

Abstract
Due to the discovery of more and more roles of cellular noncoding RNAs, the approaches for introducing RNAs including small interfering RNA (siRNA), micro RNA (miRNA), ribozyme, and riboswitch into cells for regulating cell life cycle and for the treatment of diseases have become routine practice. The understanding of RNA folding, degradation, and intracellular half-life after entering the cell is an intriguing question in biology and pharmacology. Currently, methods to detect RNA folding, degradation, and half-life in real time within the cell is extremely challenging. The common assay method to measure RNA half-life and degradation in vivo is the use of radioactive markers or fluorescence RNA labeling. The challenge is, after RNA becomes degraded or misfolded, the isotope or the fluorescence is still present in the cell, thus the signals are not a true indication of the presence of the RNA in the cell. The alternate method commonly used to measure RNA life is to isolate RNA from cells and distinguish between intact and degraded RNA by gel, chromatography, or capillary electrophoresis. However, when a cell is breaking down, ribonucleases (RNases) will be released from cell compartments, and degradation of small RNA in cell lysates occurs immediately after cell lysis. Here we report a method to monitor RNA degradation in real time in living cells using fluorogenic RNA in combination with RNA nanotechnology (Guo, 2010; Guo et al., 2012). The RNA aptamer that binds malachite green (MG), the ribozyme that cleaves the hepatitis virus genome, and a siRNA for firefly luciferase were all fused to the bacteriophage phi29 packaging RNA (pRNA) 3-way junction (3WJ) motif to generate RNA nanoparticles. The MG aptamer, the hepatitis B virus ribozyme, and the luciferase siRNA all retained their function independently after fusion into the nanoparticles. When the RNA nanoparticle is degraded, denatured, or misfolded, the fluorescence disappears. MG, which is not fluorescent by itself, is capable of binding to its aptamer and emitting fluorescent light only if the RNA remains folded in the correct conformation. Therefore, the MG aptamer fluorescence (in the presence of MG dye) can be used as a measure of the degradation and folding of RNA nanoparticles, the siRNA, the aptamer, and the ribozyme in the cell in real time using epifluorescence microscopy and fluorescence spectroscopy without lysing the cells. We show that the half-life (t1/2) of the electroporated MG aptamer containing RNA nanoparticle was 4.3 hours after electroporation into cells.
Introduction
Nanotechnology research is based on the manipulation, engineering, modification, or assembly of materials on the nanometer scale. Like DNA, RNA molecules can be designed, manipulated, and constructed with the same level of simplicity, but RNA also possesses flexibility of both structure and function similar to proteins. For this reason, RNA has become a powerful tool in nanotechnological applications (Guo et al., 1998; Shu et al., 2003; Hansma et al., 2003; Shu et al., 2004; GUO, 2010; Guo et al., 2012). The term RNA nanotechnology was coined more than a decade ago (Guo et al., 1998; Zhang et al., 1998; Jaeger and Leontis, 2000; Jaeger et al., 2001; Shu et al., 2004; Chworos et al., 2004; GUO, 2005; Jaeger and Chworos, 2006; GUO, 2010). In 1998, reengineered, natural RNA nanoparticles were assembled into dimeric, trimeric, and hexameric RNA nanoparticles through self-assembly (Guo et al., 1998). The field of RNA nanotechnology has grown significantly in recent years due to the many uses of RNA nanoparticles such as the treatment of viral infection, genetic diseases, and cancer (GUO, 2010; Guo et al., 2012).
Many RNA-based therapeutic approaches down-regulate specific gene expression in cancerous or viral-infected cells using RNA molecules such as ribozymes (Kruger et al., 1982; Guerrier-Takada et al., 1983; Sarver et al., 1990; Nava Sarver et al., 1990; Chowrira et al., 1991) and small interfering RNA (siRNA) (Fire et al., 1998; Li et al., 2002; Brummelkamp et al., 2002; CARMICHAEL, 2002). Through the combination of ribozymes and/or siRNA with RNA aptamers capable of recognizing and binding to specific ligands such as organic compounds, nucleotides, or peptides (Ellington and Szostak, 1990; Tuerk and Gold, 1990), targeted delivery of RNA nanoparticles has led to a heightened interest in the development of siRNA-based therapeutics. One of the most difficult challenges for RNA therapeutics is the effective delivery of RNA to specific cells in vivo. There are, however, several methods for gene silencing with both high specificity and efficacy in vitro. The pRNA of the bacteriophage phi29 has been used to deliver siRNA to specific target cells (Guo et al., 2005; Khaled et al., 2005; Guo et al., 2006). Monomer pRNA possesses left and right interlocking loops that can interact to form dimers and trimers, which allows for the delivery of RNA therapeutic to specific cells (Guo et al., 1987; Guo et al., 1998; Chen et al., 1999).
One of the major challenges associated with RNA therapeutics is the thermodynamic and chemical stability of RNA nanoparticles. In a recent report, the 3WJ region of pRNA was used to generate highly stable RNA nanoparticles that were resistant to 8M urea denaturation and remained intact at extremely low concentrations (Shu, D., et al., 2011; Haque et al., 2012). In addition, each arm of the 3WJ was capable of carrying therapeutic RNA molecules such as siRNA, ribozymes, or receptor-binding aptamers. One example was the use of a MG binding aptamer as an assay for the proper folding and function of RNA nanoparticles (Afonin et al., 2010; Shu, D., et al., 2011; Haque et al., 2012). In solution, MG dye has a nearly negligible fluorescence; however, when bound to its specific RNA aptamer, the fluorescence increases by nearly 2,000-fold. This has made the MG aptamer a powerful tool for label-free sensors (Xu and Lu, 2010; Stead et al., 2010).
