increased viability, treatment with low concentration of Nec-1 (20 nM) did not alter CACO-2 growth when compared with cells treated with CsA alone (Fig. 6B). Moreover, treatment with CsA and Nec-1 at 50 nM led to a reduction in size of CACO-2 cells when compared with cells treated with CsA alone, comparable to that following Rapa treatment, as shown by the reduced violet crystal staining of similar numbers of cells (Fig. 6B). Taken together, our data suggest that even though treatment of colon carcinoma cells with CsA leads to a decrease in viability due to induction of necroptosis, this type of cell death is not accounting for the reduced cellular accumulation in culture.
Altered survival upon CsA treatment is not due to mitochondrial instability
It is known that CsA can disrupt the assembly of the mitochondrial transition pore, an effect commonly associated with the protection of the cell against apoptosis induction.37 Even though we observe a reduction in cellular viability associated with CsA treatment (Fig. 6A), we show that the treatment does not induce apoptosis (Fig. 4A), but necroptosis instead. In cases where apoptosis is blocked, exposure of cells to extrinsic apoptosis triggers start the necroptosis program, we went on to test the mitochondrial homeostasis in the presence of CsA, FK506 and Rapa. Our data show that, as expected, treatment with CsA lead to mitochondrial hyperpolarization (Fig. 7). Rapa did not alter mitochondrial membrane potential when compared with control cells (Fig. 7). Surprisingly, FK506 treatment of CACO-2 cells led to mitochondrial hypepolarization to the levels seen upon treatment with CsA (Fig. 7). Consequently, CsA and FK506 hyperpolarize mitochondria to a similar degree, but cell growth is reduced in the presence of CsA and not FK506. Even though these are unexpected results, they suggest that altered mitochondrial homeostasis is not generating the stress that initiates the programmed necrosis observed upon treatment of CACO-2 cells with CsA.
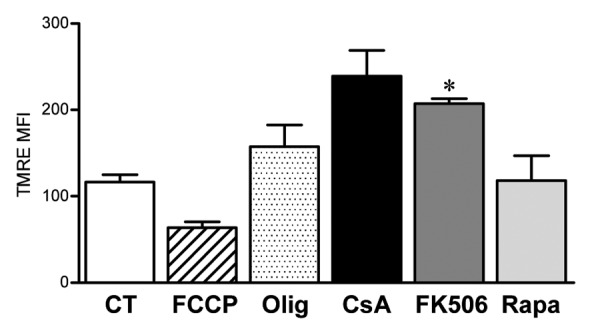
Figure 7. Altered mitochondrial membrane potential is not the cause of reduced CACO-2 viability. CACO-2 cells were cultured in the presence of vehicle, CsA (2 μM), FK506 (2 μM) or Rapa (20 nM) for 2 d. Control cells were then treated with the respiratory chain uncoupler FCCP or the ATP synthase inhibitor Oligomycin (Olig) for 15 min. All samples were stained with the fluorescent dye TMRE and analyzed by FACS. Results are the average of two independent experiments. * marks p < 0.05 when comparing to CT group.
The NFAT signaling pathway is not important for colon carcinoma cell line growth
We have been showing divergent results upon treatment of colon carcinoma cell lines with the calcineurin inhibitors CsA and FK506, suggesting that the cause of the growth arrest and reduced viability of these cells upon CsA treatment is independent of its effect on the activation of NFAT. To check if this is true, we treated CACO-2 cells with CsA or FK506 and looked for NFAT activation. We observed that both inhibitors blocked NFAT1 de-phosphorylation in a dose-dependent manner (Fig. 8A) and were able to suppress the transactivation capacity of NFAT family members in a luciferase reporter assay (Fig. 8B). We tested if Rapa was capable of modulating the activity of NFAT in our model by treating CACO-2 cells with this drug and stimulating them with phorbol-ester (PMA) and calcium ionophore (Ionomycin). Our data show that neither the NFAT1 de-phosphorylation nor NFAT transactivation capacity were inhibited by Rapa treatment, suggesting that mTOR and its complexes do not alter NFAT activity in CACO-2 cells (Fig. 8A and B).

Figure 8. Efficient inhibition of NFAT1 activation by CsA and FK506. (A) CACO-2 cells were treated with inhibitors at concentrations depicted for 30 min at 37°C. Cells were then kept unstimulated as control (CT) or stimulated with 2 μM of Iono for 3 min, lysed and the total protein extract was analyzed by SDS-PAGE. Western blot for NFAT1 followed. (B) CACO-2 cells were transfected with pGL4.30-NFAT-Luc and pRL-TK, and treated with CsA (2 μM), FK506 (2 μM) or Rapa (20 nM) for 24 h. Cells were stimulated for the last 6 h of culture with PMA (20 nM) and Iono (2 μM), lysed and luciferase activity was measured. Results are the mean of four independent experiments. * marks p < 0.05 when comparing to unstimulated group. (C) CACO-2 cells were transfected with pGL4.30-NFAT-Luc and pRL-TK, with or without pEGFP-VIVIT. Starting at 24 h after transfection (0 h), cells were stimulated for the last 6 h of culture with PMA (20 nM) and Iono (2 μM), lysed and luciferase activity was measured. Data represents the average of two independent experiments. (D) CACO-2 cells were transfected with pEGFP-VIVIT or mock-treated. Twenty-four hours after transfection cells were replated, and growth was analyzed by staining with violet crystal. Results are the mean of five independent experiments.
