

Abstract
Autosomal dominant polycystic kidney disease (ADPKD) and other forms of PKD are associated with dysregulated cell cycle and proliferation. Although no effective therapy for the treatment of PKD is currently available, possible mechanism-based approaches are beginning to emerge. A therapeutic intervention targeting aberrant cilia-cell cycle connection using CDK-inhibitor R-roscovitine showed effective arrest of PKD in jck and cpk models that are not orthologous to human ADPKD. To evaluate whether CDK inhibition approach will translate into efficacy in an orthologous model of ADPKD, we tested R-roscovitine and its derivative S-CR8 in a model with a conditionally inactivated Pkd1 gene (Pkd1 cKO). Similar to ADPKD, Pkd1 cKO mice developed renal and hepatic cysts. Treatment of Pkd1 cKO mice with R-roscovitine and its more potent and selective analog S-CR8 significantly reduced renal and hepatic cystogenesis and attenuated kidney function decline. Mechanism of action studies demonstrated effective blockade of cell cycle and proliferation and reduction of apoptosis. Together, these data validate CDK inhibition as a novel and effective approach for the treatment of ADPKD.
Keywords: animal model, therapy, cell division, cell cycle, proliferation, apoptosis, cyclin-dependent kinases
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most common human-inherited disorder with an estimated incidence of 1:400–1:1,000. ADPKD is mainly characterized by formation and progressive enlargement of fluid-filled renal cysts that ultimately lead to end stage renal disease (reviewed in refs. 1–3). Cysts often form in liver, pancreas and other organs.4 The disease is caused by mutations in either PKD1 or PKD2 genes, encoding the proteins polycystin-1 and polycystin-2, responsible for 85% and 15% of ADPKD cases, respectively.4-6 Presently, there is no effective treatment for ADPKD, although significant progress has been made in recent years in identifying potential treatments.7,8
Cystic growth is associated with abnormalities in multiple cellular processes such as basement membrane thickening, increased proliferation and apoptosis, loss of cellular differentiation and polarity and acquisition of secretory phenotype by cyst-lining epithelia.4,9-12 Several dysregulated molecular pathways that contribute to these cellular abnormalities were identified and exploited for targets of therapeutic intervention. Multiple compounds targeting cAMP accumulation, proliferation, fluid secretion and mTOR pathway have been tested in preclinical studies and entered clinical trials.13-20 Unfortunately, completed trials with mTOR inhibitors were not successful, underscoring the importance of developing alternative therapeutic targets.
New insights into molecular mechanisms of cystogenesis transpired from the observation that polycystins and other proteins responsible for multiple forms of PKD localize to the primary cilium/centrosome.21-23 The cilium is resorbed during cell division, and centrosomes can serve as organizers of the mitotic spindle. Disruption of proteins associated with cilia or centrosomes could lead to alterations in the cell cycle and proliferation as seen in cystic disease. It has been shown that polycystins can directly affect cell cycle progression and centrosome duplication.24 Polycystin-1 was shown to directly regulate the cell cycle by inhibiting CDK2 activity through upregulation of p21waf1, arresting cells in G0/G1 phase and controlling terminal differentiation of tubular epithelial cells.25 Polycystin-2 is capable of binding helix-loop-helix protein Id2 and preventing its translocation to the nucleus, thus blocking cell cycle progression.26 The translocation of Id2 in cells with mutated polycystins is linked to downregulation of p21 leading to increase of CDK2 activity and cell cycle progression.
We have shown previously that pharmacological inhibition of cell cycle progression with the cyclin-dependent kinase (CDK) inhibitor R-roscovitine effectively attenuates cystogenesis in jck and cpk models of PKD.27-30 Mechanistic studies demonstrated that roscovitine inhibited cystogenesis through cell cycle arrest, transcriptional regulation and inhibition of apoptosis. Importantly, roscovitine treatment suppressed cAMP and aquaporin 2 in the cystic kidneys, suggesting that CDK inhibition targets the most proximal step in cystogenesis.31
To further validate CDK inhibition as an approach to treat ADPKD, preclinical efficacy needs to be established in an orthologous model. The goals of this study were to confirm efficacy of R-roscovitine in an orthologous mouse model of ADPKD with a conditionally inactivated Pkd1 gene (Pkd1 cKO)32 and to assess the efficacy of the second generation analog of roscovitine, S-CR8, a more potent and selective CDK inhibitor.33 We demonstrate effective inhibition of both renal and hepatic cystogenesis with R-roscovitine and S-CR8 compounds. Mode of action studies demonstrate that both compounds act through blockade of cell cycle and proliferation and attenuation of apoptosis.