Another challenge associated with RNA therapeutics is the stability of the RNA in vivo both within the body and within cells. Chemical modifications to RNA have been shown to be useful in extending the half-life of RNA in the body (Abdelmawla et al., 2011; Shu, Y., et al., 2011) however there are very few methods to monitor RNA folding and degradation inside of living cells. The most common methods to assay RNA entry into cells are based on the use of fluorescently labeled or radioactively labeled RNA. In both cases, the function and folding of the RNA within the cells is unknown, because even if the RNA is misfolded or degraded, the radioactive or fluorescent label is still present within the cell. In these cases, the measured signal is not dependent on the function of the RNA. Another method used to monitor the presence of the functional RNA in cells is to use separation techniques such as gel chromatography or capillary electrophoresis on cell lysates. Although this allows for the determination of the functional and properly folded RNA, the analysis takes a significant amount of time making real-time detection of the RNA impossible. In addition, when cells are lysed, the contents of the cells and cell compartments are released, which results in the interaction between ribonucleases (RNAses) and the target RNA. This makes accurately determining the amount of RNA in the cells very challenging.
In this report, we present a method to monitor RNA folding and degradation in real time in living cells. A MG binding aptamer (MGapt) is conjugated onto one arm of the 3WJ core of a pRNA molecule. The resulting pRNA-3WJ-(MGapt) strand benefits from the thermodynamic stability of the pRNA 3WJ core motif while possessing the ability to bind MG and show enhance its fluorescence. In addition, the fluorescence of the pRNA-3WJ-(MGapt) in various solutions such as cell lysates or serum is also monitored over time to demonstrate the use of the MG aptamer to monitor RNA degradation. Finally, living cells are electroporated with the pRNA-3WJ-(MGapt), and the fluorescence of the cell suspensions was monitored in real time in order to observe the degradation of the pRNA-3WJ-(MGapt) in living cells. Using the plots of fluorescence versus time, the half-life of the RNA in the cells was calculated to be 4.3 hours. This simple method to monitor RNA degradation has the potential to be used with nearly any RNA construct and any cell line. In the future, this method could be a valuable tool in tracking RNA folding and degradation in living cells in real time.
Materials and Methods
In vitro synthesis of RNA nanoparticles
All RNA nanoparticles were made by first using polymerase chain reaction (PCR) to generate double-stranded DNA templates followed by in vitro transcription by T7 RNA polymerase as described previously (Shu et al., 2003). After transcription, all RNA particles were purified using 8% urea polyacrylamide gel electrophoresis (PAGE). For the construction of multistrand RNA nanoparticles, the purified individual strands were mixed together at an equimolar ratio, annealed, and purified in an 8% native PAGE gel. For the construction of 2′fluoro-modified RNA (2′F-RNA) nanoparticles, 2′-fluoro-pyrimidines and 2′-fluoro-purines (Trilink Biotechnologies) were included in the in vitro transcription reaction solution in place of their normal counterparts. Any combination of unmodified ribonucleotide triphosphates (rNTPs) and 2′F-rNTPs could be used to make different 2′F-RNA nanoparticles. For fluorescence labeling of the RNA strands, the fluorescent dye Cy3 was labeled on the pRNA-3WJ-(MGapt) using the Label IT siRNA Tracker Intracellular Localization Kit, Cy3™ (Mirus Bio LLC).
The chemical stability of the RNA nanoparticles was monitored by PAGE with RNA mixed in the presence of fetal bovine serum (FBS). Prior to gel loading, the RNA samples were mixed at a concentration of 1 μM in the presence of Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% FBS and incubated in a 37°C water bath for the specified amount of time (Fig. 2). The gels were stained with either ethidium bromide or 4 μM MG dye to simultaneously monitor the presence of the RNA and the binding activity of the MG aptamer.
FIG. 2.

(A) 8% Native-polyacrylamide gel electrophoresis (PAGE) gel of unmodified pRNA-3WJ-(MGapt) and pRNA-3WJ-(MGapt) made with 2′F ATP and CTP in the presence of RPMI 1640 medium containing 10% fetal bovine serum (FBS) stained with both ethidium bromide and MG dye to show that both forms of the aptamer are capable of binding MG but are rapidly degraded in the presence of FBS. (B-C) 8% Native-PAGE gels showing pRNA (B) or pRNA-3WJ-(MGapt) (C) made with either 2′F UTP and CTP or 2′F ATP and CTP in the presence of RPMI 1640 medium containing 10% FBS for up to 4 hours. In both cases, the unmodified RNA is degraded within the first hour of exposure to FBS, whereas the 2′F-modified strands remain stable. For all gels, lane 6 contains a DNA ladder. RPMI, Roswell Park Memorial Institute.
Cell culture
Human nasopharyngeal carcinoma KB cells (American Type Culture Collection) were continuously grown and subcultured in RPMI 1640 medium (Invitrogen) containing 10% FBS. All cell cultures were incubated in a 37°C incubator with a 5% CO2, humidified atmosphere.
Dual luciferase assay
The dual luciferase assay was performed using the Dual-Luciferase Reporter Assay System (Promega). KB cells were seeded on a 24-well plate and cotransfected with 1.25 nM pRNA-3WJ nanoparticles and both plasmid pGL3 and pRL-TK coding for firefly and renilla luciferase respectively. A total of 24 hours after transfection, the cells were washed with phosphate buffer saline (PBS) and lysed with passive lysis buffer. The plates were shaken for 20 minutes at room temperature and 20 μL were added to 3 wells of a 96-well plate. Each well was mixed with 100 μL of firefly luciferase assay reagent, measured for firefly luciferase activity, and mixed with 100 μL of Stop & Glo reagent, followed by the measurement of the renilla luciferase activity. The firefly luciferase activity data was normalized relative to the renilla luciferase internal control.
Hepatitis B virus ribozyme activity assay
Hepatitis B virus (HBV) substrate was labeled with Cy3 as described above and incubated with the nanoparticle harboring the HBV ribozyme at 37°C for 1 hour in a buffer containing 20 mM MgCl2, 20 mM NaCl, and 50 mM Tris-HCl (pH 7.5). pRNA-HBV ribozyme was used as a positive control, and a 3WJ with a mutated HBV ribozyme was used as a negative control. After incubation the products were run in an 8M Urea, 10% PAGE gel for fluorescence imaging.