To confirm that NFAT inhibition is not the mechanism behind the cytostactic/cytotoxic effects of CsA on colon carcinoma cells, we transfected CACO-2 cells with a plasmid coding for the peptide VIVIT, which was designed to compete with NFAT family members for the interaction site in calcineurin.39 Expression of VIVIT leads to the inhibition of NFAT de-phosphorylation and therefore activation, as confirmed by evaluation of NFAT1 de-phosphorylation by western blot (data not shown) and the transactivation of a NFAT-responsive reporter gene at different times after cellular transfection (Fig. 8C). When the growth of these cells were evaluated, we observed that even though the expression of VIVIT was able to inhibit NFAT transactivation capacity throughout the time course assessed (Fig. 8C), there was no difference in the growth of CACO-2 measured by violet crystal (Fig. 8D). We can therefore conclude that the cytostactic and toxic effects of CsA on human colon carcinoma cell lines is not due to the inhibition of NFAT family members.
Effect observed upon CsA treatment does not involve modulation of NFκB
Even though calcineurin is the common target of CsA and FK506, their effect in other signaling pathways have been shown, situations where they can have differential modulatory capacities. Among those, the nuclear factor κB (NFκB) pathway can be inhibited by FK506 in hepatocytes, but is not altered by treatment with CsA.40 Even though constitutive activation of this transcription factor has more often been associated with cellular transformation,41 a biphasic role for NFκB has been shown in ovarian cancer cell lines, where it can also act as a tumor suppressor and its inhibition promotes cellular growth.42 Since we observe a tendency toward overgrowth of colon carcinoma cell lines upon treatment with FK506 in vitro (Figs. 1 and 2), we hypothesized that modulation of this transcription factor could be playing a role in the phenotype we are observing. We therefore tested the capacity of CsA, FK506 and Rapa to modulate the transactivation capacity of NFκB in CACO-2 cells. We observed that neither of the inhibitors altered the capacity of NFκB to induce the expression of the luciferase reporter gene controlled by a NFκB-responsive artificial promoter, whereas MG132, a proteasome inhibitor, prevented NFκB transactivation (Fig. 9). We conclude that the mechanism behind the decreased viability of colon carcinoma cells induced by CsA does not depend on altered NFκB signaling.
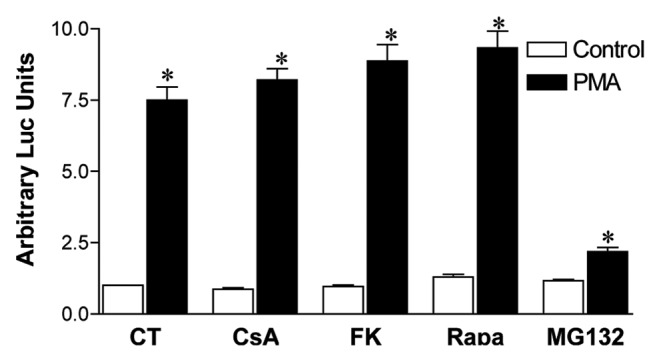
Figure 9. CsA blockade of CACO-2 cell growth is not mediated by altered NFκB activity. CACO-2 cells were transfected with pGL3–6xNFκB-Luc and pRL-TK. Twenty-four hours after transfection, cells were treated overnight with CsA (2 μM), FK506 (2 μM), Rapa (20 nM), Ly294002 (30 μM) or MG132 (20 μM). Cells were stimulated for the last 6 h of culture with PMA (20 nM), lysed and luciferase activity was measured. Results are the mean of two independent experiments. * marks p < 0.05 when comparing to unstimulated group.
CsA does not act upon the PI3-K/mTOR pathway
We have shown that CsA and Rapa do not synergize in the inhibition of CACO-2 cell growth (Fig. 3C). It is therefore possible that these two drugs are acting upon similar pathways. Since Rapa does not alter the de-phosphorylation or transactivation capacity of NFAT (Fig. 8), we checked if treatment with CsA modified the activation of the PI-3K/mTOR pathway following nutrient sensing. First, we assayed the impact of PI-3K inhibition on the growth of human colon carcinoma cell lines. Treatment of these cells with the PI-3K inhibitor Ly294002 had a dramatic impact on cellular growth, observed both by crystal violet staining and in clonogenic assays (Fig. 10A and B). We went on to look at the phosphorylation status of p70S6K and p80S6K, mTORC1 targets, when the cells were treated with calcineurin inhibitors and those of the PI-3K pathway. We observed that even though treatment of CACO-2 cells with Ly294002 or Rapa completely inhibited the phosphorylation of S6K in response to nutrient exposure, CsA and FK506 had no effect (Fig. 10C). We therefore conclude that the reduced growth of colon carcinoma cells exposed to CsA is not due to an effect of this drug on the PI-3K/ mTOR pathway.

