

Abstract
CD59 is a glycosylphosphatidylinositol (GPI)-anchored membrane regulator of complement expressed on blood cells as well as peripheral tissues. It protects host cells from complement injury by inhibiting formation of the membrane attack complex. Recent studies in mice have suggested also a role of CD59 in T cell immune response that was mechanistically independent of complement. In the present study, we investigated the function of CD59 in the MRL/lpr model of murine lupus. We backcrossed the Cd59a knockout (Cd59a−/−) mouse onto the MRL/lpr background and compared Cd59a+/+-MRL/lpr and Cd59a−/−-MRL/lpr littermates for the development of systemic autoimmunity. We found that CD59a deficiency significantly exacerbated the skin disease and lymphoproliferation characteristic of MRL/lpr mice. It also increased autoantibody titers and caused a higher level of proteinuria in male MRL/lpr mice. Bone marrow transfer experiments indicated that CD59a expression on both BM-derived cells and peripheral tissues played a role in lymphoproliferation, whereas the skin disease phenotype is determined mainly by local CD59a expression. Importantly, C3 gene deletion or C5 neutralization with a blocking mAb in Cd59a−/−-MRL/lpr mice did not rescue the pro-autoimmune phenotype associated with CD59a deficiency. These results together suggest that CD59a inhibits systemic autoimmunity in MRL/lpr mice through a complement-independent mechanism.
Introduction
Systemic lupus erythematosus (SLE4) is an autoimmune disorder characterized by high levels of autoantibody production and multi-organ tissue damage including kidney and skin involvement (1, 2). The complement system has been shown to play an important but paradoxical role in the pathogenesis of SLE. On the one hand, genetic deficiencies of classical pathway components such as C1q, C2 and C4 strongly increase the risk of developing a monogenetic form of SLE, suggesting an essential role of the classical pathway complement in preventing the onset of disease, possibly due to their role in clearance of apoptotic cells and auto-antigens. On the other hand, autoantibody- and immune complex-mediated end organ injury is considered to involve complement activation as an effector pathway (3). The role of complement as a double-edged sword in SLE pathogenesis and development has also been demonstrated in transgenic mouse studies. Mice lacking C1q or C4, like humans with similar genetic deficiencies, are predisposed to developing an SLE-like autoimmune phenotype (4–7). In contrast, deletion of the key alternative pathway complement proteins factor B or factor D ameliorated aspects of the autoimmune disease phenotype of MRL/lpr mice (8, 9). Deletion of C3 did not offer much protection in MRL/lpr mice, possibly reflecting a zero sum effect of C3 in contributing to inflammation as well as to immune complex solubilization and clearance (10).
Under physiological conditions, host tissues are protected from complement-mediated autologous injury because of the presence of a number of cell membrane-linked and fluid phase complement regulatory proteins (11, 12). Two central membrane-bound complement regulators in mammals are the decay-accelerating factor (DAF) and CD59, both of which attach to the cell surface via a glycosylphosphatidylinositol (GPI) anchor. DAF inhibits complement activation by preventing the formation and accelerating the decay of C3 and C5 convertases, whereas CD59 inhibits the formation of the membrane attack complex (MAC) and thus serves as a key regulator of the terminal complement pathway on host cells (12). Additionally, both DAF and CD59 have been implicated in T cell-mediated immune responses that were either complement-dependent or complement-independent (13–16). It has been well documented that mutations in membrane or fluid phase complement regulators in humans are associated with alternative pathway complement-mediated rare, but potentially life-threatening, diseases such as paroxysmal nocturnal hemoglobinuria, C3 glomerulopathy and atypical hemolytic uremic syndrome (17–19). The role of complement regulatory proteins in the pathogenesis and manifestation of human systemic autoimmune diseases in general, and SLE in particular, has not been well studied.
Using the MRL/lpr murine model of SLE, we previously have obtained evidence for a role of DAF in mitigating SLE development in ways which were both complement dependent and complement independent (20). We showed that lack of DAF exacerbated the autoimmune manifestations in MRL/lpr mice by causing increased lymphoproliferation, anti-chromatin autoantibody production and dermatitis (20). In the present study, we have investigated the role of CD59 in the MRL/lpr model of murine SLE. Unlike humans, the mouse has two Cd59 genes (Cd59a and Cd59b) which are located in tandem on chromosome 2 (21, 22). Both genes encode functionally active GPI-anchored proteins but only the Cd59a gene, like human Cd59 gene, is widely expressed across different tissues whereas Cd59b expression is restricted to the mouse testis (23). Thus, CD59a is considered to be functionally more representative of human CD59. We backcrossed the CD59a deficient mouse onto the MRL/lpr background. We found that CD59a deficiency significantly exacerbated several aspects of the autoimmune diseases phenotype of MRL/lpr mice, including lymphoproliferation, dermatitis, autoantibody production, and nephritis. Interestingly, the effect of CD59a was determined to be independent of C3 and C5. Thus, our data suggest an activity of CD59 in regulating SLE-like systemic autoimmunity through a novel and complement-independent mechanism.
Materials and Methods
Mice
The generation of the CD59a knockout (Cd59a−/−) mouse has been described previously (24). Cd59+/− mice were crossed with MRL/lprIghb mice (MRL/lpr mice congenic for the Ig heavy chain b allele, maintained in our animal colony) for a total of 9 generations. Nine-time backcrossed Cd59+/−-MRL/lpr mice were then intercrossed to derive Cd59+/+-MRL/lpr and Cd59−/−-MRL/lpr as littermates. Cd59a−/−C3−/−-MRL/lpr mice were obtained by crossing Cd59a−/−-MRL/lpr mice with C3−/−-MRL/lpr mice previously described (10). The generation of Daf−/−Crry−/−C3−/− mice on a mixed C57BL/6 and 129J background, was described previously (25). The Cd59a genotype was screened by FACS analysis of erythrocytes as described previously (24). C3 genotyping was performed by polymerase chain reaction (PCR) of tail DNA (13). Mice were monitored for autoimmune disease until they were 5 months old, at which time they were sacrificed for pathological evaluation. Mice were kept in a specific pathogen-free barrier facility, and all animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Generation of BM chimera mice
Bone marrow (BM) chimera mice were generated between male Cd59a−/−-MRL/lpr and Cd59a+/+-MRL/lpr mice. BM cells were flushed out from the bones (femur and tibia) of donor mice. After filtration through nylon mesh and clearing of erythrocytes with red blood cell lysing buffer (ACK buffer), the single-cell suspension was depleted of mature T and B cells by incubating with anti-CD4, CD8, CD19 and CD90 antibodies conjugated to magnetic microbeads (Miltenyi Biotec, Auburn, CA), followed by processing on a fully automated Clinimacs device (Miltenyi) equipped with TS separation columns. Recipient mice were lethally irradiated with two doses of 525 rads spaced 3 hours apart (totally 1050 rads). Irradiated recipient mice then received T/B cell-depleted donor mouse BM cells (1×107 cells/mouse) through the tail vein. Repopulation of the immune system was monitored by flow cytometric analysis of the blood using rat anti-mouse CD59a. In the chimeric mice, more than 95% of the T cells, B cells and myeloid cells were derived from donor bone marrow. Mice were studied for autoimmunity for 5 months after BM transfer.