Transfection of RNA nanoparticles into living cells by electroporation
Living KB cells were removed from culture flacks via trypsinization and resuspended in PBS (pH 7.4) containing 10 mM MgCl2 at a concentration of 2.0–5.0×106 cells/mL. For each cell sample, 200 μL of cells in PBS was mixed with RNA to a final concentration of 5 μM RNA in a 0.4-cm-wide gene-pulser electroporation cuvette (Bio-Rad). Electroporation was achieved by a single pulse of 1 kV at a capacitance of 25 μF using a Gene-pulser system (Bio-Rad). After electroporation, the cells were incubated with 1 mg/mL RNase for 10 minutes followed by 3 rinses with PBS buffer. The cell suspensions were then diluted to a final concentration of 1×105 cells/mL in medium with 10% FBS and incubated at 37°C before analysis for MG binding. At each time point, a 50-μL aliquot of cells was removed, centrifuged, and resuspended in 50 μL of MG-binding buffer containing 10 μM MG dye for analysis by either epifluorescence microscopy or fluorescence spectrometry.
Fluorescence microscopy imaging
The cell samples were electroporated and prepared as described above. After incubation with the MG dye, a 20-μL aliquot of the cells in MG dye was placed on a glass coverslip for microscopic imaging. All images were acquired at 20×magnification on an inverted Olympus IX-71 microscope. For fluorescence imaging (Fig. 3), the Cy3 fluorescence channel was used to image the Cy3-pRNA-3WJ-(MGapt), and the Cy5 channel was used to image MG binding and fluorescence inside of the living cells.
FIG. 3.

White light and fluorescence images of KB cells with and without electroporation of either 0.5 μM Cy3-labeled pRNA-3WJ-(MGapt) or 5 μM pRNA-3WJ-(MGapt) in the presence of 10 μM MG. The MG aptamer fluorescence can be clearly visualized in the pRNA-3WJ-(MGapt) electroporated sample. As a positive control for electroporation Cy3- pRNA-3WJ-(MGapt) can be seen inside of the cells with electroporation but not in the non-electroporated sample.
MG binding aptamer fluorescence assay
The function of the MG binding aptamer within the various forms of the pRNA-3WJ-(MGapt) was measured by mixing the RNA at a concentration of 100 nM with 4 μM MG dye in a binding buffer (100 mM KCl, 5 mM MgCl2, and 10 mM HEPES at pH 7.4). The solutions were allowed to bind at room temperature for 10 minutes. The fluorescence emission spectrum (630–750 nm) of the mixtures was measured using a fluorospectrometer (Horiba Jobin Yvon) with an excitation of 615 nm. For the assays of the pRNA-3WJ-(MGapt) degradation in various solutions (Fig. 4), the same procedures were used with a few modifications. A total of 500 μL of each solution containing 100 nM pRNA-3WJ-(MGapt) in MG-binding buffer in the selected solution were incubated at 37°C and at each time-point, a 50-μL aliquot of the solution was removed and MG was added to a concentration of 4 μM. The MG dye was allowed to bind for 10 minutes before measurement of the fluorescence spectra.
FIG. 4.

Emission spectra, acquired at various times, of 100 nM pRNA-3WJ-(MGapt) with 2 μM MG dye in the presence of MG binding buffer (A), cell lysates (B), RPMI 1640 medium (C), and RPMI 1640 with 10% FBS (D). A summary of the fluorescence intensities at 650 nm versus time for all samples is shown in (E).
Calculation of RNA half-life
To determine the half-life of the RNA in the cell, a linear fit was performed on the fluorescence versus time data for the cell samples electroporated with pRNA-3WJ-(MGapt). The intensity of fluorescence (If) after half of the RNA has been degraded (If(1/2)) must first be calculated using the following equation:
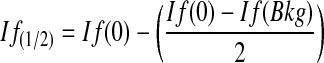 |
where If(1/2) is the intensity of fluorescence after half of the RNA has been degraded, If(0) is the intensity of fluorescence at 0 hours (y-intercept of fit equation) and If(Bkg) is the initial intensity of fluorescence of the negative control sample. The If(1/2) can then be substituted into the linear fit equation for the specific sample.
Results and Discussion
Phi29 pRNA-3WJ motif as scaffold to construct fluorogenic RNA nanoparticles harboring an MG binding aptamer
A large portion of RNA therapeutics research focuses on discovering various approaches to introduce small noncoding RNA such as siRNA, micro RNA (miRNA), ribozyme, and/or riboswitches into living cells. The long-term goal is to be able to deliver these RNAs to specific cells in vivo to perform a specific function inside the target cells without adverse effects to other cells in the body. An important aspect of this goal is the ability to monitor the folding and degradation of the RNA in the target cells. Recently, a major focus within RNA therapeutics has been on increasing the stability of RNA (Liu et al., 2010; Shu, D., et al., 2011). We have recently demonstrated the construction of thermodynamically and chemically stable 3WJ-pRNA nanoparticles harboring multiple functional modules (Shu, D., et al., 2011; Haque et al., 2012). Each arm of the 3WJ harboring a functional module is able to perform its specific function without affecting the folding and function of other modules or the core scaffold. In this report, we focus on developing a method to monitor the function and degradation of an RNA molecule in living cells using an RNA aptamer specific for the binding of MG that is known to increase the fluorescence of the MG dye (Baugh et al., 2000).