Figure 10. CsA is not altering signaling via the PI-3K/mTOR pathway. (A) Colon carcinoma cell lines were cultured in the presence of Rapa (20 nM) or Ly294002 (30 μM) for 96 h and stained with violet crystal. Results are the mean of three to six independent experiments. * marks p < 0.05 when comparing to CT group. (B) Clonogenic assay of colon carcinoma cell lines cultured in the presence of specific inhibitors as in panel A. Results are representative of three independent experiments. (C) Colon carcinoma cell lines were plated, followed by overnight culture without serum. Cells were left untreated or received CsA (2 μM), FK506 (2 μM), Rapa (20 nM) or Ly294002 (30 μM) 30 min prior to addition of 10% FBS. Control cells were left without treatment or stimulation (NS). Cells were then lysed, and the total protein extract was analyzed by SDS-PAGE. Western blot for p70S6K and GAPDH followed.
Toxicity of CsA is transformation- and tissue-specific
We have observed that treatment with CsA leads to an impairment in cell growth in all human colon carcinoma cell lines tested (Figs. 1 and 2), suggesting a general toxic effect. We therefore tested if the concentration of CsA herein used (2 μM) would affect the growth of non-transformed human cells. We cultured normal, immortalized human esophagus and lung cell lines for 96 h in control conditions or in the presence of CsA or FK506 and accessed cell growth by crystal violet staining. Our data shows that treatment of these non-transformed cell lines with CsA at concentrations that reduced the growth of colon adenocarcinoma cells did not alter their growth (Fig. 11A). Since the specific target of CsA in our model has not yet been identified, and we were not able to find non-transformed human colon epithelial cells, these results could be explained by a tissue-specific effect of CsA. We therefore tested the effect of CsA on esophagus adenocarcinoma cells, as well as breast cancer cells. Treatment of OE33 and OE19 esophagus adenocarcinoma cell lines with CsA led to a significant decrease in cell growth, as measured by crystal violet staining (Fig. 11B). Interestingly, the human breast adenocarcinoma cell line MDA-MB-231 and the human breast-invasive ductal carcinoma cells MCF-7 and MN1 were not sensitive to CsA treatment and grew normally in the presence of this drug (Fig. 11C). None of the cell lines were sensitive to FK506 treatment (Fig. 11), as shown for the colon adenocarcinoma tested (Figs. 1 and 2). We therefore conclude that the toxicity of CsA is not generalized. Instead, the calcineurin-independent cell growth arrest induced by CsA is transformation- and cell type-specific.
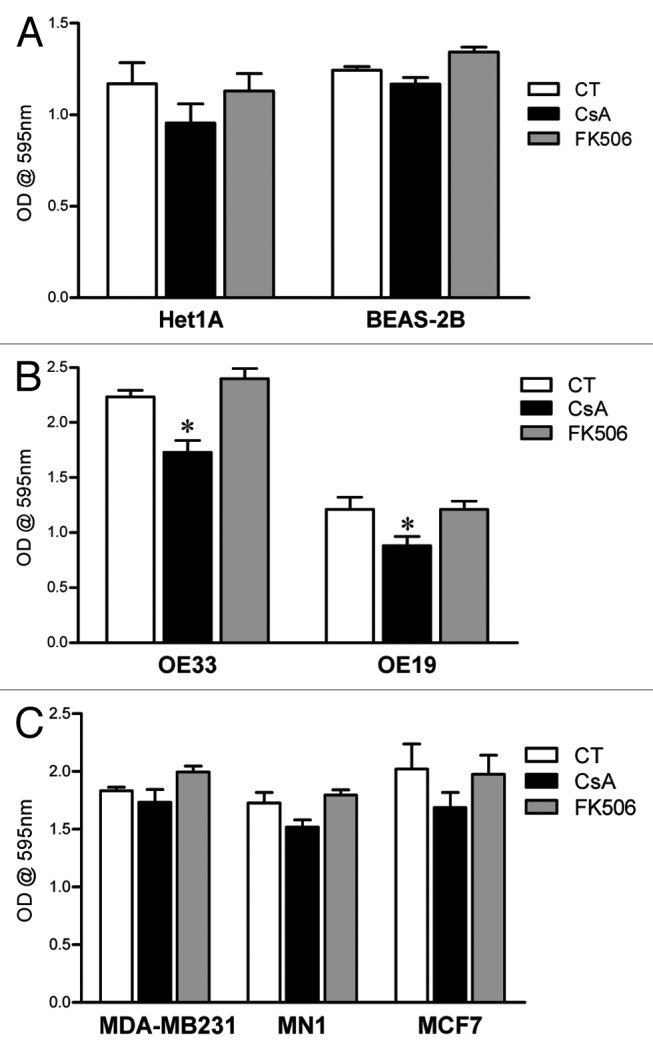
Figure 11. Differential susceptibility of human cell lines to CsA treatment in vitro. (A) Not-transformed human cell lines of esophagus and lung were cultured in control conditions or treated with CsA or FK506 (2 μM) for 96 h, when they were fixed and stained with violet crystal. Results are the mean of three to five independent experiments. (B) Human esophagus adenocarcinoma cell lines were treated as in (A). Results are the mean of three to five independent experiments. * marks p < 0.05 when comparing to CT group. (C) Human breast adenocarcinoma cell lines were treated as in (A). Results are the mean of three to five independent experiments. * marks p < 0.05 when comparing to CT group.