Results
CDK inhibitor S-CR8 potently inhibits cystogenesis in vitro
To improve drug-like properties of R-roscovitine (metabolic stability, potency and selectivity), extensive medicinal chemistry studies identified a new and improved analog S-CR8, shown in Figure 1A.33,34 We have used a standard assay of MDCK cystogenesis in vitro to assess potency of S-CR8 as described previously.29,35 R-roscovitine was tested in parallel for comparison. MDCK cysts were grown in 96-well plates containing collagen gel with FBS-containing media for 4 d. Increasing concentrations of compounds were added to cysts and incubated for additional 4 d. Percent of inhibition of cystogenesis by each compound was measured by standard Alamar Blue assay (Fig. 1B) and confirmed by visual observation of cultured cysts under light microscope (not shown). The assay showed that both R-roscovitine and S-CR8 compounds reduce cyst formation in vitro in a dose-dependent manner with an IC50 of 16 μM and 0.2 μM, respectively. These data indicate that S-CR8 is approximately 80-fold more potent than R-roscovitine in cellular assay. This observation is in agreement with previously published data suggesting greater anti-tumor potency for S-CR8 compared with R-roscovitine in multiple cell lines (100-fold on the average of more than 65 cell lines).33
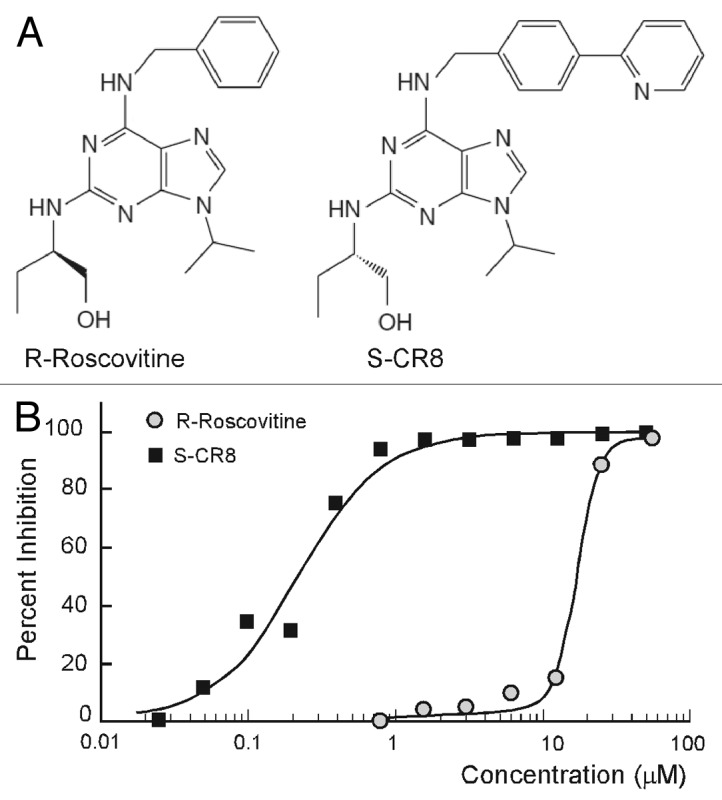
Figure 1. Comparative analysis of inhibitory activities of CDK inhibitors S-CR8 and R-roscovitine on cystogenesis in vitro. (A) Chemical structures of R-roscovitine and its derivative, S-CR8. (B) In vitro inhibition of cystic growth in MDCK 3D collagen-based assay. Values were measured in quadruplets in two independent experiments.
R-roscovitine and S-CR8 effectively inhibit renal cystic disease progression in Pkd1-conditional knockout mice
To determine whether CDK inhibition is an effective approach for the treatment of ADPKD, we sought to demonstrate the effect of R-roscovitine in an orthologous mouse model. This model has a germline Pkd1-null allele (Pkd1tm1Gzbd), a conditional knockout allele with lox sites flanking exons 21–23 (Pkd1tm1Gztn) and a tamoxifen-inducible Cre-gene.32 As shown previously, conditional inactivation of the Pkd1 gene at day 5 results in a rapid onset PKD that is gender-independent.32 Cysts in the liver are also observed in this model. Similar to other models with conditionally inactivated Pkd1 gene, the majority of cysts originate from distal nephron segments and collecting ducts.17 In the current study, cystogenesis was induced with tamoxifen at postnatal day 5. Animals received daily injections of either R-roscovitine (100 mg/kg IP, once a day) or vehicle control from day 7–33 (Fig. 2A). The R-roscovitine-treated group showed a significant inhibition of PKD, evident by a decrease in kidney to body weight ratio, cystic volume and blood urea nitrogen (BUN) (Fig. 2B and Table 1). Effective reduction of cystic tissue in a representative R-roscovitine treated kidney is illustrated in Figure 2C.
