

Abstract
Aging has been associated with mitochondrial DNA damage. P66Shc is an age-related adaptor protein that has a substantial impact on mitochondrial metabolism through regulation of the cellular response to oxidative stress. Our study aimed to establish a D-galactose (D-gal)-induced inner ear aging mouse model and to investigate the potential role of p66Shc and its serine 36-phosphorylated form in the inner ear during aging by using this model. Real-time PCR was performed to detect the mtDNA 3873-bp deletion and the level of p66Shc mRNA in the cochlear lateral wall. Western blot analysis was performed to analyze the total and mitochondrial protein levels of p66Shc and the level of Ser36-P-p66Shc in the cochlear lateral wall. Immunofluoresence was performed to detect the location of the Ser36-P-p66Shc expression in the cochlear lateral wall. The results showed that the accumulation of the mtDNA 3873-bp deletion, total and mitochondrial protein levels of p66Shc and level of Ser36-P-p66Shc were significantly increased in the cochlear lateral wall of the D-gal-treated group when compared to the control group and that Ser36-P-p66Shc was mainly localized in the cytoplasm of the cells in the stria vascularis. During aging, the oxidative stress-related increase of p66Shc and Ser36-P-p66Shc might be associated with the accumulation of the mtDNA 3873-bp deletion in the inner ear.
Introduction
Age-related hearing loss (ARHL) or presbycusis is the most common sensory deficit in the elderly, and it has become a severe social and health problem. Although many theories underlying presbycusis have been proposed, the overall mechanisms remain speculative. Among these theories, many studies support the idea that a mitochondrial malfunction plays an important role in presbycusis [1]–[3]. The mitochondrion is the organelle that generates the high energy intermediate ATP and is also a major source and major target of reactive oxygen species (ROS) [4], [5]. ROS that originates from the mitochondrial respiratory chain can damage macromolecules, especially mitochondrial DNA (mtDNA) [6]. Mammalian mtDNA is a closed circular molecule of approximately 16 kb and encodes many critical components of the oxidative phosphorylation pathway, which is responsible for generating ATP for the cell. Thus, a deletion in the mtDNA would lead to a mitochondrial malfunction. The 3867-bp deletion in mice that corresponds to the mtDNA 4977-bp deletion in humans and the mtDNA 4834-bp deletion in rats, which are also called common deletions (CD), is the most common mtDNA damage associated with aging [7]–[9]. According to the NCBI reference sequence for mtDNA (NC_005089.1), there are 6 bp added to the mtDNA 3867-bp deletion. Thus, it is more accurately a mtDNA 3873-bp deletion. The mtDNA 3873-bp deletion occurs within nt9089– nt12961 or nt9104– nt12976, which are flanked by a 15-bp direct-repeat sequence (AGCCCTACTAATTAC), and results in a partial to complete loss of the following mtDNA genes coding for components of oxidative phosphorylation (OXPHOS): COX3, ND3, ND4L, ND4, ND5 and 5 tRNAs (tRNA-Gly, tRNA-Arg, tRNA-His, tRNA-Ser and tRNA-Leu). Previous studies have suggested that the mtDNA deletion is associated with pathological disorders of presbycusis in human beings, rats and mice [10]–[13]. We have previously utilized overdoses of D-galactose (D-gal) to induce an aging model of rats, which harbor increased amounts of the mtDNA 4834-bp deletion in the central and peripheral auditory system [14]–[19]. Moreover, we found that below a certain level, the mtDNA 4834-bp deletion might not directly lead to a hearing impairment but rather act as a predisposing factor that can greatly enhance the sensitivity of the inner ear to aminoglycoside antibiotics [17]. These findings suggest that the mtDNA deletion plays an important role in presbycusis. As the inner ear tissue of live humans is inaccessible and the genetic and environmental background of individuals with hearing loss is nonhomogeneous, studies on presbycusis are limited. The mouse is one of the mammals, after humans, whose genome has been sequenced. Additionally, approximately 99% of human genes have homologs in the mouse [20]. Thus, compared to the D-gal-induced aging rat, the D-gal-induced aging mouse would provide a more ideal model to explore the potential mechanisms involved in inner ear aging.