In order to be used to track the function and folding of an RNA nanoparticle inside of cells, the function of the MG aptamer first had to be verified on a construct containing functional therapeutic moieties held together in a single RNA nanoparticle by the pRNA 3WJ scaffold. An example RNA nanoparticle, shown in Fig. 1A, contains the MG aptamer, a HBV-cleaving ribozyme (Hoeprich et al., 2003), and a siRNA strand targeting the luciferase gene. The function of each RNA moiety was assayed individually to confirm that each was able to function while bound to the pRNA-3WJ scaffold. The binding and fluorescence of MG dye to the MG aptamer in the nanoparticle is shown in Fig. 1B. The HBV ribozyme on the nanoparticle was able to cleave its substrate, as shown in Fig. 1C. The silencing function of the siRNA for firefly luciferase is demonstrated in Fig. 1D. The low ratio of firefly to renilla luciferase expression relative to a scrambled siRNA control indicates proper siRNA function by the nanoparticle.
FIG. 1.

(A) Design and sequence of an RNA nanoparticle harboring a malachite green (MG) aptamer, hepatitis B virus (HBV) ribozyme, and small interfering RNA(siRNA) targeting firefly luciferase. (B) Functional assay of the MG aptamer incorporated in the 3-way junction (3WJ) nanoparticle. MG fluorescence was measured using 615 nm excitation. (C) Catalytic activity of the HBV ribozyme. Cleaved products are boxed. (D) Dual-luciferase assay for target gene knockdown of the firefly luciferase gene. Relative luciferase activity is obtained by normalizing the firefly luciferase expression to the renilla luciferase internal control. Error bars represent the standard deviation of 3 trials. The RNA sequence and structure of the wild type packaging RNA (pRNA) (E) and pRNA-3WJ-(MGapt) (F). (G-I) The binding and fluorescence spectra of pRNA-3WJ-(MGapt) made with various 2′F-modified rNTPs (ATP, CTP, GTP, UTP) including a comparison of UTP and CTP binding (G); 2′F-ATP, 2′F-GTP, and 2′F-CTP (H); as well as the structures with multiple modified nucleotides (I). This shows that the modification of UTP or GTPs deactivates the MG aptamer and no binding of MG occurs.
The MG aptamer provides a method to monitor the degradation and misfolding of RNA nanoparticles by monitoring the loss of function of the MG aptamer. To this end, we designed an RNA nanoparticle, pRNA-3WJ-(MGapt), which benefits from the strong folding of the pRNA 3WJ motif and contains an aptamer capable of binding MG and generating fluorescence.
The primary RNA used in this work to monitor the degradation of the RNA in real time is made up of two major components. The structure of pRNA is shown in Fig. 1E with the 3WJ core region shown in the box. In the experiments presented in this report, pRNA is used as a negative MG binding control because it contains the pRNA 3WJ core but does not have the MG binding aptamer and is therefore unable to bind MG and show fluorescence.
Due to its high thermodynamic stability, we have used this sequence as a scaffold to build fluorogenic RNA nanoparticles containing an aptamer that is known to specifically bind malachite green (MG). MG is nonfluorescent until it binds to this specific aptamer which allows the MG to fluorescence. The sequence of the pRNA-3WJ-(MGapt) is shown in Fig. 1F. The construct uses the right arm of a pRNA 3WJ to hold the MG aptamer in the proper confirmation for MG binding. Although not required for binding, the pRNA-3WJ core provides additional thermodynamic stability for the MG aptamer. We have made the assumption that if MG aptamer is degraded, then the pRNA-3WJ scaffold is also degraded at the same time. Since any modifications would affect both the MG aptamer and the pRNA-3WJ scaffold, we assume that they would be degraded at the same time.
One of the other critical considerations before the pRNA-3WJ-(MGapt) could be used to monitor the stability of RNA in vitro is its chemical stability. Recently, the use of various chemical modifications, such as the 2′F, has become routine for increasing the resistance of RNA to RNases (Liu et al., 2010). In this case the 2′-OH of the ribose backbone is replaced with a fluorine on selected bases. Generally, 2′F pyrimidines are used, therefore the binding of MG by pRNA-3WJ-(MGapt) was first evaluated using 2′-fluoro-pryrimidines. The goal was to determine if MG binding was retained by the 2′F-pRNA-3WJ-(MGapt). Figure 1G shows the MG fluorescence spectra of different forms of the pRNA-3WJ-(MGapt) made with single nucleotide modifications and both 2′F-uridine triphosphate (UTP) and 2′F-cytidine triphosphate (CTP) in the presence of 2 μM MG in MG binding buffer. The only construct that showed binding was the 2′F-CTP-pRNA-3WJ-(MGapt) which is evidenced by the presence of the peak in fluorescence at 650 nm. If 2′F-UTP was used, the binding and fluorescence of MG was eliminated (Fig. 1G).
To further evaluate the effect of 2′F nucleotides on the binding of the MG aptamer, 2′F purines were also used to make the pRNA-3WJ-(MGapt). Figure 1H shows the fluorescence of pRNA-3WJ-(MGapt) made with single 2′F nucleotide substitutions. The fluorescence of the pRNA-3WJ-(MGapt) made with 2′F-adenosine triphosphate (ATP) is consistent with that of unmodified pRNA-3WJ-(MGapt) and its 2′F-CTP counterpart. The binding and fluorescence of 2′F-guanosine triphosphate (GTP)-pRNA-3WJ-(MGapt), however, is greatly reduced. When double nucleotide replacements are used, only the 2′F-CTP-ATP- pRNA-3WJ-(MGapt) shows binding similar to the unmodified pRNA-3WJ-(MGapt). If either 2′F-UTP or 2′F-GTP is used, the fluorescence of the aptamer is eliminated as shown in Fig. 1I.
With the knowledge of which modifications allow for binding of MG, the next issue to investigate was the RNase stability. To assay the chemical stability of the 2′F-ATP-CTP- pRNA-3WJ-(MGapt), a sample of the RNA was mixed with RPMI 1640 medium with 10% FBS for a specific amount of time and then run in an 8% native-PAGE gel. In this case, the stability of the RNA was monitored over 4 hours. Figure 2A shows an 8% Native-PAGE gel of both unmodified and 2′F-ATP-CTP- pRNA-3WJ-(MGapt). Lanes 1 and 7 show a band resulting from the native form of the pRNA-3WJ-(MGapt); however, upon mixing with RPMI 1640 containing 10% FBS, the bands rapidly disappear. The binding of MG by both forms of the pRNA-3WJ-(MGapt) in the gel is confirmed by staining the gel with MG dye (Fig. 2A). The red fluorescence is only visible in the samples with no FBS. This indicates that neither the unmodified nor 2′F-ATP-CTP-pRNA-3WJ-(MGapt) are stable in the presence of FBS.