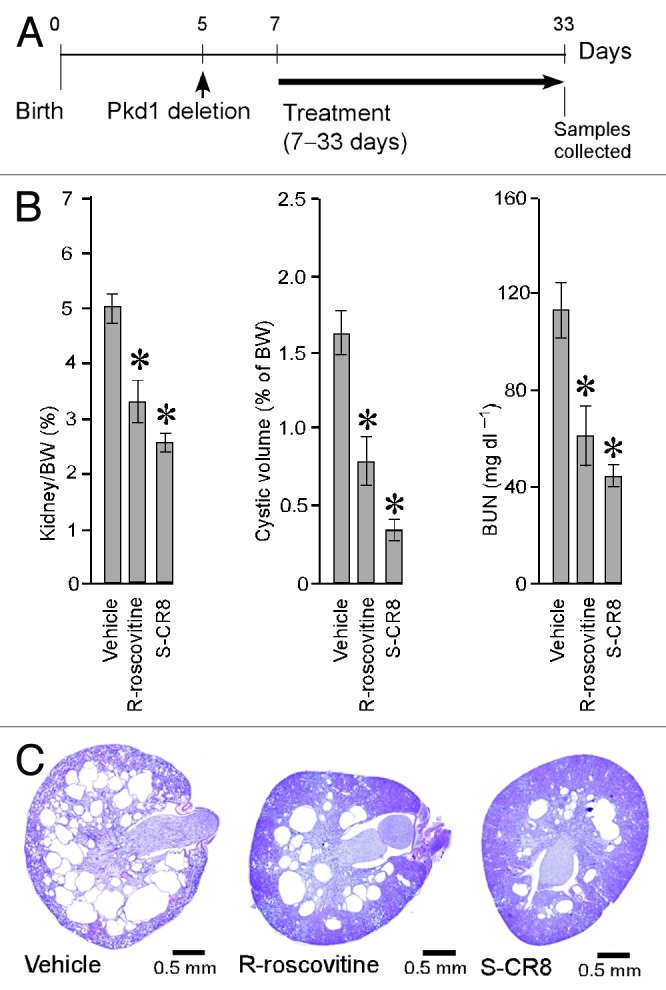
Figure 2. CDK inhibitors R-roscovitine and S-CR8 inhibit renal cystogenesis in Pkd1-conditional knockout mice. (A) Time frame of induction of Pkd1 deletion with tamoxifen and schedule of treatment with R-roscovitine and S-CR8. (B) Quantitative analysis of effect of R-roscovitine and S-CR8 on cystogenesis in kidney measured as kidney/body weight (BW) ratio, cystic volume and blood urea nitrogen (BUN); * p < 0.05 compared with vehicle control. Error bars indicate SEM; (C) Representative kidney sections (H&E staining) from treated mice and vehicle control suggest preservation of kidney parenchyma in animals treated with CDK inhibitors as compared with vehicle-treated group.
Table 1. Anti-cystic effect of CDK inhibitors R-roscovitine and S-CR8 in Pkd1 cKO mice .
| No | No of animals |
K/BW ratio (%) | Kidney cystic volume (%BW) |
BUN (mg dl–1) |
Liver cystic tissue (%) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
Vehicle |
27 |
4.98 |
± |
0.26 |
1.63 |
± |
0.14 |
113 |
± |
11 |
4.62 |
± |
0.53 |
| 2 |
R-roscovitine |
14 |
3.29 |
± |
0.39* |
0.80 |
± |
0.16* |
61 |
± |
12* |
2.25 |
± |
0.25* |
| 3 | S-CR8 | 19 | 2.55 | ± | 0.19* | 0.34 | ± | 0.07* | 45 | ± | 4* | 1.48 | ± | 0.19* |
indicates p < 0.05 for reduction in drug-treated vs. vehicle-treated animals. Values expressed as mean ± SEM.
In parallel, we also investigated the in vivo effect of S-CR8, a more potent and selective CDK inhibitor with a similar CDK inhibitory profile. Administration of S-CR8 (2.5 mg/kg IP, twice daily) was highly effective in inhibiting PKD progression in the conditional Pkd1-knockout model (Fig. 2A–C). Both compounds were equally well-tolerated in the course of treatment. As expected, S-CR8 was more dose-potent and showed greater PKD inhibition compared with R-roscovitine. Overall, these data support the conclusion that CDK inhibitors R-roscovitine and S-CR8 are effective in attenuating the progression of renal cystogenesis and improving kidney function in an orthologous Pkd1-linked mouse model of ADPKD.
Assessment of CDK inhibitors effect on hepatic cystogenesis
Because our Pkd1 cKO mice develop liver cysts in addition to PKD, we next examined the effect of CDK inhibition on hepatic cystogenesis. In contrast to kidney cystic disease, hepatic cystogenesis appears to be much less severe under conditions we used to induce Pkd1 gene deletion (see Fig. 2A). At postnatal day 33, the sporadic surface cysts are usually visible in the vehicle-treated group with a cystic area accounting for less than 5% of the total hepatic area (Table 1). To assess the percentage of hepatic cysts, liver sections of animals treated with either vehicle or CDK inhibitors R-roscovitine and S-CR8 were H&E stained, scanned with light microscopy and digitized followed by Metamorph analysis. Percentage of cystic liver tissue in mice treated with R-roscovitine and S-CR8 was significantly decreased as compared with the vehicle-treated group (Fig. 3A and B; Table 1). Similar to the effects on kidney cysts, S-CR8 was more effective in reducing liver cyst growth relative to R-roscovitine.