Discussion
The contribution of chronic inflammation to cellular transformation and the establishment of cancer has been shown in diverse models.2,3 Specifically for colorectal tumors, continuous, prophylactic administration of aspirin, a non-steroidal immunosuppressor, reduces the incidence of colon cancer, not only due to a reduction in the tissue-associated inflammation, but also because it inhibits Cox-2 in the transformed epithelial cells.12,15 Deregulated expression of Cox-2 has been associated with cellular transformation,14 and its inhibition reduces the proliferation and growth of colon carcinoma cells.12,15 Cox-2 is one of the genes regulated by Ca2+ signaling through the activation of NFAT, which directly binds to the cox-2 promoter and transactivates the gene.25,43
To a similar extent, mutations that enhance the PI-3K/mTOR pathway, the signaling pathway activated upon nutrient sensing, have been shown to contribute to tumorigenesis of epithelial cells, which include colon carcinoma.26,27 Most commonly, reduced PTEN activity, which leads to a prolonged PI-3K signaling window, or mutations that constitutively activate the PI3K catalytic subunit have been found in colorectal tumor cells and, when induced, can promote cellular transformation.26,27
In this study we went on to test if, as shown for NFAT3 in fibroblasts,22 mTOR was capable of regulating the activity of NFAT family members in cancer carcinoma cells. Specifically, that of NFAT1, which functions as a tumor suppressor and, in the case of being inactivated by mTOR, could directly contribute to the transformed phenotype of cells in which the PI3K/mTOR pathway is enhanced. We show that treatment of the human colon carcinoma cell lines CACO-2, HCT-116, HT-29 and LOVO with CsA or Rapa leads to a decrease in cell growth without inducing cell death by apoptosis. We observe an arrest in cell cycle progression following the treatment with these drugs. In addition, treatment with CsA leads to a decrease in cellular viability compatible with the induction of programmed necrosis, which can be inhibited by treatment with the RIP1/RIP3 inhibitor Nec-1. Surprisingly, this rescue does not restore cellular growth and accumulation in culture, suggesting that several pathways are involved in the growth arrest induced by CsA. Furthermore, this effect cannot be ascribed to the inhibition of calcineurin or NFAT family members, since treatment of these cells with FK506 leads to opposite results in terms of cellular progression in culture. Corroborating these observations, blockade of calcineurin does not lead to production of TGFβ by colon cancer cells, and blockade of this growth factor does not alter the effect of CsA in culture. The impact of CsA treatment is dominant over that of FK506. However, it is not due to altered modulation of NFκB activity or to mitochondrial instability, since neither drug changes NFκB transactivation capacity, and both lead to mitochondrial hyperpolarization.
The reduction in colon carcinoma cell growth following CsA treatment has been previously reported.44 However, because only this calcineurin inhibitor was used at the time, its effects were attributed to the lack of this phosphatase's activity. The data herein presented clarifies this issue by showing that it is not the absence of calcineurin activity that impairs the growth of these cells. In fact, FK506 treatment tends to promote cell growth and survival. Therefore, the deleterious effect following CsA treatment is independent of calcineurin.
We were not able to determine the specific pathway being targeted by CsA in this model. We have, though, clearly shown that CsA is not acting through its usual or previously described targets. An important suspect, the NFκB pathway, can be inhibited by FK506 but not by CsA in hepatocytes,40 and has been shown to promote either tumor cell death or growth depending on the characteristics of the transformed cell.41,42 However, we show here that its activity is not modified by treatment of human colon carcinoma cells with CsA or FK506, demonstrating that FK506 does not act upon NFκB in this cell type and excluding an altered NFκB activity as the mechanism behind the reduced cell viability and growth seen upon CsA treatment. The effect of CsA on mitochondrial homeostasis through the impaired formation of the transition pore was also assayed and was shown not to be the mechanism behind our results, since similar data showing mitochondrial hyperpolarization are obtained upon FK506 treatment without the consequence of a reduced viability. We also demonstrate that combining CsA and Rapa does lead to an additive effect in culture, suggesting at first that both are acting on the same pathway in this model. The data suggesting that both drugs lead to cell cycle arrest in the G0/G1 phase corroborates this idea. Speaking against this hypothesis are the observations that treatment with Rapa but not CsA leads to a reduction in cell size, whereas CsA but not Rapa activates the necroptosis pathway. Indeed, we did not identify a direct cross-talk between the two signaling pathways. Rapa did not change the phosphorylation status of NFAT1 or the transactivation capacity of NFAT family members, and CsA is not modulating the activity of mTORC1, as seen by similar phosphorylation of its target S6K when compared with the control group. Interestingly, combination of CsA with the currently used genotoxic chemotherapeutic drugs oxaliplatin or 5-fluorouracil did not change the sensitivity of CACO-2 cells to these drugs (data not shown). Similar to what was observed with the combination of CsA with Rapa, these data suggest a common mechanism between these treatments, specificities of which remain to be shown.