To confirm that 2′F ATP and 2′F CTP together are not able to increase RNA stability, both pRNA and pRNA-3WJ-(MGapt) were made using 2′F-UTP and 2′F-CTP or 2′F-ATP and 2′F-CTP and were run in an 8% native-PAGE gel under the same conditions as in Fig. 2A. Figure 2B shows a native gel with two different 2′F nucleotide combinations of pRNA and Fig. 2C shows two different forms of the 2′F-pRNA-3WJ-(MGapt). In both gels, lanes 1–5 show that the 2′F-RNA is not degraded over a 4-hour period in the presence of FBS but lanes 7–11 show that both the 2′F-ATP-CTP modified form of both pRNA and pRNA-3WJ-(MGapt) are degraded within minutes of exposure to the FBS. This indicates that the use of 2′F-ATP and 2′F-CTP does not result in an increase in RNase resistance. It should be noted that the low mobility band present in all of the gels is produced by the FBS in the medium/RNA mixture and does not interfere with the RNA in any way. The bands are consistent in both their position in the gels and their brightness, indicating that the RNA samples do not interact with that band. In addition we have tested FBS in a gel with the same conditions and the same band was observed (data not shown). Overall, the data suggests that the 2′F-modification on U in RNA molecules plays a particularly important role in all 2′F-RNA's resistance to RNase.
Imaging of fluorogenic RNA nanoparticles in living cells
The pRNA-3WJ-(MGapt) was then applied to living cells. The first objective was to determine if pRNA-3WJ-(MGapt) could be transfected into living cells and imaged by fluorescence microscopy using MG with the cells. For transfection of RNA into cells, electroporation was used. Unlike transfection using cationic lipids such as Lipofectamine 2000, electroporation does not rely on trapping the RNA in liposomes. Liposomes protect RNA from interaction with the outside solution which, in this case, means that the RNA would be protected from degradation by RNases inside of the cells. Since the goal is to monitor RNA as it is being degraded in real time, liposomes would interfere with accurately monitoring the RNA, because the interaction between the RNA and RNase in the cell would be a function of liposomal breakdown. Another reason is that electroporation is virtually an instantaneous process. The electric field across the cell solution causes openings in the cell membrane that allows reagents outside of the cells to diffuse into the cells. After the pulse, the cell membrane rapidly closes leaving the RNA inside of the cell. Therefore, the time at which the RNA is inserted into the cells is known precisely and the cells can be monitored at specific times afterward.
There is a critical difference between the method demonstrated here and the new method of monitoring RNA folding and localization in living cells reported recently (Paige et al., 2011). In the case of the new RNA mimics of green fluorescent protein (GFP), the plasmid containing the RNA aptamer sequence must be first transfected into the cells so that the cells can express the RNA. Because the cells are constantly expressing the RNA, it would be impossible to monitor the degradation of the RNA in real time.
We demonstrated that electroporation was able to transfect pRNA-3WJ-(MGapt) into living cells using KB cells. KB cells were grown normally, trypsinized, and resuspended in PBS buffer. Each sample contained 2×105 to 3×105 cells/mL. For verification of electroporation, a Cy3 whole chain labeled version of the pRNA-3WJ-(MGapt) was constructed. The Cy3- pRNA-3WJ-(MGapt) showed significantly less binding of MG than the pRNA-3WJ-(MGapt), therefore Cy3- pRNA-3WJ-(MGapt) was not used to verify MG binding and fluorescence but was used only as an indicator that the electroporation method worked. Figure 3 shows white light, Cy3 fluorescence, and MG fluorescence images of KB cells incubated with 500 nM Cy3 pRNA-3WJ-(MGapt) or 5 μM pRNA-3WJ-(MGapt) with and without electroporation and imaged in the presence of 10 μM MG dye. The 500 nM Cy3 pRNA-3WJ-(MGapt) was used to avoid overexposing the camera because Cy3 dye is brighter than MG bound to the MG aptamer. Since the Cy3-pRNA-3WJ-(MGapt) shows reduced MG binding, there is no way to make a quantitative comparison of brightness in the MG channel, so the optimal imaging concentrations for RNA constructs were used. The non-electroporated samples show only dim background fluorescence levels in both the Cy3 and MG channels. The electroporated samples show fluorescence corresponding to the dye used for imaging. The Cy3 pRNA-3WJ-(MGapt) shows bright fluorescence inside of the cells in the Cy3 channel. The pRNA-3WJ-(MGapt) electroporated sample shows MG fluorescence inside of the cells in the MG channel with minimal background fluorescence in the Cy3 channel. Overall, this shows that electroporation is able to transfect pRNA-3WJ-(MGapt) RNA into living KB cells and that fluorescence microscopy can be used to image MG binding and fluorescence in living cells. It should be noted that the Cy3-pRNA-3WJ-(MGapt) was used in these experiments only to verify that the electroporation of the RNA is successful.