The p66Shc protein is a Src homologue and collagen homologue (Shc) adaptor protein, which has the ability to interact with other proteins that mediate cell signaling [21]. It is encoded by the mammalian shcA gene, which also encodes the two other ShcA adaptor proteins p46Shc and p52Shc that are named for their molecular masses [22]. However, only the p66Shc isoform participates in mitochondrial ROS generation and acts as an oxidative signal regulator, which translates oxidative signals to the mitochondria [23], [24]. P66shc-null mice display an increased lifespan and enhanced resistance to oxidative stress [25]. Upon oxidative stress, p66Shc is phosphorylated at a serine residue (Ser36), and the phosphorylation of Ser36 is necessary for the cellular response to oxidative stress [25]. A portion of p66Shc partially localizes within the membrane space of the mitochondria where it oxidizes cytochrome c (Cytc), thus producing ROS [23]. Since the publication by Migliaccio, the p66Shc protein has been recognized as an important element of the free radical theory of aging [24], and many age-related diseases were suggested to associate with p66Shc [26]–[29]. However, no study of p66Shc in presbycusis has previously been reported.
As the stria vascularis is an essential structure of the inner ear that contains abundant mitochondria and has the highest rate of aerobic oxidation in the inner ear [30], [31], we focused on the cochlear lateral wall as the major subject of our study. We established a D-gal-induced aging mouse model that harbored increased levels of the mtDNA 3873-bp deletion in the cochlear lateral walls and investigated the total and mitochondrial levels of p66Shc and the Ser36-phosphorylated form of p66Shc (Ser36-P-p66Shc) in this aging model. Our results suggested that p66Shc and Ser36-P-p66Shc might play a critical role in inner ear aging and the formation and accumulation of mtDNA damage in the inner ear.
Materials and Methods
Animals and Treatment
One hundred and two male Kunming mice (median body weight of 22 g; 7–8 weeks old) with normal hearing were purchased from the Laboratory Animal Center of Tongji Medical College of Huazhong University of Science and Technology. The Kunming mouse strain is an outbred strain of the Swiss mouse strain, which came to Kunming, China in 1944 from the Indian Haffkine Institute, and was widely used for the D-galactose-induced aging mice [32]–[36]. The mice were acclimated to the new environment (23°C; 12-h light/dark cycle) for at least one week before the start of the experiments. All the animals had free access to water and food. The animals were randomly assigned to three groups (n = 34 for each group) depending on the dose of D-gal (Sigma Chemical, St. Louis, MO), as follows: a low dose (LD) group, high dose (HD) group and control group. Mice were treated with a daily subcutaneous injection of the D-gal solutions (LD group, 800 mg/kg of D-gal; HD group, 1000 mg/kg of D-gal) or an equal volume of normal saline (NS) (for control group) in the neck for 10 weeks. The animal studies were performed in accordance with the guidelines for the care and use of laboratory animals that were prepared by the Institution of Laboratory Animals of Huazhong University of Science and Technology. The protocol was approved by the Committee on the Ethics of Animal Experiments of Huazhong University of Science and Technology (Permit Number: S244). All surgeries were performed under a mixture of ketamine and chlorpromazine anesthesia, and all efforts were made to minimize suffering.