We show that the effect of CsA on cellular growth is not unrestricted, but may work in a transformation-specific fashion, since CsA has no effect on non-transformed esophagus and lung cell lines. Furthermore, among transformed cells, colon and esophagus adenocarcinoma cell lines are sensitive while breast cancer cells are refractory to this drug. These observations suggest an increased susceptibility of digestive track-associated tumors to the cytostatic effects of CsA when compared with breast tumors. Even though it is possible that this susceptibility is due to tissue-specific characteristics maintained by the transformed cells, it is equally possible that the chronic inflammatory milieu responsible for the initiation and maintenance of esophagus and colon cancer imprints an epigenetic signature that relies on pathways targeted by CsA. If the former hypothesis stands, studies comparing the expression profile of cancer cells from different inflammation-associated tumors may determine good, more broadly specific therapeutic targets to be used in the clinics as well as help uncovering the signaling pathway modified by CsA in this context.
The mechanism underpinning the effect of CsA on the growth of human colon adenocarcinoma cell lines remains to the determined. Nevertheless, we confirm its effect on reducing cell growth and clearly show that it is not acting through its most common target calcineurin; therefore, inhibition of NFAT activity does not change the physiology of these cells. Moreover, treatment of colon carcinoma cells with Rapa leads to differential cell viability and growth, suggesting that even though we did not observe statistically different growth rates, concomitant treatment of tumors with these two drugs may be beneficial to the host. These suggestions are in agreement with experimental data that show reduced side effects and risk of tumor formation in combination with efficacious immune suppression when these two drugs are combined.45-47 Chronic inflammation and specifically inflammatory bowel disease can lead to tumorigenesis,2,3,48 conditions for which the treatment includes immunosuppressive therapy. Keeping in mind that our data show that the two calcineurin inhibitors, CsA and FK506, are not interchangeable in the context of colon tumor growth, it will be of interest to check if cohorts of inflammatory bowel disease patients receiving CsA as immunosuppressive therapy show reduced incidence of colon cancer.
Materials and Methods
Cell culture
The human colon carcinoma cell lines CACO-2, HCT-116, HT-29 and LOVO cells, the human esophagial adenocarcinoma cell lines OE19 and OE33, the human breast adenocarcinoma cell line MDA-MB-231, the human breast-invasive ductal carcinoma MCF-7 and MN1 and the normal human bronchial epithelium cells BEAS-2B were cultured in RPMI-1610 supplemented with 10% FCS, L-glutamine, streptomycin-penicillin, sodium pyruvate and 2-mercaptoethanol (Invitrogen) at 37°C in 5% of CO2. The human immortalized esophageal epithelial cells Het-1A were cultured in BEGM™ Bronchial Epithelial Cell Growth Medium (Lonza) supplemented with streptomycin-penicillin in tissue culture flasks and plates coated with a mixture of 0.01 mg/ml fibronectin, 0.03 mg/ml bovine collagen type I and 0.01 mg/ml bovine serum albumin dissolved in culture medium, at 37°C in 5% of CO2.
Cell proliferation studies
CACO-2, HCT-116, HT-29, LOVO, Het1A, BEAS-2B, OE33, OE19, MDA-MB231, MN1 or MCF7 cells were plated in triplicate in 96-well microtiter plates at the concentrations of 2.5, 5.0, 10, 10, 20, 20, 10, 20, 10, 20 or 10 × 103 cells per well, respectively. Cell proliferation was analyzed at the indicated times by violet crystal as previously described.20 Briefly, cells were fixed with ethanol, stained with 0.05% violet crystal in 20% ethanol, washed with distilled water and solubilized with methanol. The plate was read on a spectrometer at 595 nm. When indicated, cells were treated with 10 μg/ml of αTGFβ (R&D Systems).
Clonogenic assay
Six-well plates were seeded in triplicate with 200, 200, 400 or 400 CACO-2, HCT-116, HT-29 or LOVO cells, respectively. Cells were allowed to grow until colonies in the control well were easily distinguishable, fixed with ethanol, stained with 0.05% violet crystal in 20% ethanol and washed with distilled water. Quantification of colony size and area was performed by analyzing each picture using the colony quantification method in ImageQuant TL sofware (GE Healthcare) after images were saved as gray scale TIFF files using the Photoshop CS4 software (Adobe).
Cell cycle and sub-G1 analyses
Six-well plates were seeded with 1.0, 2.0, 4.0 or 4.0 × 105 CACO-2, HCT-116, HT-29 or LOVO cells, respectively, and stained proceeded as previously described.20 Briefly, after the treatment described in each figure legend and on the indicated day, the cells were trypsinized and washed once with phosphate-buffered saline (PBS). The cells were then stained with propidium iodide (75 μM) in the presence of NP-40. Analysis of the DNA content was done by collecting 10,000 events for cell cycle analysis in linear scale or 20,000 events for sub-G1 analysis in logarithmic using a FACScalibur flow cytometer (BD Biosciences). Data was analyzed using the FlowJo software (Tree Star Inc.).