Figure 3. CDK inhibitors R-roscovitine and S-CR8 effectively inhibit hepatic cystogenesis in Pkd1 cKO mice. (A) Quantitative assessment of effects of R-roscovitine and S-CR8 on percentage of liver cysts. * p < 0.05 compared with vehicle control. Hepatic cystic areas are decreased in R-roscovitine and S-CR8-treated animals. Data are expressed as means ± SEM. (B) Representative light microscopic (H&E staining) images of liver sections from animals treated with R-roscovitine, S-CR8 and vehicle. Treatment with R-roscovitine and S-CR8 reduced hepatic cystogenesis. Scale bars, 100 μm
CDK inhibitors target dysregulated cell cycle and apoptosis in cystic kidneys
To elucidate the mechanisms by which CDK inhibitors affect PKD in an orthologous mouse model, we probed key pathways of cystogenesis using western blotting of kidney lysates from Pkd1 cKO mice treated with CDK inhibitors R-roscovitine and S-CR8. First, we examined the effects of CDK inhibitors on cell cycle progression. As shown in Figure 4A, Rb phosphorylation (p-Rb) was significantly increased in Pkd1 cKO kidneys (indicating cell cycle activation) and decreased in samples treated with either R-roscovitine or S-CR8 (suggesting G1/S cell cycle inhibition). We also observed significant upregulation of cyclinD3 (cycD3) expression and decreased cyclinD1 phosphorylation at Thr286 (p-cycD1) in vehicle-treated cystic kidneys, indicative of cell cycle activation and reversal of these patterns in samples treated with CDK inhibitors. Activation of Erk1/2, known to regulate cyclinD1, was effectively inhibited in treated samples. PCNA levels were decreased in treated samples, indicating inhibition of proliferation. Overall, S-CR8 showed more effective cell cycle blockade compared with R-roscovitine (Fig. 4A).
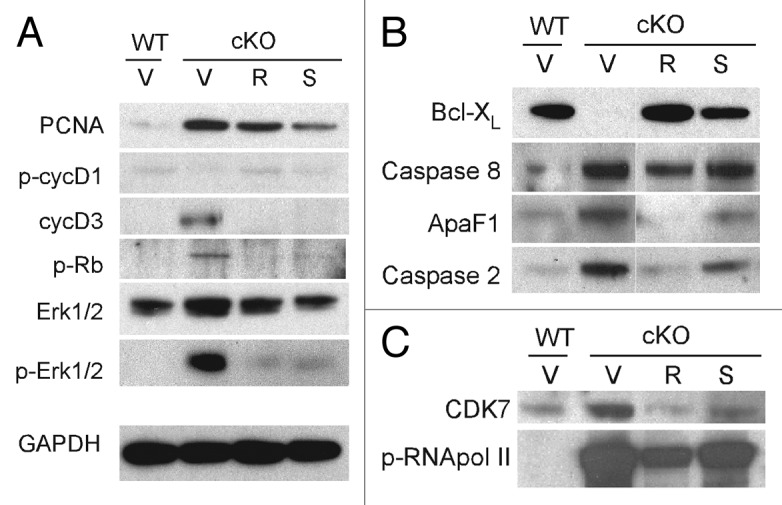
Figure 4. Molecular pathways of cystogenesis affected by CDK inhibitors R-roscovitine and S-CR8 in kidneys from Pkd1 cKO mice. Representative immunoblots show expression of markers in total kidney extracts from wild-type controls (WT) and Pkd1 cKO animals (cKO) treated with vehicle (V), R-roscovitine (R) or S-CR8 (S). Shown are effects of CDK inhibitors on cell cycle markers (A), apoptotic pathway (B) and transcriptional inhibition (C).
Next, we tested the effect of CDK inhibitors on apoptosis in treated kidneys. As shown in Figure 4B, we detected increased apoptosis in Pkd1 ThcKO kidneys evident by induction of caspase 2, an initiator of the mitochondrial apoptotic pathway; caspase 8, an activator of extrinsic apoptotic pathway; as well as decreased Bcl-XL expression and increase in ApaF1, as compared with wild-type (WT) kidneys. Interestingly, aberrant expression of apoptotic markers was normalized in kidneys treated with R-roscovitine and to a lesser extent in kidneys treated with S-CR8.
We also examined the effect of R-roscovitine and S-CR8 on the level of RNA polymerase II-dependent transcription, as shown in Figure 4C. Both compounds decreased CDK7 expression levels and effectively inhibited RNA pol II phosphorylation in treated kidneys.
Discussion
A significant progress in understanding the molecular mechanisms responsible for the development and progression of ADPKD has been made in recent years, providing the foundation for translation of preclinical therapeutic approaches into clinical development.7,36 Of particular interest is a recent discovery that proteins disrupted in ADPKD and other forms of cystic diseases map to a common site: primary cilia.21-23 There is a direct link between cilia, centrosomes and cell cycle dysregulation in PKD as reviewed in refs. 37 and 38. Through use of the CDK inhibitor R-roscovitine, we have recently shown that treatment of jck and cpk mice with slowly progressive and aggressive forms of PKD, respectively, resulted in a striking inhibition of cystic disease and improvement of renal function.29 Because these models do not carry mutations in Pkd1 or Pkd2 genes that are responsible for human ADPKD, therapeutic benefits of targeting the dysregulated cell cycle need to be validated in orthologous models. In fact, it has been previously shown that polycystins directly arrest cell cycle progression in G1 through mechanisms that converge on the induction of p21 and CDK2 inhibition and are important in maintaining centrosome integrity.24-26 Herein, we demonstrate effective blockade of PKD in an orthologous model of ADPKD with a conditionally inactivated Pkd1 gene using the CDK inhibitors R-roscovitine and S-CR8, a more potent and selective analog. Unlike many of the first generation CDK inhibitors that lack specificities and target multiple families of kinases, roscovitine is a highly selective, orally bioavailable compound targeting a small subset of CDKs that have been tested in clinical settings.39,40 The need to further improve its relatively low potency and metabolic stability has led to the generation of an improved, highly potent derivative, S-CR8.33
Mechanistic studies showed effective blockade of the cell cycle, attenuation of apoptosis and transcriptional inhibition with both R-roscovitine and S-CR8. Notably, both compounds displayed similar protein kinase selectivity profiles characterized by targeting CDK2/cyclinA, CDK2/cyclinE, CDK5/p25, CDK7/cyclinH and CDK9/cyclinT with approximately 2–3-fold greater potency of S-CR8 for each individual target.33 On the other hand, S-CR8 showed ~80-fold greater potency over R-roscovitine in cellular assay. This remarkably potent effect of S-CR8 in cellular cystic assay parallels previous findings on the survival of several tumor cell lines.34 It is possible that cellular effects of S-CR8 represent cumulative or synergistic effects on a subset of specific CDKs. In addition, target accessibility within a cell, permeability and subcellular compartment localization may also contribute to the observed differences in cellular assays.