Auditory Brainstem Response
At the beginning and at the end of the treatment, the hearing functions of the mice were tested by auditory brainstem responses (ABR). Mice (n = 10 for each group) were anaesthetized with a mixture of ketamine (120 mg/kg) and chlorpromazine (20 mg/kg) and placed on a heated mat in an audiometric chamber. The sound delivery tube of the insert earphone was tightly fitted into the external auditory canal. Electrodes were placed subdermally over the vertex (active), right mastoid (reference) and left mastoid (ground). The ABR responses were measured with a tone burst stimulus at 4, 8, 16 and 32 kHz using a computer-based signal averaging system (Tucker-Davis Technology, Alachua, FL, USA), as Yu described previously [37]. The threshold was verified by two unblinded investigators.
Tissue Sample Preparation
Mice were killed after the last subcutaneous injections. All animals were euthanized under deep anesthesia with chlorpromazine and ketamine hydrochloride, and the temporal bones and plasma were collected. For immunofluorescence experiments, the cochleae from 12 animals (n = 4 from each group) were perfused slowly with cold 4% paraformaldehyde in phosphate-buffered saline (PBS) and then kept in the same fixative at 4°C overnight. Following fixation, the cochleae were decalcified with disodium EDTA. Then, the decalcified cochleae were paraffin embedded, and 5-µm sections were cut and mounted on glass slides for analysis of immunofluorescent labeling. For experiments involving DNA, total RNA and total or mitochondrial protein detection, the cochlear lateral wall, which included the stria vascularis and spiral ligament, was isolated under a stereomicroscope (Zeiss, Germany). Then, the DNA and total RNA from 18 animals (n = 6 from each group) were extracted with a genomic DNA Kit (Tiangen, China) and RNAiso Plus reagent (TaKaRa, Japan), respectively. cDNA was reverse transcribed using a PrimeScript® RT reagent kit (Perfect Real Time; TaKaRa, Japan). The total protein from 18 animals (n = 6 from each group) was isolated with RIPA lysis buffer (Beyotime, China). The mitochondrial proteins from 36 animals (n = 12 from each group) were isolated with the Tissue Mitochondria Isolation Kit (Beyotime, China). Protein concentrations were determined with the BCA Protein Assay Kit (Beyotime, China), with BSA as the standard.
SOD Activity and MDA Assay of Plasma
The SOD (Superoxide Dismutase) activity and MDA (Malondialdehyde) levels in the plasma were quantified with colorimetric kits (Jiancheng, China) according to the manufacturer’s instructions [17].
Real-time PCR
To evaluate the mtDNA damage induced by D-gal, a sequence designed according to the description of Brossas et al. [38], which represented the new fusion sequence (NFS) that is present in only the deleted mtDNA, was used as a measurement of the mtDNA 3873-bp deletion. Because the D-loop region of mtDNA is rarely deleted, it can represent the conserved segment and be used to determine the copy number of mtDNA. All gene-specific primers used in this study were designed and synthesized at TaKaRa Biotechnology (Dalian) Co. Ltd. and were previously tested for optimal efficiency. The primer pairs for the mtDNA 3873-bp deletion and D-loop were as follows: NFS forward, 5′-CGAAACCACATAAATCAAGCCCTAC-3′; NFS reverse, 5′-AATGATTCGTATGCTGTACATAGCTGTT-3′; D-loop forward, 5′-GGGCTGATTAGACCCGATACCAT-3′; and D-loop reverse, 5′-TACCATCCTCCGTGAAACCAACA-3′. The primer pairs for p66Shc and the internal standard (β-actin) were as follows: p66Shc forward, 5′-CCGACTACCTGTGTTCCTTCTT-3′; p66Shc reverse, 5′-CCCATCTTCAGCAGCCTTTCC-3′; β-actin forward, 5′-CATCCGTAAAGACCTCTATGCCAAC-3′; and β-actin reverse, 5′-ATGGAGCCACCGATCCACA-3′. The DNA and cDNA products were subjected to real-time PCR with the ABI StepOnePlus™ instrument equipped with StepOne software (ABI) for 40 cycles (95°C for 30 s, 95°C for 5 s and 60°C for 5 s) by using the SYBR® Premix Ex Taq™ (Tli RNaseH Plus) kit (TaKaRa, Japan). Each reaction was performed in duplicate and analyzed by melting curves at the end to confirm specificity. The products obtained with primers for detection of the mtDNA 3873-bp deletion were excised from a gel. Then, the products were gel purified, cloned and analyzed using an ABI Prism 377 DNA sequencer (Applied Biosystems, Foster City, CA). The ΔCt method (Ctdeletion-CtD-loop) was used to calculate the abundance of the mtDNA deletion. The relative amount of the mtDNA 3873-bp deletion and the mRNA levels of p66Shc were calculated with the 2−ΔΔCt method.