Assessment of dermatitis
Mice were inspected monthly for the development of dermatitis in the usual body site, and the age at which open skin lesions developed was recorded and lesion size measured with a ruler.
ELISA for autoantibodies and their isotypes
Serum was collected by tail vein bleeding at 3, 4 and 5 months of age, and frozen at −20°C until use. Sandwich ELISA was used for determining the titers of Abs. For total serum IgM, IgG and IgG2a, plates were coated with the appropriate capture Ab. Diluted sera, as well as serially diluted IgM, IgG and IgG2a standards, were added to the plates and allowed to bind overnight. The plate was incubated with biotin-conjugated anti-mouse IgM, IgG and IgG2a Ab (Jackson ImmunoResearch Laboratories, West Grove, PA) for 2 hours at 4°C. Avidin-alkaline phosphatase (Sigma, St Louis, CA) was added, followed by para-nitrophneylphosphate (PNPP) (Sigma) for color development. OD405 was measured using a microplate reader (Molecular device, Sunnyvale CA). Similar procedures were used for determining Ag-specific autoantibodies, except that the plates were coated with the target Ag, chromatin or double-stranded DNA. Quantification was achieved with either a standard curve (total IgM, IgG, IgG2a) or a positive serum sample from a 5 month old MRL/lpr mouse as an internal standard (Ag-specific autoantibodies) for inter-plate comparison. Ig concentrations or relative OD 405 values for serum from each animal were plotted individually.
Assessment of Nephritis
Urine samples were collected in metabolic cages at monthly intervals starting at 3month until sacrifice. Urinary albumin concentration was measured by a mouse albumin ELISA kit (Bethyl Laboratories, Montagomery, TX), and normalized to urinary creatinine. At the time of sacrifice, one kidney was fixed in 10% buffered formalin and processed for paraffin embedding and sectioning, followed by hematoxylin and eosin (H&E) staining and histological evaluation. The other kidney was frozen in OCT medium and processed for immunofluorescence staining of IgG and C3 with fluorescent isothiocyanate-conjugated goat anti-mouse IgG and C3 F(ab′)2 fragments (used at 1:75 for anti-IgG and 1:500 for anti-C3; Fisher/ICN, Durham, NC). The presence and severity of nephritis was determined as previously described (20). A blinded observer (M. Madaio) evaluated and scored independently the severity of glomerular and interstitial nephritis (0–4 scale) by light microscopy (26). Similarly, the presence of glomerular or tubular basement membrane deposits of IgG and C3 was graded on a 0–3 scale by immunofluorescence microscopy (27). Multiple sections at a minimum of two different levels were observed. Each section typically involved evaluation of >25 glomeruli, >10 blood vessels, and the interstitium contained within two to three longitudinal sections of kidney.
Assessment of lymphoproliferation
At the time of sacrifice, spleen and cervical, axillary, inguinal, and mesenteric lymph nodes (LN) were weighed, meshed, and cleared of erythrocytes by ACK buffer. After the spleen and LN cells were counted by cell counter (Beckman Coulter, Fullerton, CA), the cell suspension (1.5 × 106 cells) was incubated with antibodies to the following cell-surface markers: Thy1.2-PE, CD4-PE, CD8-PE, B220-FITC, CD19-FITC, CD1-FITC (Pharmingen, San Diego, CA). Flow cytometry analysis was performed by using FACScan with CellQuest data acquisition software (Becton Dickinson Immunocytometry Systems, San Jose, CA)
Analysis of expression of CD59a
CD59a expression on erythrocytes or lymphocytes from spleen, lymph nodes and thymus was evaluated by flow cytometry using FITC-conjugated anti-mouse Cd59a mAb (Hycult, Plymouth Meeting, PA). Cells were co-stained for a panel of surface markers including CD3, CD4, CD8, B220, CD19 and Thy1.2 to identify relevant cell populations. Fluorescence labeled mAbs against the cell surface markers were from Pharmingen.
Anti-C5 mAb treatment
The source and preparation of mouse anti-mouse C5 mAb (BB5.1) was described previously (14). An isotype control mAb (MOPC21, IgG1, lyophilized protein) was from Sigma-Aldrich. BB5.1 or MOPC21 mAbs (1 mg/kg i.p.) were injected twice weekly for 3 months (from 2 to 5 month old).
Hemolytic assays
Hemolytic assay was performed as described (28). Briefly, erythrocytes from Daf−/−Crry−/−C3−/− mice (25) were incubated (37°C, 30 minutes) with 50% serum from anti-mouse C5 mAb or isotype control mAb-treated Cd59a−/−-MRL/lpr mice (taken 2 weeks after twice weekly mAb treatment), in the presence of 5μM of a recombinant mouse factor H short consensus repeat (SCR) 19–20 (28). At the end of the incubation period, cold EDTA-PBS was added to stop the reaction. Samples were centrifuged and the supernatant OD values were measured at 405nm. Percent hemolysis was calculated by dividing the OD405 value with that of a sample in which total hemolysis was induced by hypotonic shock (28).
Statistics
Data were analyzed by Student t test (for normally distributed data), Mann-Whitney test (for nonparametric data) or Fisher’s exact test (for incidence of dermatitis) as specified in the text and significant difference is defined as p < 0.05.