Unlike tumorigenesis, cystogenesis is accompanied by an increase in apoptosis shown to be causally linked to cystic transformation: deletion of anti-apoptotic Bcl-2 and AP-2β genes in mice results in cystic disease.41,42 Also, in vitro formation of cysts by MDCK cells grown in collagen matrix is accompanied by increased apoptosis.43 Direct inhibition of apoptosis in vivo was shown to ameliorate PKD in mouse models.44 It has been proposed that an imbalance between pro-apoptotic and pro-proliferative factors plays a critical role in the development of cystic kidney disease.10 Our data suggest that anti-proliferative mechanistic effects of R-roscovitine and S-CR8 combined with their anti-apoptotic effects seen in Pkd1 cKO-treated kidneys are responsible for therapeutic efficacy.
Cystic epithelial cells in multiple forms of PKD are characterized by increased rates of both proliferation and apoptosis. Given the structural similarity between R-roscovitine and S-CR8 and similar kinase-inhibitory profiles, it is expected that they induce similar molecular responses in treated cells. Indeed, both compounds affected cell cycle and apoptosis in treated samples. At the same time, we observed some subtle differences between the two compounds. Specifically, S-CR8 showed a mechanistically stronger effect on cell cycle machinery, but a somewhat milder effect on attenuation of apoptosis relative to R-roscovitine. Notably, S-CR8 demonstrated better in vivo efficacy than R-roscovitine in reducing cystogenesis and preserving renal function, suggesting that dysregulated cell cycle and proliferation rather than apoptosis may play a pivotal role in promoting cystogenesis in the Pkd1 cKO model.
The relationship between proliferation and apoptosis in ADPKD is complex and not completely understood. While caspase inhibition slowed cystic disease progression in Han:SPRD rats, effective rapamycin treatment of the orpk-rescue mouse model and Pkd1 cKO model was associated with increased apoptosis.17,44,45 It has been shown recently that Cdc25A plays an important role in driving renal and hepatic cystogenesis in several animal models, providing further support for a key role of cell cycle dysregulation and proliferation in polycystic kidney and liver diseases.46 The authors showed elevated expression of Cdc25A phosphatase, a regulator of the cell cycle in cystic cholangiocytes of PKD patients, PCK rats and Pkd2ws25/- mice. Also, genetic and pharmacological inhibition of Cdc25A suppressed renal and hepatic cystogenesis in animal models.46
In summary, we have established efficacy of the CDK inhibitor R-roscovitine and its more potent derivative, S-CR8, in an orthologous model of ADPKD. Importantly, both CDK inhibitors significantly suppressed kidney cystic disease progression and functional decline as well as liver cystogenesis. Mechanistic studies showed effective inhibition of cell cycle, proliferation and apoptosis in treated kidneys. Taken together, our study provides further experimental support for the use of potent and selective CDK inhibitors for the treatment of ADPKD.
Material and Methods
Mouse colony handling and treatment
Mice were maintained and treated in accordance with Genzyme Institutional Animal Care and Use Committees (IACUC) guidelines. The generation and genotyping of Pkd1 cKO mice was done as described previously.32 Briefly, we crossed females homozygous for a Pkd1-conditional knockout allele (Pkd1tm1Gztn) with males heterozygous for a Pkd1 germline mutation (Pkd1tm1Gzbd)47 and homozygous for a tamoxifen-inducible Cre allele.48 Resulting animals heterozygous for the Cre allele and the Pkd1tm1Gztn-conditional allele were either heterozygous for Pkd1tm1Gzbd germline allele (mutants) or carried wild-type Pkd1 allele (wild-type controls). Disease was induced by intraperitoneal (IP) injection of nursing females with tamoxifen (250 mg/kg of body weight) to deliver it to pups with milk at day 5 after birth. R-roscovitine and S-CR8 were synthesized as previously described.49,50 Treatment with R-roscovitine was performed by IP injection of 100 mg/kg once daily from day 7–33 after birth. Treatment with S-CR8 was performed twice daily by IP injection with 2.5 mg/kg starting from day 7–33 after birth. Two and a half percent propylene glycol/10% Cerestar in water was used as a vehicle. Mice were euthanized by CO2 asphyxiation, and tissues were harvested for histological examination. Blood urea nitrogen levels were measured using a VetACETM analyzer (Alfa Wasserman).