Western Blot
We detected the total protein level of p66Shc, p52Shc and p46Shc by using an antibody against SHC (Abcam, USA), which detects the three isoforms simultaneously by binding to a similar domain in all three proteins. The total protein level of SHC and the level of Ser36-P-p66Shc (p66Shc phosphorylated at serine 36) were measured with the corresponding antibodies and normalized to the level of glyceraldehyde-3-phosphate dehydrogenase (GADPH). The mitochondrial p66Shc levels were measured with the SHC antibody and normalized to the voltage-dependent anion channel (VDAC1). For this analysis, 20 µg of protein was separated by 10% or 12% SDS-PAGE and then transferred to nitrocellulose membranes. The SHC and Ser36-P-p66Shc levels were detected after incubation with primary antibodies (SHC, Ser36-P-p66Shc and VDAC1, 1∶1000, Abcam, USA; GAPDH, 1∶5000, Kangchen, China), which was followed by an incubation with a horseradish peroxidase (HRP)-conjugated goat anti-rabbit (1∶5000, Santa Cruz, USA) or goat anti-mouse (1∶2000, Santa Cruz, USA) immunoglobulin, as appropriate. The immunoreactivity was determined using the ECL reaction kit (Beyotime, China) according to the manufacturer’s instructions by exposure on medical film. The band density was quantified with the Image-Pro Plus 6.0 software (Media Cybernetics, Inc., USA).
Immunofluorescent Labeling
After deparaffinization, rehydration, antigen retrieval and nonspecific antigen site blocking, immunofluorescence was performed on the sections overnight at 4°C with the monoclonal anti-Ser36-P-p66Shc antibody (1∶40, Abcam, USA), which were then incubated for 1 h with the fluorescently tagged secondary antibody. Control staining was performed without the primary antibody. Images were taken with a laser scanning confocal microscope (Nikon, Japan).
Statistical Analysis
All data are presented as the mean±standard deviation (Mean±S.D.). Statistical analyses were performed with the SPSS 13.0 software (IBM, Armonk, NY, USA). One-way analysis of variance (ANOVA) was used for comparisons of the relative expression levels for the different groups. The least significant difference (LSD) post-hoc test was used to compare differences between two of the groups. Differences with a p-value<0.05 were considered to be statistically significant.
Results
D-gal did not Induce Hearing Loss
To determine the effect of the mtDNA 3873-bp deletion on hearing, the auditory status was evaluated with ABR after the last D-gal administration. There was no significant difference in the ABR threshold (Table 1) among the three experimental groups (p>0.05).
Table 1. The ABR threshold (dB SPL).
| Frequency | NS group (n = 10) | LD group (n = 10) | HD group (n = 10) |
| 4 kHz | 39.0±3.94 | 38.5±5.80 | 38.0±6.75 |
| 8 kHz | 30.0±4.83 | 30.5±5.50 | 27.5±4.86 |
| 16 kHz | 23.0±4.22 | 23.0±4.83 | 22.5±4.25 |
| 32 kHz | 34.5±4.38 | 37.0±4.25 | 36.0±3.16 |
The data are expressed as mean±standard deviation. (NS, the group treated with normal saline; LD, the group treated with 800 mg/kg D-gal; HD, the group treated with 1000 mg/kg D-gal. p>0.05 when compared to the NS group.).