Results
To assess if CD59a plays a role in the pathogenesis of lupus-like disease in mice, we backcrossed a Cd59a−/− mouse (originally on a mixed C57BL/6-129/SvEv background)(24) onto the MRL/lpr genetic background for a total of 9 generations. Ninth generation backcrossed Cd59a+/− -MRL/lpr mice were then intercrossed to obtain Cd59a−/− -MRL/lpr and Cd59a+/+-MRL/lpr littermates. In total, we studied 24 Cd59a−/− -MRL/lpr mice (16 males and 8 females) and 24 Cd59a+/+-MRL/lpr mice (14 males and 10 females). These mice were monitored for autoimmune disease development until 5 months of age.
MRL/lpr mice have a mutation in the Fas ligand, leading to impaired apoptosis of lymphocytes (29). They spontaneously develop splenomegaly and lymphadenopathy due to accumulation of lymphocytes expressing an activated phenotype in these organs (30). This lymphoproliferation affects all lymphocytes subtypes but is mostly composed of CD4−CD8− aberrant αβT cells that are B220+/Thy1.2+ (31). While the role of these aberrant T cells in the disease phenotype is still not clearly elucidated, they have a possible counterpart in SLE patients and express high amounts of IL-17 (32). To assess if CD59a deficiency affected lymphoproliferation in MRL/lpr mice, we examined the size, cell number and composition of spleens and lymph nodes (LN) of Cd59a+/+-MRL/lpr and Cd59a−/− -MRL/lpr mice at 5 month of age. As shown in Figure 1, we found that the degree of splenomegaly and lymphadenopathy was significantly increased in Cd59a−/− -MRL/lpr mice compared with their Cd59a+/+-MRL/lpr littermates. FACS analysis showed that CD59a−/−-MRL/lpr mouse LNs contained a higher percentage of aberrant B220+/Thy1.2+ T cells (Fig 1C). This increase in the aberrant T cell population was mirrored by corresponding decreases in normal T cells of various developmental stages including CD4+ and CD8+ T cells (Fig 1D), as well as other cell populations (e.g. B cells: B220+/CD19+ and IgM+/CD21+, data not shown). There were also significant differences between Cd59a+/+-MRL/lpr and Cd59a−/− -MRL/lpr mice in the total number of LN and spleen cells, as well as various cell subsets (CD4+ or CD8+ T cells, aberrant B220+/Thy1.2+ T cells in the LNs, aberrant B220+/Thy1.2+ T cells, B220+/CD19+ and IgM+/CD21+ B cells in the spleen) (Fig 1E, F).
Figure 1.

CD59a deficiency increases lymphoproliferation in MRL/lpr mice. CD59a−/−-MRL/lpr (CD59a−/−) mice had significantly larger spleens (A) and lymph nodes (B) than CD59a+/+-MRL/lpr (CD59a+/+) mice. Flow cytometry analysis showed that CD59a−/− MRL/lpr mouse lymph nodes contained a higher percentage of aberrant B220+/Thy1.2+ T cells (C) and a lower percentage of combined single positive (CD4+ or CD8+) T cells (D). Cellularity of LNs in 5 month old Cd59a−/− MRL/lpr and Cd59a+/+ MRL/lpr mice (E). Cellularity of spleens in 5 month old Cd59a−/− MRL/lpr and Cd59a+/+ MRL/lpr mice (F). 24 Cd59a−/− -MRL/lpr mice (16 males and 8 females) and 24 Cd59a+/+-MRL/lpr mice (14 males and 10 females) were used in A-F. Each data point in A–D represents a mouse and cell numbers in E and F are averaged (mean ± SED). * P < 0.01, ** P< 0.05, Student t test.
Starting at 3 months of age, both male and female Cd59a−/− -MRL/lpr mice developed severe autoimmune dermatitis, which reached an incidence of about 50% at the time of sacrifice. The wild type Cd59a+/+ -MRL/lpr cohort showed only a gradual onset of very mild skin disease (Fig 2). Thus, 5 of 16 male Cd59a−/− -MRL/lpr mice at 3 month of age, and 11 of 16 at 5 month of age, developed severe skin disease whereas only 1male of the 24 Cd59a+/+ -MRL/lpr mice was found to have a small open skin lesion at 5 month of age. It is of interest that susceptibility to skin disease development in CD59a−/− -MRL/lpr mice had a gender bias towards males. One third of male Cd59a−/− -MRL/lpr mice already had skin disease at 3 month of age but only 1 of 8 female Cd59a−/− -MRL/lpr mice developed skin disease at 4 month of age (Fig 2).
Figure 2.

CD59a−/− -MRL/lpr mice developed autoimmune dermatitis with high incidence and severity. Representative pictures of a CD59a+/+-MRL/lpr mouse at 5 month of age (A) showing the lack of skin disease and of an age-matched CD59a−/− -MRL/lpr mouse (B) showing severe skin disease. (C) Percentage of Cd59−/−-MRL/lpr (CD59a−/−) and Cd59+/+-MRL/lpr (CD59a+/+) mice with visible open skin lesions at 3, 4, and 5 months of age. Numbers of mice are indicated on the graph. * P<0.05, ** P<0.01, compared with male Cd59+/+-MRL/lpr mice. Fisher’s exact test. (D). Average size of open skin lesions (mean ± SED). Data are only for mice with open skin lesions (numbers indicated above the columns) among the same mice studied in C.
To further examine the immunological changes in Cd59a−/− -MRL/lpr mice, the level and nature of serum Igs and autoantibodies were evaluated at 5 months. We found that serum levels of total IgG and IgM, as well as anti-chromatin autoantibody titers, were significantly elevated in male Cd59a−/− -MRL/lpr mice compared with male Cd59a+/+ -MRL/lpr mice (Fig 3). Significant increases, however, were not observed in the female Cd59a−/− -MRL/lpr group. In fact, total IgG levels in female Cd59a−/− -MRL/lpr mice were actually lower than that of female Cd59a+/+ -MRL/lpr mice. There was no significant difference in the titers of serum total IgG2a and anti-dsDNA antibodies between Cd59a−/− -MRL/lpr and Cd59a+/+ -MRL/lpr mice (data not shown).