Viability analyses
Six-well plates were seeded with 1.0 × 105 CACO-2 cells and treated with vehicle, CsA (2 μM), FK506 (2 μM), Rapa (20 nM), CsA (2 μM) + Necrostatin 1 (Nec-1; 20 μM) or CsA (2 μM) + Nec-1 (50 μM) for 48 h. Cells were then trypsinized, washed once with phosphate-buffered saline (PBS) and ressuspent in 5 μg/ml PI in PBS buffer prior to acquisition of data at FACSCalibur (BD Biosciences) and analysis using the FlowJo software (Tree Star Inc.).
Quantification of human LAP
The amount of human latency-associated peptide (hLAP) produced by CACO-2 cells upon treatment with vehicle, CsA (2 μM), FK506 (2 μM) or Rapa (20 nM) was quantified by ELISA, following manufacturer’s protocols (R&D Systems).
Measurement of mitochondrial membrane potential
Six-well plates were seeded with 7.5 × 104 CACO-2. The following day CsA (2 μM), FK506 (2 μM), Rapa (20 nM) or vehicle was added to the wells and cells were treated for 48 h prior to analysis. On the day of the analysis, treated cells were trypsinized and ressuspent in conditioned media containing the respective drugs, control cells were left untreated, treated with the proton ionophore FCCP (1 μM; Sigma) or with the F1F0 ATP-synthase inhibitor Oligomicin (2 μg/ml; Sigma) for 15 min at 37°C prior to addition of TMRE (100 nM; Fluka Analytical). Cells were incubated with TMRE at 37°C for 15 min, samples were run at FACSCalibur (BD Biosciences), and data was analyzed using the FlowJo software (Tree Star Inc.).
Western blot for NFAT1
CACO-2 at 2 × 106 cells/ml were treated with CsA, FK506, Rapa or vehicle for 30 min at 37°C with periodical homogenization. Cells were then left unstimulated or stimulated with 2 μM Ionomycin (Iono) for 3 min. After a quick spin, cells were lysed in ressuspension buffer [40 mM Tris (pH 7.5), 60 mM NaPPi, 10 mM EDTA] containing 5% SDS. Total protein extracts were analyzed by electrophoresis on 6% SDS-PAGE, and blotted with polyclonal antibody 67.1 anti-NFAT1 (kind gift from Dr. A. Rao, Harvard University) as described.49
Western blot for mTOR target
Six-well plates were seeded with 2.0, 4.0, 8.0 or 8.0 × 105 CACO-2, HCT-116, HT-29 or LOVO cells, respectively. Twenty-four hours later, wells were washed and cells were left in medium without serum overnight. The following day cells were treated with CsA (2 μM), FK506 (2 μM), Rapa (20 nM) or vehicle for 30 min, followed by stimulation with serum at a final concentration of 10% for 30 min. Cells were immediately lysed in warm ressuspension buffer and total protein extracts analyzed by electrophoresis as described above. Anti-GAPDH (Santa Cruz Biotechnology) and anti-p70S6K (Cell Signaling Inc.) were used following manufacturer’s protocols.
Transfection and reporter assay
Ten centimeter plates were seeded with 5.0 × 105 CACO-2 cells and transfected by CaCl2 precipitation with pGL4.30-NFAT-Luc and pRL-TK with or without pEGFP-VIVIT, or with pGL3–6xNFκB-Luc and pRL-TK. Twenty-four hours after transfection, cells were washed, trypsinized and seeded at 7.5 × 104 cells/well. Cells were treated with CsA (2 μM), FK506 (2 μM), Rapa (20 nM) or MG132 (20 μM) for 30 min, and stimulated for 6 h with PMA (20 nM) and Iono (2 μM). Cells were lysed in Lysis buffer and luciferase activity was measured by Dual Luciferase-Reporter Assay System (Promega) in a Veritas Microplate luminometer (Turner BioSystems).
Statistics
Data was analyzed using Microsoft Excel software and the two-tailed Student’s t test. The null hypothesis was rejected, and differences were assumed to be significant at a p value < 0.05.
Acknowledgments
We are grateful to Dr. Anjana Rao for kindly providing the NFAT1 reagents. This work was supported by grants from CNPq (to M.B.F.W., P.T.B. and J.P.B.V.), FAPERJ (to P.T.B. and J.P.B.V.) and INCT-Cancer (to P.T.B. and J.P.B.V.). M.B.F.W. was supported by a PNPD fellowship from CAPES/FAPERJ.