Cell culture
MDCK kidney epithelial cells (ATCC) were grown in MEM/10% FBS. To test drug potency in vitro, the standard MDCK cyst assay was used. MDCK cysts were grown in 3D collagen I gel as previously described.35 Briefly, cysts from MDCK cells were grown in collagen I gel in 96-well plates for 4–5 d until cystic lumens were fully formed. Serial dilutions of drugs were added to cysts in culture followed by incubation for 4 d. Inhibition of cystic growth was quantified using Alamar Blue assay (BioSource). Fluorescence was measured with the microplate reader Spectra Max Gemini (Molecular Devices). Values were measured in quadruplets in two independent experiments.
Histology and quantitative analysis of cystogenesis
Percentage of cystic tissue was quantified as described previously.29 Briefly, longitudinal and cross kidney or liver sections (4 μm) were stained with hematoxylin and eosin (H&E) with a Tissue Tek® 2000 processor (Sakura-Finetek). Slides were scanned and digitized with an ACIS® system (Clarient) followed by processing with the Metamorph Imaging Series® software (Molecular Devices Corp.). Cystic volume was measured as cystic percentage (ratio of cystic area to a total section area) normalized by body weight.
Western blot analysis
Total kidney lysates were prepared in RIPA buffer containing 1 mM DTT, 5 mM EDTA, 2 mM NaF, 1 mM Na3VO4, with PefablockTM and complete protease-inhibitor cocktail (Roche Applied Science). Protein concentration was determined with BCA assay (Pierce). Proteins (40 μg) were resolved by SDS-PAGE using 4–12% NuPage gradient gels (Invitrogen) and blotted with semi-dry apparatus (Genomic Solutions) as previously described.29 Membranes were blocked with 5% non-fat milk in PBS and incubated overnight with primary antibodies at 4°C. Membranes were washed with Tris-buffered saline (TBS) containing 0.1% of Tween-20. Secondary antibodies (Promega) conjugated with horseradish peroxidase were used at 1:5,000 dilution. Immunoreactive proteins were detected by enhanced chemiluminescence (GE Healthcare). Equal loading of protein was controlled by anti-GAPDH staining. We used primary antibodies: GAPDH, PCNA, CDK7 (US Biological), ApaF1 (BD Pharmingen), Bcl-XL (BD Biosciences), caspase 2 (Chemicon), caspase 8, p-Rb, p-cycD1 (Cell Signaling), cycD3 (BD Transduction), p-RNApol II (Abcam).
Statistics
Data are expressed as means ± SEM. Comparisons were made by two-tailed t-test with GraphPad Prism software (GraphPad Software, Inc.), and p < 0.05 was considered statistically significant.
Acknowledgments
The authors would like to thank Hervé Husson and Kathy W. Klinger for stimulating discussions. L.M. was supported by grants from the Polycystic Kidney Foundation and from “la Foundation PKD France.”
Glossary
Abbreviations:
- PKD
polycystic kidney disease
- ADPKD
Autosomal Dominant PKD
- MDCK
Madin Darby canine kidney
- BUN
blood urea nitrogen
- IP
intraperitoneal
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- PCNA
proliferating cell nuclear antigen
- Rb
retinoblastoma protein
- cyc
cyclin
- CDK
cyclin-dependent kinase
- Bcl-XL
Bcl-2-like protein-1
- ApaF1
apoptotic peptidase activating factor-1
- mTOR
mammalian target of rapamycin
- Id2
DNA-binding protein inhibitor ID-2
- Erk1/2
extracellular signal-regulated kinases
- Cdc25A
cell division cycle 25 homolog A
Disclosure of Potential Conflicts of Interest
L.M. is co-inventor on the roscovitine patent. L.M., H.G. and N.O. are co-inventors of the CR8 patent. L.M. and H.G. are co-founders of ManRos Therapeutics, which has an exclusive license on the CR8 patent.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/22375
References
- 1.Grantham JJ, Mulamalla S, Swenson-Fields KI. Why kidneys fail in autosomal dominant polycystic kidney disease. Nat Rev Nephrol. 2011;7:556–66. doi: 10.1038/nrneph.2011.109. [DOI] [PubMed] [Google Scholar]
- 2.Takiar V, Caplan MJ. Polycystic kidney disease: pathogenesis and potential therapies. Biochim Biophys Acta. 2011;1812:1337–43. doi: 10.1016/j.bbadis.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torres VE, Bankir L, Grantham JJ. A case for water in the treatment of polycystic kidney disease. Clin J Am Soc Nephrol. 2009;4:1140–50. doi: 10.2215/CJN.00790209. [DOI] [PubMed] [Google Scholar]
- 4.Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. 2009;60:321–37. doi: 10.1146/annurev.med.60.101707.125712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millán JL, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995;10:151–60. doi: 10.1038/ng0695-151. [DOI] [PubMed] [Google Scholar]
- 6.Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–42. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- 7.Chang MY, Ong AC. Mechanism-based therapeutics for autosomal dominant polycystic kidney disease: recent progress and future prospects. Nephron Clin Pract. 2012;120:c25–34, discussion c35. doi: 10.1159/000334166. [DOI] [PubMed] [Google Scholar]