Figure 3.
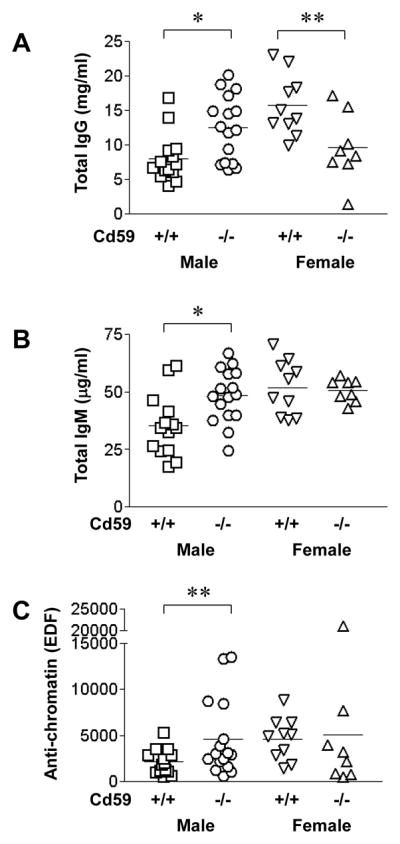
ELISA assay of serum antibodies in CD59a−/−- and CD59a+/+-MRL/lpr mice at 5 months of age. Serum total IgG (A), IgM (B) and anti-chromatin autoantibody (C) levels in male CD59a−/−-MRL/lpr (CD59a−/−) mice were significantly elevated compared with male CD59a+/+-MRL/lpr (CD59a+/+) mice. * p<0.01, ** p<0.05, Student t test for A and B, Mann-Whitney test for C.
We also studied the impact of CD59a deficiency on the development of autoimmune nephritis in MRL/lpr mice. Lupus nephritis is characterized by IgG and C3 deposition, proteinuria and increased nephritis score. We measured proteinuria at 5 months of age prior to sacrifice and examined renal pathology for nephritis under light microscopy, and IgG and C3 deposition by immunofluorescence. We found that male Cd59a−/−-MRL/lprmice had significantly higher levels of urine albumin to urine creatinine ratios compared with gender-matched Cd59a+/+-MRL/lpr mice (Fig 4A). Although as a group female Cd59a−/−-MRL/lprmice did not show a statistical significant increase in proteinuria, 2 of the 8 female Cd59a−/−-MRL/lprmice, but none of the 10 female Cd59a+/+-MRL/lpr mice, developed heavy proteinuria (Fig 4A). We detected no significant difference between Cd59a−/−-MRL/lpr and Cd59a+/+-MRL/lpr groups in the semi-quantitative histopathological analysis of nephritis and C3 deposition (Fig 4), but glomerular IgG deposition was higher in Cd59a−/−-MRL/lprmice, particularly in the male cohort (Fig 4C).
Figure 4.

Effect of CD59a deficiency on renal disease of MRL/lpr mice. Male Cd59a−/−MRL/lpr (CD59a−/−) mice developed significantly increased proteinuria (A) and glomerular/tubular IgG deposition (C) compared with male Cd59a+/+ MRL/lpr (CD59a+/+) littermates. ** p<0.05, Mann-Whitney test. There was no significant difference in glomerular nephritis scores (B) and glomerular/tubular C3 staining (D) between Cd59a−/−- and Cd59a+/+-MRL/lpr mice. Scores for IgG and C3 deposition represent composite scores of glomerular/tubular staining (maximal score 6).
To determine if CD59a expression on BM-derived cells or peripheral tissues is important for regulating the autoimmunity phenotype in MRL/lpr mice, we generated BM chimera mice between male Cd59a−/−- and Cd59a+/+-MRL/lpr mice. Because increased lymphoproliferation and development of skin disease were the more striking phenotypes associated with CD59a deficiency, we used these disease features as our readouts for this experiment. By using CD59a expression as a measurement, we determined that >95% of blood cells were derived from donor BM (data not shown). We found that in general CD59a expression on BM-derived cells as well as peripheral tissues had an impact on lymphoproliferation (Fig 5A, B). Thus, chimeric mice generated using Cd59a−/−- MRL/lpr mice as BM donors had greater lymphoproliferation than those using Cd59a+/+-MRL/lpr as BM cell donors. However, peripheral CD59a expression also affected the degree of lymphoproliferation, as for a given BM cell donor, Cd59a+/+- MRL/lpr recipients tended to have smaller spleens and lymph nodes (Fig 5A, B). For the skin disease phenotype, however, we found that CD59a expression on peripheral tissues clearly played a dominant role. Regardless of the donor BM cell genotype, chimeric mice generated using Cd59a−/−- MRL/lpr mice as recipients developed skin disease with higher incidence and increased severity (Fig 5C, D). Thus, skin disease development in Cd59a−/−-MRL/lpr mice was mainly exacerbated by Cd59a deficiency on peripheral tissues.
Figure 5.

Bone marrow chimera experiment between Cd59a−/− MRL/lpr (CD59a−/−) and Cd59a+/+ MRL/lpr (CD59a+/+) mice. Assessment of splenomegaly (A) and lymphadenopathy (B) of the chimeric mice at 5 months after BM transfer showed a greater degree of lymphoproliferation when the BM donors were Cd59a−/− MRL/lpr mice. However, peripheral Cd59a also had an impact on lymphoproliferation [compare Cd59a+/+→Cd59a+/+ and Cd59a+/+→Cd59a−/− groups in A, and Cd59a−/−→Cd59a−/− and Cd59a−/−→Cd59a+/+ groups in B). In contrast, only peripheral CD59a expression was important for preventing skin disease development in MRL/lpr mice. *p<0.01, ** p<0.05, Student t test for A and B. (C) Percentage of mice with visible open skin lesions at 2–5 months after BM transfer. Chimeras with Cd59a−/− MRL/lpr (CD59a−/−) mice as recipients (filled symbols) appeared to be more prone to develop skin disease than those with Cd59a+/+ -MRL/lpr (CD59a+/+) mice as recipients (open symbols). *** p<0.05 vs Cd59a+/+→Cd59a+/+, Fisher’s exact test. Numbers of mice are indicated on the graph. (D) The average size of open skin lesions (mean ± SED) in chimeras at 3–5 months after BM transfer. Data plotted are only for mice with open skin lesions (numbers indicated above the columns) among the same mice studied in C. Group b and c mice had clearly less skin disease as there were less than 2 mice with open skin lesions at any time points. No statistical difference between group a and d mice at 4 and 5 months (n=3–5, Mann-Whitney test).