Glossary
Abbreviations:
- NFAT
nuclear factor of activated T cells
- mTOR
mammalian target of rapamycin
- Rapa
rapamycin
- CsA
cyclosporin A
- Nec-1
necrostatin 1
- PI-3K
phosphoinositide 3-kinase
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22222
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trinchieri G. Cancer and Inflammation: An Old Intuition with Rapidly Evolving New Concepts. Annu Rev Immunol. 2011;30:677, 706. doi: 10.1146/annurev-immunol-020711-075008. [DOI] [PubMed] [Google Scholar]
- 4.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–37. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sonnemann KJ, Bement WM. Wound repair: toward understanding and integration of single-cell and multicellular wound responses. Annu Rev Cell Dev Biol. 2011;27:237–63. doi: 10.1146/annurev-cellbio-092910-154251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–7. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 8.Qazi BS, Tang K, Qazi A. Recent advances in underlying pathologies provide insight into interleukin-8 expression-mediated inflammation and angiogenesis. Int J Inflam. 2011:908468. doi: 10.4061/2011/908468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131:33–45. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garrett WS, Punit S, Gallini CA, Michaud M, Zhang D, Sigrist KS, et al. Colitis-associated colorectal cancer driven by T-bet deficiency in dendritic cells. Cancer Cell. 2009;16:208–19. doi: 10.1016/j.ccr.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuo SH, Yeh KH, Wu MS, Lin CW, Hsu PN, Wang HP, et al. Helicobacter pylori eradication therapy is effective in the treatment of early-stage H pylori-positive gastric diffuse large B-cell lymphomas. Blood. 2012;119:4838–44. doi: 10.1182/blood-2012-01-404194. [DOI] [PubMed] [Google Scholar]
- 12.Chan AT, Ogino S, Fuchs CS. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N Engl J Med. 2007;356:2131–42. doi: 10.1056/NEJMoa067208. [DOI] [PubMed] [Google Scholar]
- 13.Chan AT, Ogino S, Fuchs CS. Aspirin use and survival after diagnosis of colorectal cancer. JAMA. 2009;302:649–58. doi: 10.1001/jama.2009.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010;10:181–93. doi: 10.1038/nrc2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, et al. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–9. doi: 10.1016/S0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 16.Tedesco D, Haragsim L. Cyclosporine: a review. J Transplant. 2012;2012:230386. doi: 10.1155/2012/230386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–84. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- 18.Müller MR, Rao A. NFAT, immunity and cancer: a transcription factor comes of age. Nat Rev Immunol. 2010;10:645–56. doi: 10.1038/nri2818. [DOI] [PubMed] [Google Scholar]
- 19.Mancini M, Toker A. NFAT proteins: emerging roles in cancer progression. Nat Rev Cancer. 2009;9:810–20. doi: 10.1038/nrc2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robbs BK, Cruz AL, Werneck MB, Mognol GP, Viola JP. Dual roles for NFAT transcription factor genes as oncogenes and tumor suppressors. Mol Cell Biol. 2008;28:7168–81. doi: 10.1128/MCB.00256-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science. 1997;275:1930–4. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- 22.Yang TT, Yu RY, Agadir A, Gao GJ, Campos-Gonzalez R, Tournier C, et al. Integration of protein kinases mTOR and extracellular signal-regulated kinase 5 in regulating nucleocytoplasmic localization of NFATc4. Mol Cell Biol. 2008;28:3489–501. doi: 10.1128/MCB.01847-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Araki K, Ellebedy AH, Ahmed R. TOR in the immune system. Curr Opin Cell Biol. 2011;23:707–15. doi: 10.1016/j.ceb.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Powell JD, Delgoffe GM. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity. 2010;33:301–11. doi: 10.1016/j.immuni.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iñiguez MA, Martinez-Martinez S, Punzón C, Redondo JM, Fresno M. An essential role of the nuclear factor of activated T cells in the regulation of the expression of the cyclooxygenase-2 gene in human T lymphocytes. J Biol Chem. 2000;275:23627–35. doi: 10.1074/jbc.M001381200. [DOI] [PubMed] [Google Scholar]
- 26.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–62. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 27.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–44. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bjornsti MA, Houghton PJ. The TOR pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4:335–48. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- 29.Fernandez-Zapico ME, Ellenrieder V. NFAT transcription factors, the potion mediating “Dr. Jekill-Mr. Hyde” transformation of the TGFβ pathway in cancer cells. Cell Cycle. 2010;9:3838–9. doi: 10.4161/cc.9.19.13413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gooch JL, Gorin Y, Zhang BX, Abboud HE. Involvement of calcineurin in transforming growth factor-beta-mediated regulation of extracellular matrix accumulation. J Biol Chem. 2004;279:15561–70. doi: 10.1074/jbc.M308759200. [DOI] [PubMed] [Google Scholar]