- 8.Leuenroth SJ, Crews CM. Targeting cyst initiation in ADPKD. J Am Soc Nephrol. 2009;20:1–3. doi: 10.1681/ASN.2008101118. [DOI] [PubMed] [Google Scholar]
- 9.Wilson PD, Goilav B. Cystic disease of the kidney. Annu Rev Pathol. 2007;2:341–68. doi: 10.1146/annurev.pathol.2.010506.091850. [DOI] [PubMed] [Google Scholar]
- 10.Goilav B. Apoptosis in polycystic kidney disease. Biochim Biophys Acta. 2011;1812:1272–80. doi: 10.1016/j.bbadis.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 11.Gallagher AR, Germino GG, Somlo S. Molecular advances in autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17:118–30. doi: 10.1053/j.ackd.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76:149–68. doi: 10.1038/ki.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reif GA, Yamaguchi T, Nivens E, Fujiki H, Pinto CS, Wallace DP. Tolvaptan inhibits ERK-dependent cell proliferation, Cl⁻ secretion, and in vitro cyst growth of human ADPKD cells stimulated by vasopressin. Am J Physiol Renal Physiol. 2011;301:F1005–13. doi: 10.1152/ajprenal.00243.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Torres VE, Wang X, Qian Q, Somlo S, Harris PC, Gattone VH., 2nd Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat Med. 2004;10:363–4. doi: 10.1038/nm1004. [DOI] [PubMed] [Google Scholar]
- 15.Higashihara E, Torres VE, Chapman AB, Grantham JJ, Bae K, Watnick TJ, et al. TEMPOFormula and 156-05-002 Study Investigators Tolvaptan in autosomal dominant polycystic kidney disease: three years’ experience. Clin J Am Soc Nephrol. 2011;6:2499–507. doi: 10.2215/CJN.03530411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hogan MC, Masyuk TV, Page LJ, Kubly VJ, Bergstralh EJ, Li X, et al. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol. 2010;21:1052–61. doi: 10.1681/ASN.2009121291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shillingford JM, Piontek KB, Germino GG, Weimbs T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J Am Soc Nephrol. 2010;21:489–97. doi: 10.1681/ASN.2009040421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walz G, Budde K, Mannaa M, Nürnberger J, Wanner C, Sommerer C, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:830–40. doi: 10.1056/NEJMoa1003491. [DOI] [PubMed] [Google Scholar]
- 19.Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:820–9. doi: 10.1056/NEJMoa0907419. [DOI] [PubMed] [Google Scholar]
- 20.Sweeney WE, Jr., von Vigier RO, Frost P, Avner ED. Src inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol. 2008;19:1331–41. doi: 10.1681/ASN.2007060665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, et al. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol. 2000;151:709–18. doi: 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002;13:2508–16. doi: 10.1097/01.ASN.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- 23.Hildebrandt F, Otto E. Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease? Nat Rev Genet. 2005;6:928–40. doi: 10.1038/nrg1727. [DOI] [PubMed] [Google Scholar]
- 24.Battini L, Macip S, Fedorova E, Dikman S, Somlo S, Montagna C, et al. Loss of polycystin-1 causes centrosome amplification and genomic instability. Hum Mol Genet. 2008;17:2819–33. doi: 10.1093/hmg/ddn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhunia AK, Piontek K, Boletta A, Liu L, Qian F, Xu PN, et al. PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell. 2002;109:157–68. doi: 10.1016/S0092-8674(02)00716-X. [DOI] [PubMed] [Google Scholar]
- 26.Li X, Luo Y, Starremans PG, McNamara CA, Pei Y, Zhou J. Polycystin-1 and polycystin-2 regulate the cell cycle through the helix-loop-helix inhibitor Id2. Nat Cell Biol. 2005;7:1202–12. doi: 10.1038/ncb1326. [DOI] [PubMed] [Google Scholar]
- 27.Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, et al. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem. 1997;243:527–36. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- 28.Meijer L, Bettayeb K, Galons H. Roscovitine (CYC202, Seliciclib). In: Yue E, Smith PJ, eds. Monographs on enzyme inhibitors: CDK inhibitors and their potential as anti-tumor agents: CRC Press, Taylor & Francis, 2006:187-226. [Google Scholar]
- 29.Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O. Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature. 2006;444:949–52. doi: 10.1038/nature05348. [DOI] [PubMed] [Google Scholar]
- 30.Moreno S, Ibraghimov-Beskrovnaya O, Bukanov NO. Serum and urinary biomarker signatures for rapid preclinical in vivo assessment of CDK inhibition as a therapeutic approach for PKD. Cell Cycle. 2008;7:1856–64. doi: 10.4161/cc.7.12.6055. [DOI] [PubMed] [Google Scholar]
- 31.Ibraghimov-Beskrovnaya O. Targeting dysregulated cell cycle and apoptosis for polycystic kidney disease therapy. Cell Cycle. 2007;6:776–9. doi: 10.4161/cc.6.7.4047. [DOI] [PubMed] [Google Scholar]