Decreased SOD Activity and Increased MDA Levels in Plasma Induced by D-gal
The SOD activity in the plasma of mice treated with the low and high doses of D-gal was 170.67±19.11 U/ml and 146.52±13.42 U/ml, respectively, which was significantly lower than that of the control group (198.62±10.35 U/ml) (p<0.01). The MDA levels in the plasma of mice treated with the low and high doses of D-gal were 2.52±0.49 µmol/ml and 3.25±0.47 µmol/ml, respectively, which were significantly higher than that of the control group (1.22±0.20 µmol/ml) (p<0.01) (Table 2).
Table 2. The plasma SOD activity and MDA levels.
| NS group (n = 6) | LD group (n = 6) | HD group (n = 6) | |
| T-SOD (U/ml) | 198.62±10.35 | 170.67±19.11** | 146.52±13.42**# |
| MDA (µmol/ml) | 1.22±0.20 | 2.52±0.49** | 3.25±0.47**∧ |
The data are expressed as the mean±standard deviation. (NS, the group treated with normal saline; LD, the group treated with 800 mg/kg D-gal; HD, the group treated with 1000 mg/kg D-gal. **, p<0.01 when compared to the NS group; #, p<0.05 when compared to the LD group; ∧, p<0.01 when compared to the LD group.).
Accumulation of the Mitochondrial DNA 3873-bp Deletion Induced by D-gal
The amount of mtDNA 3873-bp deletion was significantly increased in the cochlear lateral wall of the D-gal-treated groups. Compared to the control group, the accumulation of the mtDNA 3873-bp deletion in the low and high dose D-gal groups was increased by approximately 1.70-fold and 2.30-fold, respectively (Fig. 1). The products obtained with the primers for the mtDNA 3873-bp deletion were sequenced to prove the presence of the mtDNA 3873-bp deletion (Fig. 2).
Figure 1. The effect of D-gal on the amount of mtDNA 3873-bp deletion in the cochlear lateral wall of the experimental groups.

The accumulation of the mtDNA 3873-bp deletion in the D-gal-treated groups was significantly higher when compared to the control group. (NS, the group treated with normal saline; LD, the group treated with 800 mg/kg D-gal; HD, the group treated with 1000 mg/kg D-gal. **, p<0.01 when compared to the NS group.).
Figure 2. Schematic diagram and sequences of the mtDNA 3873-bp deletion (nt9089–nt12961 or nt9104–nt12976).

Bold black letters indicate the nucleotide sequences flanking the breakpoints of the deleted mtDNA, and shaded letters indicate the direct repeats. The sequences of the PCR products are shown below the schematic diagram. The arrowheads point to the possible breakpoints.
Increased mRNA Levels of p66Shc Induced by D-gal
The mRNA levels were significantly increased in the cochlear lateral wall of the D-gal-treated groups (Fig. 3). Compared to the control group, the relative expression levels in the low and high dose D-gal-treated groups were increased approximately 1.53-fold and 2.12-fold, respectively.
Figure 3. The effect of D-gal on the mRNA levels of p66Shc in the cochlear lateral wall of the experimental groups.
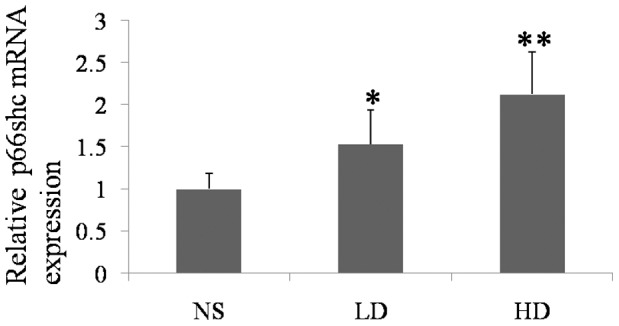
With the increasing doses of D-gal, the mRNA levels of p66Shc were increased. (NS, the group treated with normal saline; LD, the group treated with 800 mg/kg D-gal; HD, the group treated with 1000 mg/kg D-gal. *, p<0.05 when compared to the NS group. **, p<0.01 when compared to the NS group.).