- 31.Singh G, Singh SK, König A, Reutlinger K, Nye MD, Adhikary T, et al. Sequential activation of NFAT and c-Myc transcription factors mediates the TGF-beta switch from a suppressor to a promoter of cancer cell proliferation. J Biol Chem. 2010;285:27241–50. doi: 10.1074/jbc.M110.100438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li B, Sehajpal PK, Khanna A, Vlassara H, Cerami A, Stenzel KH, et al. Differential regulation of transforming growth factor beta and interleukin 2 genes in human T cells: demonstration by usage of novel competitor DNA constructs in the quantitative polymerase chain reaction. J Exp Med. 1991;174:1259–62. doi: 10.1084/jem.174.5.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minguillón J, Morancho B, Kim SJ, López-Botet M, Aramburu J. Concentrations of cyclosporin A and FK506 that inhibit IL-2 induction in human T cells do not affect TGF-beta1 biosynthesis, whereas higher doses of cyclosporin A trigger apoptosis and release of preformed TGF-beta1. J Leukoc Biol. 2005;77:748–58. doi: 10.1189/jlb.0904503. [DOI] [PubMed] [Google Scholar]
- 34.Prashar Y, Khanna A, Sehajpal P, Sharma VK, Suthanthiran M. Stimulation of transforming growth factor-beta 1 transcription by cyclosporine. FEBS Lett. 1995;358:109–12. doi: 10.1016/0014-5793(94)01382-B. [DOI] [PubMed] [Google Scholar]
- 35.Stover DG, Bierie B, Moses HL. A delicate balance: TGF-beta and the tumor microenvironment. J Cell Biochem. 2007;101:851–61. doi: 10.1002/jcb.21149. [DOI] [PubMed] [Google Scholar]
- 36.Akool S, Doller A, Babelova A, Tsalastra W, Moreth K, Schaefer L, et al. Molecular mechanisms of TGF beta receptor-triggered signaling cascades rapidly induced by the calcineurin inhibitors cyclosporin A and FK506. J Immunol. 2008;181:2831–45. doi: 10.4049/jimmunol.181.4.2831. [DOI] [PubMed] [Google Scholar]
- 37.Azzolin L, von Stockum S, Basso E, Petronilli V, Forte MA, Bernardi P. The mitochondrial permeability transition from yeast to mammals. FEBS Lett. 2010;584:2504–9. doi: 10.1016/j.febslet.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–14. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 39.Aramburu J, Garcia-Cózar F, Raghavan A, Okamura H, Rao A, Hogan PG. Selective inhibition of NFAT activation by a peptide spanning the calcineurin targeting site of NFAT. Mol Cell. 1998;1:627–37. doi: 10.1016/S1097-2765(00)80063-5. [DOI] [PubMed] [Google Scholar]
- 40.Kaibori M, Sakitani K, Oda M, Kamiyama Y, Masu Y, Nishizawa M, et al. Immunosuppressant FK506 inhibits inducible nitric oxide synthase gene expression at a step of NF-kappaB activation in rat hepatocytes. J Hepatol. 1999;30:1138–45. doi: 10.1016/S0168-8278(99)80270-0. [DOI] [PubMed] [Google Scholar]
- 41.Perkins ND. The diverse and complex roles of NF-κB subunits in cancer. Nat Rev Cancer. 2012;12:121–32. doi: 10.1038/nrc3204. [DOI] [PubMed] [Google Scholar]
- 42.Yang G, Xiao X, Rosen DG, Cheng X, Wu X, Chang B, et al. The biphasic role of NF-kappaB in progression and chemoresistance of ovarian cancer. Clin Cancer Res. 2011;17:2181–94. doi: 10.1158/1078-0432.CCR-10-3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Corral RS, Iñiguez MA, Duque J, López-Pérez R, Fresno M. Bombesin induces cyclooxygenase-2 expression through the activation of the nuclear factor of activated T cells and enhances cell migration in Caco-2 colon carcinoma cells. Oncogene. 2007;26:958–69. doi: 10.1038/sj.onc.1209856. [DOI] [PubMed] [Google Scholar]
- 44.Masuo T, Okamura S, Zhang Y, Mori M. Cyclosporine A inhibits colorectal cancer proliferation probably by regulating expression levels of c-Myc, p21(WAF1/CIP1) and proliferating cell nuclear antigen. Cancer Lett. 2009;285:66–72. doi: 10.1016/j.canlet.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 45.Basu A, Banerjee P, Contreras AG, Flynn E, Pal S. Calcineurin inhibitor-induced and Ras-mediated overexpression of VEGF in renal cancer cells involves mTOR through the regulation of PRAS40. PLoS One. 2011;6:e23919. doi: 10.1371/journal.pone.0023919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Basu A, Banerjee P, Pal S. Critical role of mTOR in calcineurin inhibitor-induced renal cancer progression. Cell Cycle. 2012;11:633–4. doi: 10.4161/cc.11.4.19276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Piao SG, Bae SK, Lim SW, Song JH, Chung BH, Choi BS, et al. Drug interaction between cyclosporine and mTOR inhibitors in experimental model of chronic cyclosporine nephrotoxicity and pancreatic islet dysfunction. Transplantation. 2012;93:383–9. doi: 10.1097/TP.0b013e3182421604. [DOI] [PubMed] [Google Scholar]
- 48.Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology. 2011;140:1807–16. doi: 10.1053/j.gastro.2011.01.057. [DOI] [PubMed] [Google Scholar]
- 49.Loh C, Shaw KT, Carew J, Viola JP, Luo C, Perrino BA, et al. Calcineurin binds the transcription factor NFAT1 and reversibly regulates its activity. J Biol Chem. 1996;271:10884–91. doi: 10.1074/jbc.271.18.10884. [DOI] [PubMed] [Google Scholar]