Real time determination of the half-life (t1/2) of fluorogenic RNA within living cells
Before testing the degradation of pRNA-3WJ-(MGapt) in living cells, the MG binding and fluorescence of the strand was first tested in a series of solutions to determine if the use of fluorescence spectroscopy would be suitable to monitor the degradation of pRNA-3WJ-(MGapt) in real time. Four different solutions were tested, MG binding buffer, RPMI 1640 medium, RPMI 1640 medium with 10% FBS, and KB cell lysates. Each sample contained 100 nM pRNA-3WJ-(MGapt) in the specific solution. At each time point, 50 μL of the sample was removed and MG binding buffer and MG dye was added to a final concentration of 2 μM. The fluorescence emission spectrum was then acquired and compared over time. Figure 4 shows the fluorescence emission spectra for pRNA-3WJ-(MGapt) in different solutions over time. In each case there is a decrease in the fluorescence over time, however, the most pronounced decrease is observed in the KB cell lysate solution and RPMI 1640 medium containing 10% FBS. Figure 4E shows the 650 nm fluorescence intensity versus time for each solution. The pRNA-3WJ-(MGapt) in the RPMI 1640 medium with 10% FBS is clearly degraded within the first 2 hours which is consistent with the gel analysis in Fig. 2. In KB cell lysates the degradation of the pRNA-3WJ-(MGapt) is more gradual but after 30 hours, the fluorescence from the MG aptamer is gone indicating that the pRNA-3WJ-(MGapt) is fully degraded. The other solutions show a small decrease over time but still have significant levels of fluorescence after 30 hours. Overall, this demonstrates that the fluorescence from the pRNA-3WJ-(MGapt) with MG can be monitored over time and is an indicator of RNA degradation.
The same principles were then applied to a similar analysis in suspensions of living KB cells electroporated with different RNA strands. KB cells were electroporated with different RNA strands including pRNA-3WJ-(MGapt) or pRNA (as a negative control). As additional controls, non-electroporated samples were also monitored. Immediately after electroporation, the cell samples were washed twice with PBS and incubated with RNase in PBS for 10 minutes before being resuspended in RPMI 1640 medium supplemented with 10% FBS. The fluorescence spectrum of each cell suspension was acquired initially (40 minutes after electroporation) and at several time-points afterward. Before each spectrum was acquired, a portion of the cells were resuspended in MG binding buffer with 10 μM MG dye and incubated in the dark for 10 minutes to allow the MG to bind to the aptamer.
Representative fluorescence spectra of KB cell suspensions 40 minutes after electroporation with various RNA strands are shown in Fig. 5A. From the graph it is clear that the cell samples electroporated with pRNA-3WJ-(MGapt) both with and without RNase treatment (–RNase) show the highest fluorescence at 650 nm as a result of containing pRNA-3WJ-(MGapt) which is able to bind MG. The other cell samples show consistent background fluorescence from MG which is a result of nonspecific binding of the MG to DNA and RNA in the cells. The fluorescence background from nonspecific MG binding is consistent with previously published work (Paige et al., 2011). Although there is significant fluorescence relative to cells without MG, the fluorescence is consistent among the cell suspensions that were not electroporated with pRNA-3WJ-(MGapt). Figure 5B shows this more clearly as the fluorescence of the pRNA-3WJ-(MGapt) electroporated sample is clearly higher than the non-electroporated sample.
FIG. 5.

(A) Representative fluorescence spectra of KB cell suspensions with and without electroporated of either pRNA or pRNA-3WJ-(MGapt) and treated with RNase for 10 minutes in the presence of 10 μM MG dye. The cell samples electroporated with pRNA-3WJ-(MGapt) show the highest initial fluorescence intensity as shown in (B). (C) Mean fluorescence intensity at 650 nm versus time of 2 repeat trials. In both cases, the pRNA-3WJ-(MGapt) electroporated sample with and without RNase show the highest initial fluorescence intensity and shows a decrease over time. By 8 hours, there is no significant difference in the intensities of the pRNA-3WJ-(MGapt) electroporated cells and the various negative controls. (D) Illustration of the calculation of the half-life of RNA within the cells.
The mean fluorescence intensity of two trials versus time for each of the cell samples is shown in Fig. 5C. For both of the samples electroporated with pRNA-3WJ-(MGapt) with and without RNase incubation, by 8 hours the mean fluorescence is not significantly different from the controls without electroporation. This indicates that the fluorescence from the pRNA-3WJ-(MGapt) bound with MG decreases over time due to the cell breaking the RNA down. An example of this analysis is shown in Fig. 5D. For the sample electroporated with pRNA-3WJ-(MGapt), the linear fit is
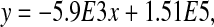 |
where y is the intensity of fluorescence (If) and x is time.
In this case, the half-life of the pRNA-3WJ-(MGapt) in the electroporated cell sample is 4.3 hours. The same analysis was performed on the sample electroporated with pRNA-3WJ-(MGapt) without RNase treatment to calculate a half-life of 4.0 hours. This small difference in half-life is a result of the greater initial fluorescence intensity of the sample that was not incubated with RNase (Fig. 5C). Without RNase treatment, there is a small amount of residual pRNA-3WJ-(MGapt) outside of the cells which is indicated by the higher fluorescence at 40 minutes. The residual pRNA-3WJ-(MGapt) is degraded by the serum outside of the cell during the incubation times between measurements while the pRNA-3WJ-(MGapt) inside of the cells is degraded by RNases inside of the cells. These 2 processes occur simultaneously, which results in a larger measured rate of decrease in the fluorescence and therefore a slightly shorter half-life.
It should be noted, however, that there is a significant background fluorescence generated by the nonspecific binding of MG to the DNA and RNA inside of the living cells. This background was also observed in the emission spectra of all cell suspensions analyzed by fluorescence spectrometry in Fig. 5. Because the background is stable for a given concentration of cells and in samples that were not electroporated with pRNA-3WJ-(MGapt), the difference between the fluorescences of the negative control samples and the pRNA-3WJ-(MGapt) electroporated cell samples could still be used to monitor the decrease in the fluorescence intensity over time, which is a direct result of the degradation of the pRNA-3WJ-(MGapt) RNA inside of the cells. It should be noted, however, that the background MG fluorescence in all cell samples is shown to decrease slowly over time. This is likely a result of changes in cell viability during the incubation process between sample measurements. The cells are maintained in suspension by mixing, which is not their native growth condition. Therefore, it was expected that a small percentage of the cells would die over time and therefore the background fluorescence from nonspecific binding would decrease slightly over time. This decrease in the background does not interfere with the half-life calculations of pRNA-3WJ-(MGapt) degradation in the cells.