To understand how CD59 protected disease progression in MRL/lprmice, we investigated the role of complement by breeding Cd59a−/−-MRL/lpr mice with C3−/−-MRL/lpr mice and by blocking C5 function with a mAb. From C3+/−-Cd59a−/− -MRL/lpr intercrosses, we generated and studied 19 (8 males and 11 females) C3−/−-Cd59a−/−-MRL/lpr mice and 17 (10 males and 7 females) C3+/+-Cd59a−/−-MRL/lpr mice as littermates. Fig 6 shows that C3 deficiency did not significantly affect lymphoproliferation in Cd59a−/−-MRL/lpr mice, nor did it reduce the incidence of skin disease. In fact, female C3−/−-Cd59a−/−-MRL/lpr mice had the highest incidence of open skin lesions at 5 months of age (Fig 6E). On the other hand, C3 deficiency reduced skin disease severity, as the average size of open skin lesions in male and female C3−/−-Cd59a−/−-MRL/lpr mice was smaller than in corresponding C3+/+-Cd59a−/−-MRL/lpr littermates (Fig 6F). C3 gene deletion affected neither autoantibody production nor nephritis severity of Cd59−/− -MRL/lpr mice (data not shown). In a further experiment, we treated 7 male Cd59a−/−-MRL/lpr mice with an anti-mouse C5 mAb and another 7 male with a control mAb. The dosage and frequency of anti-C5 mAb treatment was based on published studies (33), and the effectiveness of the anti-C5 mAb to block C5 function was confirmed by diminished hemolytic activity in the treated Cd59a−/−-MRL/lpr mice (data not shown). However, we found that anti-C5 mAb treatment had no impact on lymphoproliferation or skin disease development in Cd59a−/−-MRL/lpr mice (Fig 7).
Figure 6.

Effect of C3 gene deletion on lymphoproliferation and skin disease development in Cd59a−/− MRL/lpr mice. C3−/−-Cd59a−/−-MRL/lpr mice and Cd59a−/−-MRL/lpr mice were produced as littermates and studied for 5 months. No difference was seen in splenomegaly (A) and lymphadenopathy (B) between the two genotypes. There was also no difference between C3−/−-and C3+/+-Cd59a−/−-MRL/lpr mice in the percentage of abnormal T cells (C) or single-positive (CD4+ or CD8+) T cells (D) in their lymph nodes. C3 gene deletion did not reduce the incidence of open skin lesions (E) but reduced open skin lesion size (F) in Cd59a−/−-MRL/lpr mice. Numbers of mice for panel C are indicated on the graph. In panel E, average lesion size (mean ± SED) was only for mice with open lesions (numbers indicated above the columns) among the same mice studied in C.
Figure 7.

Lack of an effect of C5 blockade with mAb on lymphoproliferation and skin disease development in Cd59a−/− MRL mice. (A) Hemolytic activities of sera from Cd59a−/−-MRL/lpr mice treated with an anti-C5 (α-C5, n=7) or control (con, n=7) mAb. Spleen and lymph node weights (B), percentage of mice with open skin lesions (C) and average open skin lesion size (D) were not significantly different between Cd59a−/− MRL mice treated with anti-C5 (n=7) or control mAb (n=7). Average size of open skin lesions (mean ± SED) was only for mice with open skin lesions (numbers indicated above the columns) among the same mice studied in C. * p<0.0001, Student t test.
Discussion
Our present results are compatible with a protective role of CD59 in the MRL/lpr model of murine lupus. We found that deficiency of CD59a significantly increased lymphoproliferation and exacerbated skin disease development in MRL/lpr mice. The effect of CD59a deletion on systemic autoimmunity in MRL/lpr mice appeared to have a gender bias, being more striking in male mice than in female mice. Specifically, we found that only male Cd59a−/−-MRL/lpr mice, but not females, had elevated total serum IgG and IgM levels, higher anti-chromatin autoantibody titers and worsened proteinuria, when compared to Cd59a-sufficient MRL/lpr mice. Skin disease was also more prevalent and severe in male Cd59a−/−-MRL/lpr mice. This gender bias towards males is in contrast to that observed for the effect of DAF deficiency in MRL/lpr mice (20). DAF is another GPI-anchored membrane complement regulator that inhibits complement at the C3 activation step (12, 34). In a previous study, we observed that Daf1 gene deletion in MRL/lpr mice aggravated autoimmunity to a greater degree in female mice (20). The mechanism for the gender bias in either case is unknown but the fact that gender bias of opposite direction was observed in the two models argued against it being related to complement protein levels in male and female mice. It has been reported that male mice have higher complement activity than females (35, 36) and we found also that male MRL/lpr mice (Cd59a+/+ or Cd59−/−) had higher plasma C5 levels (data not shown). Rather, the gender bias may relate to interactions between Daf1 or Cd59a gene deficiency and sex hormones. Many human autoimmune diseases including lupus have a bias towards one gender or another (37). It should be noted that higher lymphoproliferation and antibody levels had a gender bias towards females in Cd59a+/+-MRL/lpr mice (Fig 1 and Fig 3), but not in Cd59a−/−-MRL/lpr mice, and this made the effect of Cd59a gene deletion on disease exacerbation to be more pronounced in the male cohort.