Increased Total Protein Levels of p66Shc and Ser36-P-p66Shc Induced by D-gal
The result showed that the total protein level of p66Shc was increased in the cochlear lateral wall of the D-gal-treated groups. However, the relative changes of the total protein levels of p52Shc and p46Shc between the groups were not statistically significant (p>0.05). The levels of Ser36-P-p66Shc were increased in the cochlear lateral wall of the D-gal-treated groups (Fig. 4A and B). Compared to the control group, the total protein levels of p66Shc in the low and high dose groups were increased approximately 2.59-fold and 3.58-fold, respectively, and the levels of Ser-P-p66Shc were increased approximately 2.48- and 3.39-fold, respectively.
Figure 4. The expression of total SHC protein and Ser36-P-p66Shc in the cochlear lateral wall of the experimental groups.

A, Western blot assessment of the protein expression of SHC and Ser36-P-p66Shc in the NS, LD and HD groups. B, Semiquantitative representation of the total protein levels of p66Shc and Ser36-P-p66Shc compared to the control group. C, Cochlea sections were subjected to immunofluorescent labeling of Ser36-P-p66Shc; Ser36-P-p66Shc was mainly expressed in the cytoplasm of cells in the stria vascularis. (NS, the group treated with normal saline; LD, the group treated with 800 mg/kg D-gal; HD, the group treated with 1000 mg/kg D-gal. STV, stria vascularis; SL, spiral ligament. **, p<0.01 when compared to the NS group.).
In addition, we analyzed the location of the Ser36-P-p66Shc expression by using immunofluorescent labeling. We found that Ser36-P-p66Shc was mainly expressed in the cytoplasm of the cells in the stria vascularis, and it was increased with the increasing doses of D-gal (Fig. 4C).
Increased Mitochondrial Protein Level of p66Shc Induced by D-gal
To detect the protein levels of p66Shc in the mitochondria, we utilized western blot analysis. The mitochondrial protein levels of p66Shc were significantly increased in the cochlear lateral wall of the D-gal-treated mice (Fig. 5A). Compared to the control group, the mitochondrial protein levels of p66Shc in the D-gal-treated groups were increased approximately 2.21-fold and 3.22-fold in the low and high dose groups, respectively (Fig. 5B).
Figure 5. The mitochondrial protein expression of p66Shc in the cochlear lateral wall of the experimental groups.

A, Western blot assessment of the protein expression of p66Shc in the NS, LD and HD groups. B, Semiquantitative representation of the relative abundance of the mitochondrial protein levels of p66Shc in D-gal-treated groups compared to the control group. (NS, the group treated with normal saline; LD, the group treated with 800 mg/kg D-gal; HD, the group treated with 1000 mg/kg D-gal. **, p<0.01 when compared to the NS group.).
Discussion
According to the oxidative stress theory by Denham Harman, the age-related loss of physiological function is due to the progressive accumulation of oxidative damage, which ultimately determines the lifespan of an organism [39]. In this study, we used an overdose of D-gal that was thought to result in the generation of superoxide anions and oxygen-derived free radicals and the formation of advanced glycation end products (AGEs) [40]–[42], which triggers excess ROS and abnormally high oxidative stress, to accelerate the aging of mice. We demonstrated that the plasma SOD activity was decreased and the MDA level in plasma was increased in the D-gal-treated mice. The changes of these two age-related biochemical markers were similar to those that occur during the natural aging process in humans and other animals [43]–[45]. The CD is an important biomarker of aging [46]–[48]. Our previous study on the D-gal-induced rats that mimic aging reported that the amount of CD in the inner ear of the high dose D-gal group was increased 3.69-fold compared to the control group [18]. Zhang et al. [13] reported that the level of the CD in the inner ear of 17- to 19-month-old mice was increased 3.32 fold compared to 7- to 10-month-old mice. In this study, we demonstrated that the amount of CD in the cochlear lateral wall of the high dose D-gal group was increased 2.30-fold compared to the control group. These data indicated that we established a mouse model of aging in the inner ear by using D-gal, which was similar to the D-gal-induced inner ear aging rat model. Corresponding to our previous study in rats [16], [17], [19], there was no significant difference in the ABR threshold among the three experimental groups. The result of the mouse aging model further confirmed the result from the rat aging model that, below a certain level, the mtDNA deletion does not directly lead to a hearing impairment [16], [17], [19].