Conclusions
In summary, we have developed a new method to monitor RNA degradation in living cells in real time using an MG binding aptamer. RNA nanoparticles were constructed using the pRNA-3WJ motif as a scaffold to harbor the MG binding aptamer. We have shown that the pRNA-3WJ-(MGapt) is unable to bind MG and fluoresce if made with 2′F UTPs or 2′F GTPs, which indicates that the 2′–OH group on the backbone of these bases within the active site play an important role in the fluorescence increase of the MG. We used electroporation to transfect pRNA-3WJ-(MGapt) into living cells and show that we are able to image the MG fluorescence by fluorescence microscopy and monitor its degradation in the cells over time using fluorescence spectroscopy without lysing or harming the cells. Finally, we demonstrated that the method is able to measure the half-life of the pRNA-3WJ-(MGapt) inside of the cells.
Acknowledgments
The research was supported by National Institutes of Health (NIH) grants EB003730 and CA151648 to P.G.
Author Disclosure Statement
PG is a cofounder of Kylin Therapeutics, Inc., and Biomotor and Nucleic Acid Nanotechnology Development Corp., Ltd.
References
- ABDELMAWLA S. GUO S. ZHANG L. PULUKURI S. PATANKAR P. CONLEY P. TREBLEY J. GUO P. LI Q.X. Pharmacological characterization of chemically synthesized monomeric pRNA nanoparticles for systemic delivery. Mol. Ther. 2011;19:1312–1322. doi: 10.1038/mt.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AFONIN K.A. BINDEWALD E. YAGHOUBIAN A.J. VOSS N. JACOVETTY E. SHAPIRO B.A. JAEGER L. In vitro assembly of cubic RNA-based scaffolds designed in silico. Nat. Nanotechnol. 2010;5:676–682. doi: 10.1038/nnano.2010.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAUGH C. GRATE D. WILSON C. 2.8 A crystal structure of the malachite green aptamer. J. Mol. Biol. 2000;301:117–128. doi: 10.1006/jmbi.2000.3951. [DOI] [PubMed] [Google Scholar]
- BRUMMELKAMP T.R. BERNARDS R. AGAMI R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- CARMICHAEL G.G. Medicine: silencing viruses with RNA. Nature. 2002;418:379–380. doi: 10.1038/418379a. [DOI] [PubMed] [Google Scholar]
- CHEN C. ZHANG C. GUO P. Sequence requirement for hand-in-hand interaction in formation of pRNA dimers and hexamers to gear phi29 DNA translocation motor. RNA. 1999;5:805–818. doi: 10.1017/s1355838299990350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN S.J. RNA folding: conformational statistics, folding kinetics, and ion electrostatics. Annu. Rev. Biophys. 2008;37:197–214. doi: 10.1146/annurev.biophys.37.032807.125957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOWRIRA B.M. BERZAL-HERRANZ A. BURKE J.M. Novel guanosine requirement for catalysis by the hairpin ribozyme. Nature. 1991;354:320–322. doi: 10.1038/354320a0. [DOI] [PubMed] [Google Scholar]
- CHWOROS A. SEVERCAN I. KOYFMAN A.Y. WEINKAM P. OROUDJEV E. HANSMA H.G. JAEGER L. Building programmable jigsaw puzzles with RNA. Science. 2004;306:2068–2072. doi: 10.1126/science.1104686. [DOI] [PubMed] [Google Scholar]
- ELLINGTON A.D. SZOSTAK J.W. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- FIRE A. XU S. MONTGOMERY M.K. KOSTAS S.A. DRIVER S.E. MELLO C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- GUERRIER-TAKADA C. GARDINER K. MARSH T. PACE N. ALTMAN S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell. 1983;35:849–857. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- GUO P. RNA nanotechnology: engineering, assembly and applications in detection, gene delivery and therapy. J. Nanosci. Nanotechnol. 2005;5(12):1964–1982. doi: 10.1166/jnn.2005.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUO P. The emerging field of RNA nanotechnology. Nat. Nanotechnol. 2010;5:833–842. doi: 10.1038/nnano.2010.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUO P. ERICKSON S. ANDERSON D. A small viral RNA is required for in vitro packaging of bacteriophage phi29 DNA. Science. 1987;236:690–694. doi: 10.1126/science.3107124. [DOI] [PubMed] [Google Scholar]
- GUO P. ZHANG C. CHEN C. TROTTIER M. GARVER K. Inter-RNA interaction of phage phi29 pRNA to form a hexameric complex for viral DNA transportation. Mol. Cell. 1998;2:149–155. doi: 10.1016/s1097-2765(00)80124-0. [DOI] [PubMed] [Google Scholar]
- GUO P. HAQUE F. HALLAHAN B. REIF R. LI H. Uniqueness, advantages, challenges, solutions, and perspectives in therapeutics applying RNA nanotechnology. Nucleic Acid Ther. 2012;22:226–245. doi: 10.1089/nat.2012.0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUO S. TSCHAMMER N. MOHAMMED S. GUO P. Specific delivery of therapeutic RNAs to cancer cells via the dimerization mechanism of phi29 motor pRNA. Hum. Gene Ther. 2005;16:1097–1109. doi: 10.1089/hum.2005.16.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUO S. HUANG F. GUO P. Construction of folate-conjugated pRNA of bacteriophage phi29 DNA packaging motor for delivery of chimeric siRNA to nasopharyngeal carcinoma cells. Gene Ther. 2006;13:814–820. doi: 10.1038/sj.gt.3302716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HANSMA H.G. OROUDJEV E. BAUDREY S. JAEGER L. TectoRNA and ‘kissing-loop’ RNA: atomic force microscopy of self-assembling RNA structures. J. Microsc. 2003;212:273–279. doi: 10.1111/j.1365-2818.2003.01276.x. [DOI] [PubMed] [Google Scholar]