Like human CD59, mouse CD59a is expressed broadly on various tissues (23, 38). However, as reported before for C57BL/6 mice (15), we found CD59a expression on developing and mature CD4+ and CD8+ T cells and abnormal B220+/Thy1.2+ T cells of MRL/lpr mice to be under the limit of FACS detection (data not shown). Thus, a possible association between T cell CD59a expression and the gender difference in the Cd59a−/−-MRL/lpr mouse phenotype could not be easily assessed. Nevertheless, Longhi and colleagues have previously demonstrated CD59a expression on mouse T cells using other methods and observed increased anti-CD3 antibody-induced proliferation with Cd59a−/− mouse CD4+ T cells (15). To dissect the respective roles of CD59a on hematopoietic cells and peripheral tissues, we generated BM chimeric mice between Cd59−/−-MRL/lpr and Cd59+/+-MRL/lpr mice. This experiment revealed that expression of CD59a on BM-derived cells played an important role in regulating the lymphoproliferation phenotype, whereas peripheral expression of CD59a is critical in preventing skin disease development. In further experiments, we evaluated if the effect of CD59a on autoimmunity in MRL/lpr mice is related to CD59a working as a terminal complement regulator. Two separate experimental approaches were used to address this question. First, by cross breeding experiment, we deleted the C3 gene from Cd59−/−-MRL/lpr mice. If the exacerbated autoimmunity phenotype was caused by increased MAC formation in the absence of CD59a, then deletion of C3 in Cd59−/−-MRL/lpr mice may prevent complement activation and MAC formation and rescue the phenotype. Of interest, we found that C3 gene deletion had no impact on lymphoproliferation (Fig 6A–D), suggesting that the regulatory function of CD59a on this aspect of autoimmunity in MRL/lpr mice was independent of complement. We also detected no reduction in skin disease incidence by C3 deficiency. One limitation of this experiment is that the C3 gene may play both a detrimental and a beneficial role in systemic autoimmunity as suggested by other studies (8, 10), and the use of an alternative pathway complement inhibitor in future studies may provide additional and more definitive insight. In this context, it is notable that C3 deficiency alleviated the severity of skin disease in Cd59−/−-MRL/lpr mice. The average size of open skin lesions in C3-deficient mice was significantly smaller than C3-sufficient littermates at 5 months of age (Fig 6F).
Another caveat in interpreting the lack of effect of C3 deficiency on lymphoproliferation and skin disease incidence is that C5 activation could have occurred in the absence of C3, as has been demonstrated in other in vivo settings (39). C3-independent cleavage and activation of C5 by proteases such as thrombin has been referred to as the extrinsic pathway of complement activation (39). If such a pathway is operative in MRL/lpr mice, then the effect of CD59a deficiency could still have been mediated by increased MAC formation even in the absence of C3. To address this possibility, we adopted a second approach and used a neutralizing mAb, given chronically twice per week, to block C5 function. Importantly, although hemolytic assays clearly confirmed the effectiveness of the mAb in blocking the terminal complement pathway, its use in Cd59−/−-MRL/lpr mice did not affect lymphoproliferation nor did it reduce skin disease incidence or severity (Fig 7). Although we did not test the involvement of C5 in Cd59a-sufficient MRL/lpr mice, the anti-C5 experiment in Cd59−/−-MRL/lpr mice showed that disease exacerbation in Cd59−/−-MRL/lpr mice was MAC-independent. That C3 deficiency, but not anti-C5 mAb treatment, reduced skin disease severity in Cd59−/−-MRL/lpr mice also suggested an effect of C3 activation products (C3a and/or C3b/iC3b) that is separate from Cd59a function. The lack of a MAC-mediated effect in Cd59−/−-MRL/lpr mice may reflect the fact that there are multiple C3 complement regulators in the mouse, and any consequence of CD59a deficiency may become evident only in the context of an impairment in C3 regulation. Indeed, we previously have found that while CD59a−/− mice did not show enhanced susceptibility to renal ischemia reperfusion injury (IRI), Daf1−/−CD59a−/− mice had markedly increased sensitivity to renal IRI, compared to either wild-type or Daf1−/− mice (40).
Several lines of evidence argue against the possibility that 129 mouse strain-derived loci that are closely linked to the Cd59a gene accounted for some or all of the pro-autoimmune phenotypes observed in Cd59a−/− -MRL/lpr mice. First, the mouse Cd59a gene is located on chromosome 2 (21), and most well-defined 129 strain-derived SLE loci are localized to mouse chromosomes 1 and 3 (41, 42). Second, our results are reminiscent of and compatible with a previously demonstrated complement-independent role of CD59a in regulating CD4+ T cell immunity in mAb blocking experiments in vitro and in a murine model of recombinant vaccinia virus infection in vivo (15). Thus, both studies supported the hypothesis that, in addition to functioning as a MAC inhibitor, CD59 works as a suppressor of T cell immunity via a novel and complement-independent mechanism. Finally, our results and that of Longhi et al (15) are consistent with the substantial literature documenting a role of CD59 in T cell activation and signal transduction, either in the capacity as a GPI-anchored protein in lipid rafts of the T cell plasma membrane (43, 44) or as a cellular ligand for the T cell co-receptor CD2 (43, 45, 46).
In summary, we have revealed in this study a prominent role for CD59a in regulating autoimmunity in MRL/lpr mice. Deletion of CD59a exacerbated a number of features of the murine SLE. The effect of CD59a was independent of complement and was conferred by its expression on both BM-derived cells and peripheral tissues. Although the precise mechanisms of this novel function of CD59a remain to be elucidated, our findings have raised the possibility of a similar role of CD59 in human SLE and suggested new avenues of investigation in the pathogenesis and therapy of human autoimmune diseases.
Footnotes
Supported by National Institutes of Health grants AI63288, AI49344 and AI44970 (to WCS) and AI036206, AR34156 (RAE).
Abbreviations used: SLE, systemic lupus erythematosus; DAF, decay-accelerating factor; MAC, membrane attack complex; BM, bone marrow; PNPP, para-nitrophneylphosphate.