- 32.Natoli TA, Smith LA, Rogers KA, Wang B, Komarnitsky S, Budman Y, et al. Inhibition of glucosylceramide accumulation results in effective blockade of polycystic kidney disease in mouse models. Nat Med. 2010;16:788–92. doi: 10.1038/nm.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bettayeb K, Oumata N, Echalier A, Ferandin Y, Endicott JA, Galons H, et al. CR8, a potent and selective, roscovitine-derived inhibitor of cyclin-dependent kinases. Oncogene. 2008;27:5797–807. doi: 10.1038/onc.2008.191. [DOI] [PubMed] [Google Scholar]
- 34.Bettayeb K, Baunbæk D, Delehouze C, Loaëc N, Hole AJ, Baumli S, et al. CDK Inhibitors Roscovitine and CR8 Trigger Mcl-1 Down-Regulation and Apoptotic Cell Death in Neuroblastoma Cells. Genes Cancer. 2010;1:369–80. doi: 10.1177/1947601910369817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O Bukanov N, Husson H, Dackowski WR, Lawrence BD, Clow PA, Roberts BL, et al. Functional polycystin-1 expression is developmentally regulated during epithelial morphogenesis in vitro: downregulation and loss of membrane localization during cystogenesis. Hum Mol Genet. 2002;11:923–36. doi: 10.1093/hmg/11.8.923. [DOI] [PubMed] [Google Scholar]
- 36.Belibi FA, Edelstein CL. Novel targets for the treatment of autosomal dominant polycystic kidney disease. Expert Opin Investig Drugs. 2010;19:315–28. doi: 10.1517/13543781003588491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee K, Battini L, Gusella GL. Cilium, centrosome and cell cycle regulation in polycystic kidney disease. Biochim Biophys Acta. 2011;1812:1263–71. doi: 10.1016/j.bbadis.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou J. Polycystins and primary cilia: primers for cell cycle progression. Annu Rev Physiol. 2009;71:83–113. doi: 10.1146/annurev.physiol.70.113006.100621. [DOI] [PubMed] [Google Scholar]
- 39.Bach S, Knockaert M, Reinhardt J, Lozach O, Schmitt S, Baratte B, et al. Roscovitine targets, protein kinases and pyridoxal kinase. J Biol Chem. 2005;280:31208–19. doi: 10.1074/jbc.M500806200. [DOI] [PubMed] [Google Scholar]
- 40.Benson C, White J, De Bono J, O’Donnell A, Raynaud F, Cruickshank C, et al. A phase I trial of the selective oral cyclin-dependent kinase inhibitor seliciclib (CYC202; R-Roscovitine), administered twice daily for 7 days every 21 days. Br J Cancer. 2007;96:29–37. doi: 10.1038/sj.bjc.6603509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fedorov LM, Schmittwolf C, Amann K, Thomas WH, Müller AM, Schubert H, et al. Renal failure causes early death of bcl-2 deficient mice. Mech Ageing Dev. 2006;127:600–9. doi: 10.1016/j.mad.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 42.Moser M, Dahmen S, Kluge R, Gröne H, Dahmen J, Kunz D, et al. Terminal renal failure in mice lacking transcription factor AP-2 beta. Lab Invest. 2003;83:571–8. doi: 10.1097/01.lab.0000064703.92382.50. [DOI] [PubMed] [Google Scholar]
- 43.Lin HH, Yang TP, Jiang ST, Yang HY, Tang MJ. Bcl-2 overexpression prevents apoptosis-induced Madin-Darby canine kidney simple epithelial cyst formation. Kidney Int. 1999;55:168–78. doi: 10.1046/j.1523-1755.1999.00249.x. [DOI] [PubMed] [Google Scholar]
- 44.Tao Y, Kim J, Faubel S, Wu JC, Falk SA, Schrier RW, et al. Caspase inhibition reduces tubular apoptosis and proliferation and slows disease progression in polycystic kidney disease. Proc Natl Acad Sci USA. 2005;102:6954–9. doi: 10.1073/pnas.0408518102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA. 2006;103:5466–71. doi: 10.1073/pnas.0509694103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Masyuk TV, Radtke BN, Stroope AJ, Banales JM, Masyuk AI, Gradilone SA, et al. Inhibition of Cdc25A suppresses hepato-renal cystogenesis in rodent models of polycystic kidney and liver disease. Gastroenterology. 2012;142:622–33, e4. doi: 10.1053/j.gastro.2011.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Natoli TA, Gareski TC, Dackowski WR, Smith L, Bukanov NO, Russo RJ, et al. Pkd1 and Nek8 mutations affect cell-cell adhesion and cilia in cysts formed in kidney organ cultures. Am J Physiol Renal Physiol. 2008;294:F73–83. doi: 10.1152/ajprenal.00362.2007. [DOI] [PubMed] [Google Scholar]
- 48.Seibler J, Zevnik B, Küter-Luks B, Andreas S, Kern H, Hennek T, et al. Rapid generation of inducible mouse mutants. Nucleic Acids Res. 2003;31:e12. doi: 10.1093/nar/gng012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oumata N, Bettayeb K, Ferandin Y, Demange L, Lopez-Giral A, Goddard ML, et al. Roscovitine-derived, dual-specificity inhibitors of cyclin-dependent kinases and casein kinases 1. J Med Chem. 2008;51:5229–42. doi: 10.1021/jm800109e. [DOI] [PubMed] [Google Scholar]
- 50.Oumata N, Ferandin Y, Meijer L, Galons H. Practical synthesis of roscovitine and CR8. Org Process Res Dev. 2009;13:641–4. doi: 10.1021/op800284k. [DOI] [Google Scholar]