- HAQUE F. SHU D. SHU Y. SHLYAKHTENKO L. RYCHAHOU P. EVERS B.M. GUO P. Ultrastable synergistic tetravalent RNA nanoparticles for targeting to cancers. Nano Today. 2012;7:245–257. doi: 10.1016/j.nantod.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOEPRICH S. ZHOU Q. GUO S. QI G. WANG Y. GUO P. Bacterial virus phi29 pRNA as a hammerhead ribozyme escort to destroy hepatitis B virus. Gene Ther. 2003;10:1258–1267. doi: 10.1038/sj.gt.3302002. [DOI] [PubMed] [Google Scholar]
- JAEGER L. CHWOROS A. The architectonics of programmable RNA and DNA nanostructures. Curr. Opin. Struct. Biol. 2006;16:531–543. doi: 10.1016/j.sbi.2006.07.001. [DOI] [PubMed] [Google Scholar]
- JAEGER L. LEONTIS N.B. Tecto-RNA: one dimensional self-assembly through tertiary interactions. Angew. Chem. Int. Ed. Engl. 2000;39:2521–2524. doi: 10.1002/1521-3773(20000717)39:14<2521::aid-anie2521>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- JAEGER L. WESTHOF E. LEONTIS N.B. TectoRNA: modular assembly units for the construction of RNA nano-objects. Nucleic Acids Res. 2001;29:455–463. doi: 10.1093/nar/29.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KHALED A. GUO S. LI F. GUO P. Controllable self-assembly of nanoparticles for specific delivery of multiple therapeutic molecules to cancer cells using RNA nanotechnology. Nano Lett. 2005;5:1797–1808. doi: 10.1021/nl051264s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRUGER K. GRABOWSKI P.J. ZAUG A.J. SANDS J. GOTTSCHLING D.E. CECH T.R. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell. 1982;31:147–157. doi: 10.1016/0092-8674(82)90414-7. [DOI] [PubMed] [Google Scholar]
- LI H. LI W.X. DING S.W. Induction and suppression of RNA silencing by an animal virus. Science. 2002;296:1319–1321. doi: 10.1126/science.1070948. [DOI] [PubMed] [Google Scholar]
- LIU J. GUO S. CINIER M. SHLYAKHTENKO L. SHU Y. CHEN C. SHEN G. GUO P. Fabrication of stable and RNase-resistant RNA nanoparticles active in gearing the nanomotors for viral DNA packaging. ACS Nano. 2010;5:237–246. doi: 10.1021/nn1024658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MITRA S. SHCHERBAKOVA I.V. ALTMAN R.B. BRENOWITZ M. LAEDERACH A. High-throughput single-nucleotide structural mapping by capillary automated footprinting analysis. Nucleic Acids Res. 2008;36:e63. doi: 10.1093/nar/gkn267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAVA SARVER N. CANTIN E.M. CHANG P.S. ZAIA J.A. LADNE P.A. STEPHENS D.A. ROSSI J.J. Ribozymes as potential Anti-HIV-1 therapeutic agents. Science. 1990;24:1222–1225. doi: 10.1126/science.2107573. [DOI] [PubMed] [Google Scholar]
- PAIGE J.S. WU K.Y. JAFFREY S.R. RNA mimics of green fluorescent protein. Science. 2011;333:642–646. doi: 10.1126/science.1207339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SARVER N.A. CANTIN E.M. CHANG P.S. ZAIA J.A. LADNE P.A. STEPHENS D.A. ROSSI J.J. Ribozymes as potential anti-HIV-1 therapeutic agents. Science. 1990;247:1222–1225. doi: 10.1126/science.2107573. [DOI] [PubMed] [Google Scholar]
- SHU D. HUANG L. HOEPRICH S. GUO P. Construction of phi29 DNA-packaging RNA (pRNA) monomers, dimers and trimers with variable sizes and shapes as potential parts for nano-devices. J. Nanosci. Nanotechnol. 2003;3:295–302. doi: 10.1166/jnn.2003.160. [DOI] [PubMed] [Google Scholar]
- SHU D. MOLL W.D. DENG Z. MAO C. GUO P. Bottom-up assembly of RNA arrays and superstructures as potential parts in nanotechnology. Nano Lett. 2004;4:1717–1723. doi: 10.1021/nl0494497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHU D. SHU Y. HAQUE F. ABDELMAWLA S. GUO P. Thermodynamically stable RNA three-way junctions as platform for constructing multifunctional nanoparticles for delivery of therapeutics. Nat. Nanotechnol. 2011;6:658–667. doi: 10.1038/nnano.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHU Y. CINIER M. FOX S.R. BEN-JOHNATHAN N. GUO P. Assembly of Therapeutic pRNA-siRNA Nanoparticles Using Bipartite Approach. Mol. Ther. 2011;19:1304–1311. doi: 10.1038/mt.2011.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEAD S.L. ASHWIN H. JOHNSTON B. DALLAS A. KAZAKOV S.A. TARBIN J.A. SHARMAN M. KAY J. KEELY B.J. An RNA-aptamer-based assay for the detection and analysis of malachite green and leucomalachite green residues in fish tissue. Anal. Chem. 2010;82:2652–2660. doi: 10.1021/ac902226v. [DOI] [PubMed] [Google Scholar]
- TUERK C. GOLD L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- XU W. LU Y. Label-free fluorescent aptamer sensor based on regulation of malachite green fluorescence. Anal. Chem. 2010;82:574–578. doi: 10.1021/ac9018473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG F. LEMIEUX S. WU X. ST.-ARNAUD S. MCMURRAY C.T. MAJOR F. ANDERSON D. Function of hexameric RNA in packaging of bacteriophage phi29 DNA in vitro. Mol. Cell. 1998;2:141–147. doi: 10.1016/s1097-2765(00)80123-9. [DOI] [PubMed] [Google Scholar]