References
- 1.Mills JA. Systemic lupus erythematosus. N Engl J Med. 1994;330:1871. doi: 10.1056/NEJM199406303302608. [DOI] [PubMed] [Google Scholar]
- 2.Kotzin BL. Systemic lupus erythematosus. Cell. 1996;85:303. doi: 10.1016/s0092-8674(00)81108-3. [DOI] [PubMed] [Google Scholar]
- 3.Manderson AP, Botto M, Walport MJ. The role of complement in the development of systemic lupus erythematosus. Annu Rev Immunol. 2004;22:431. doi: 10.1146/annurev.immunol.22.012703.104549. [DOI] [PubMed] [Google Scholar]
- 4.Einav S, Pozdnyakova OO, Ma M, Carroll MC. Complement C4 is protective for lupus disease independent of C3. J Immunol. 2002;168:1036. doi: 10.4049/jimmunol.168.3.1036. [DOI] [PubMed] [Google Scholar]
- 5.Mitchell DA, Pickering MC, Warren J, Fossati-Jimack L, Cortes-Hernandez J, Cook HT, Botto M, Walport MJ. C1q deficiency and autoimmunity: the effects of genetic background on disease expression. J Immunol. 2002;168:2538. doi: 10.4049/jimmunol.168.5.2538. [DOI] [PubMed] [Google Scholar]
- 6.Chen Z, Koralov SB, Kelsoe G. Complement C4 inhibits systemic autoimmunity through a mechanism independent of complement receptors CR1 and CR2. J Exp Med. 2000;192:1339. doi: 10.1084/jem.192.9.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cook HT, Botto M. Mechanisms of Disease: the complement system and the pathogenesis of systemic lupus erythematosus. Nat Clin Pract Rheumatol. 2006;2:330. doi: 10.1038/ncprheum0191. [DOI] [PubMed] [Google Scholar]
- 8.Watanabe H, Garnier G, Circolo A, Wetsel RA, Ruiz P, Holers VM, Boackle SA, Colten HR, Gilkeson GS. Modulation of renal disease in MRL/lpr mice genetically deficient in the alternative complement pathway factor B. J Immunol. 2000;164:786. doi: 10.4049/jimmunol.164.2.786. [DOI] [PubMed] [Google Scholar]
- 9.Elliott MK, Jarmi T, Ruiz P, Xu Y, Holers VM, Gilkeson GS. Effects of complement factor D deficiency on the renal disease of MRL/lpr mice. Kidney Int. 2004;65:129. doi: 10.1111/j.1523-1755.2004.00371.x. [DOI] [PubMed] [Google Scholar]
- 10.Sekine H, Reilly CM, Molano ID, Garnier G, Circolo A, Ruiz P, Holers VM, Boackle SA, Gilkeson GS. Complement component C3 is not required for full expression of immune complex glomerulonephritis in MRL/lpr mice. J Immunol. 2001;166:6444. doi: 10.4049/jimmunol.166.10.6444. [DOI] [PubMed] [Google Scholar]
- 11.Alexander JJ, Quigg RJ. The simple design of complement factor H: Looks can be deceiving. Mol Immunol. 2007;44:123. doi: 10.1016/j.molimm.2006.07.287. [DOI] [PubMed] [Google Scholar]
- 12.Miwa T, Song WC. Membrane complement regulatory proteins: insight from animal studies and relevance to human diseases. Int Immunopharmacol. 2001;1:445. doi: 10.1016/s1567-5769(00)00043-6. [DOI] [PubMed] [Google Scholar]
- 13.Miwa T, Maldonado MA, Zhou L, Yamada K, Gilkeson GS, Eisenberg RA, Song WC. Decay-accelerating factor ameliorates systemic autoimmune disease in MRL/lpr mice via both complement-dependent and -independent mechanisms. Am J Pathol. 2007;170:1258. doi: 10.2353/ajpath.2007.060601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu J, Miwa T, Hilliard B, Chen Y, Lambris JD, Wells AD, Song WC. The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo. J Exp Med. 2005;201:567. doi: 10.1084/jem.20040863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Longhi MP, Sivasankar B, Omidvar N, Morgan BP, Gallimore A. Cutting edge: murine CD59a modulates antiviral CD4+ T cell activity in a complement-independent manner. J Immunol. 2005;175:7098. doi: 10.4049/jimmunol.175.11.7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Longhi MP, Williams A, Wise M, Morgan BP, Gallimore A. CD59a deficiency exacerbates influenza-induced lung inflammation through complement-dependent and -independent mechanisms. Eur J Immunol. 2007;37:1266. doi: 10.1002/eji.200636755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamashina M, Ueda E, Kinoshita T, Takami T, Ojima A, Ono H, Tanaka H, Kondo N, Orii T, Okada N, et al. Inherited complete deficiency of 20-kilodalton homologous restriction factor (CD59) as a cause of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1990;323:1184. doi: 10.1056/NEJM199010253231707. [DOI] [PubMed] [Google Scholar]
- 18.Fakhouri F, Fremeaux-Bacchi V, Noel LH, Cook HT, Pickering MC. C3 glomerulopathy: a new classification. Nat Rev Nephrol. 2010;6:494. doi: 10.1038/nrneph.2010.85. [DOI] [PubMed] [Google Scholar]
- 19.Kavanagh D, Richards A, Atkinson J. Complement regulatory genes and hemolytic uremic syndromes. Annu Rev Med. 2008;59:293. doi: 10.1146/annurev.med.59.060106.185110. [DOI] [PubMed] [Google Scholar]
- 20.Miwa T, Maldonado MA, Zhou L, Sun X, Luo HY, Cai D, Werth VP, Madaio MP, Eisenberg RA, Song WC. Deletion of decay-accelerating factor (CD55) exacerbates autoimmune disease development in MRL/lpr mice. Am J Pathol. 2002;161:1077. doi: 10.1016/S0002-9440(10)64268-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Powell MB, Marchbank KJ, Rushmere NK, van den Berg CW, Morgan BP. Molecular cloning, chromosomal localization, expression, and functional characterization of the mouse analogue of human CD59. J Immunol. 1997;158:1692. [PubMed] [Google Scholar]
- 22.Qin X, Miwa T, Aktas H, Gao M, Lee C, Qian YM, Morton CC, Shahsafaei A, Song WC, Halperin JA. Genomic structure, functional comparison, and tissue distribution of mouse Cd59a and Cd59b. Mamm Genome. 2001;12:582. doi: 10.1007/s00335-001-2060-8. [DOI] [PubMed] [Google Scholar]
- 23.Qian YM, Qin X, Miwa T, Sun X, Halperin JA, Song WC. Identification and functional characterization of a new gene encoding the mouse terminal complement inhibitor CD59. J Immunol. 2000;165:2528. doi: 10.4049/jimmunol.165.5.2528. [DOI] [PubMed] [Google Scholar]
- 24.Miwa T, Zhou L, Hilliard B, Molina H, Song WC. Crry, but not CD59 and DAF, is indispensable for murine erythrocyte protection in vivo from spontaneous complement attack. Blood. 2002;99:3707. doi: 10.1182/blood.v99.10.3707. [DOI] [PubMed] [Google Scholar]
- 25.Molina H, Miwa T, Zhou L, Hilliard B, Mastellos D, Maldonado MA, Lambris JD, Song WC. Complement-mediated clearance of erythrocytes: mechanism and delineation of the regulatory roles of Crry and DAF. Decay-accelerating factor. Blood. 2002;100:4544. doi: 10.1182/blood-2002-06-1875. [DOI] [PubMed] [Google Scholar]
- 26.Chan OT, Madaio MP, Shlomchik MJ. B cells are required for lupus nephritis in the polygenic, Fas-intact MRL model of systemic autoimmunity. J Immunol. 1999;163:3592. [PubMed] [Google Scholar]
- 27.Shlomchik MJ, Madaio MP, Ni D, Trounstein M, Huszar D. The role of B cells in lpr/lpr-induced autoimmunity. J Exp Med. 1994;180:1295. doi: 10.1084/jem.180.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lesher AM, Zhou L, Kimura Y, Sato S, Gullipalli D, Ruseva MM, Pickering MC, Herbert AP, Barlow PN, Eberhardt HU, Skerka C, F Zipfel P, Hamano T, Miwa T, Tung KS, Song W-C. Lethal murine C3 glomerulonephritis resembling human dense deposit disease caused by combined fH C-terminal mutation and properdin deficiency. 2012 Submitted. [Google Scholar]
- 29.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 30.Cohen PL, Eisenberg RA. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu Rev Immunol. 1991;9:243. doi: 10.1146/annurev.iy.09.040191.001331. [DOI] [PubMed] [Google Scholar]
- 31.Morse HC, 3rd, Davidson WF, Yetter RA, Murphy ED, Roths JB, Coffman RL. Abnormalities induced by the mutant gene Ipr: expansion of a unique lymphocyte subset. J Immunol. 1982;129:2612. [PubMed] [Google Scholar]
- 32.Zhang Z, V, Kyttaris C, Tsokos GC. The role of IL-23/IL-17 axis in lupus nephritis. J Immunol. 2009;183:3160. doi: 10.4049/jimmunol.0900385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Hu Q, Madri JA, Rollins SA, Chodera A, Matis LA. Amelioration of lupus-like autoimmune disease in NZB/WF1 mice after treatment with a blocking monoclonal antibody specific for complement component C5. Proc Natl Acad Sci U S A. 1996;93:8563. doi: 10.1073/pnas.93.16.8563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lublin DM, Atkinson JP. Decay-accelerating factor: biochemistry, molecular biology, and function. Annu Rev Immunol. 1989;7:35. doi: 10.1146/annurev.iy.07.040189.000343. [DOI] [PubMed] [Google Scholar]
- 35.Holt DS, Botto M, Bygrave AE, Hanna SM, Walport MJ, Morgan BP. Targeted deletion of the CD59 gene causes spontaneous intravascular hemolysis and hemoglobinuria. Blood. 2001;98:442. doi: 10.1182/blood.v98.2.442. [DOI] [PubMed] [Google Scholar]
- 36.Beurskens FJ, Kuenen JD, Hofhuis F, Fluit AC, Robins DM, Van Dijk H. Sex-limited protein: in vitro and in vivo functions. Clin Exp Immunol. 1999;116:395. doi: 10.1046/j.1365-2249.1999.00907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whitacre CC. Sex differences in autoimmune disease. Nat Immunol. 2001;2:777. doi: 10.1038/ni0901-777. [DOI] [PubMed] [Google Scholar]
- 38.Harris CL, Hanna SM, Mizuno M, Holt DS, Marchbank KJ, Morgan BP. Characterization of the mouse analogues of CD59 using novel monoclonal antibodies: tissue distribution and functional comparison. Immunology. 2003;109:117. doi: 10.1046/j.1365-2567.2003.01628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 40.Yamada K, Miwa T, Liu J, Nangaku M, Song WC. Critical protection from renal ischemia reperfusion injury by CD55 and CD59. J Immunol. 2004;172:3869. doi: 10.4049/jimmunol.172.6.3869. [DOI] [PubMed] [Google Scholar]
- 41.Carlucci F, Cortes-Hernandez J, Fossati-Jimack L, Bygrave AE, Walport MJ, Vyse TJ, Cook HT, Botto M. Genetic dissection of spontaneous autoimmunity driven by 129-derived chromosome 1 Loci when expressed on C57BL/6 mice. J Immunol. 2007;178:2352. doi: 10.4049/jimmunol.178.4.2352. [DOI] [PubMed] [Google Scholar]
- 42.Carlucci F, Fossati-Jimack L, Dumitriu IE, Heidari Y, Walport MJ, Szajna M, Baruah P, Garden OA, Cook HT, Botto M. Identification and characterization of a lupus suppressor 129 locus on chromosome 3. J Immunol. 2010;184:6256. doi: 10.4049/jimmunol.0901463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kimberley FC, Sivasankar B, Paul Morgan B. Alternative roles for CD59. Mol Immunol. 2007;44:73. doi: 10.1016/j.molimm.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 44.Harder T, Simons K. Clusters of glycolipid and glycosylphosphatidylinositol-anchored proteins in lymphoid cells: accumulation of actin regulated by local tyrosine phosphorylation. Eur J Immunol. 1999;29:556. doi: 10.1002/(SICI)1521-4141(199902)29:02<556::AID-IMMU556>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 45.Deckert M, Kubar J, Zoccola D, Bernard-Pomier G, Angelisova P, Horejsi V, Bernard A. CD59 molecule: a second ligand for CD2 in T cell adhesion. Eur J Immunol. 1992;22:2943. doi: 10.1002/eji.1830221128. [DOI] [PubMed] [Google Scholar]
- 46.Hahn WC, Menu E, Bothwell AL, Sims PJ, Bierer BE. Overlapping but nonidentical binding sites on CD2 for CD58 and a second ligand CD59. Science. 1992;256:1805. doi: 10.1126/science.1377404. [DOI] [PubMed] [Google Scholar]