Most of the p66Shc protein is distributed throughout the cytosol, while a fraction of p66Shc localizes within the inner membrane and intermembrane space of the mitochondria; additionally, oxidative stress stimulates an increase in the mitochondrial p66Shc level [23], [49]–[51]. Protein kinase C β (PKCβ) is activated by oxidative stress and phosphorylates p66Shc on the Ser36 residue, which triggers its translocation to the mitochondria [52], [53]. Our data showed that the mRNA and total protein levels of p66Shc and levels Ser36-P-p66Shc were significantly increased in the cochlear lateral wall of the D-gal-induced aging mice. High amounts of Ser36-P-p66Shc indicate a high activation of the PKCβ pathway and higher oxidative stress in a tissue [54], [55]. Thus, the increased levels of Ser36-P-p66Shc indicated higher oxidative stress in the cochlea lateral wall of the D-gal-induced aging mice. Furthermore, we analyzed the location of the Ser36-P-p66Shc expression in the cochlear lateral wall by using immunofluorescent labeling. The results showed that Ser36-P-p66Shc was mainly expressed in the cytoplasm of the cells in the stria vascularis, which also indicates that there is higher oxidative stress in the stria vascularis. Migliaccio et al. [25] reported that their study of mouse embryo fibroblasts showed that p66Shc was activated upon exposure to agents that induce oxidative stress. Our results further established a positive correlation between the levels of oxidative stress and the amount of p66Shc in the cochlear lateral wall of mice. Consistent with the Ser36-P-p66Shc level, which was increased in the D-gal-induced aging mice, the mitochondrial protein levels of p66Shc were also increased. This result indicates that p66Shc was translocated to the mitochondria in the D-gal-induced aging mice. Pinton et al. [56] reported that phosphorylation of p66Shc at Ser36 triggers a mitochondrial accumulation of p66Shc. It was reported that before Ser36-P-p66Shc is translocated to the mitochondria, it is isomerized by the peptidyl-prolyl isomerase Pin1 and then recognized and dephosphorylated by the phosphatase PP2A [56], [57]. Thus, mitochondrial p66Shc is unphosphorylated.
In the intermembrane space of the mitochondria, p66Shc oxidizes Cytc, resulting in the generation of H2O2 via the transfer of electrons from the reduced Cytc to oxygen and, thus, increasing the intracellular ROS level [23], which has been suggested to lead to the accumulation of the mtDNA 3873-bp deletion [58]. Trinei et al. [59] reported that the mtDNA 3873-bp deletion was detected in liver samples of wildtype mice, while it was barely detectable in matched tissues from p66Shc−/− mice. This result indicates that the p66Shc expression was correlated with the level of the mtDNA 3873-bp deletion. Therefore, our data indicating that oxidative stress caused an increased expression of p66Shc and Ser36-P-p66Shc and the translocation of p66Shc to the mitochondria may be associated with the accumulation of the mtDNA 3873-bp deletion in the inner ear.
In conclusion, we established a D-gal-induced inner ear aging mouse model in this study and demonstrated the accumulation of the mtDNA 3873-bp deletion in the lateral wall of the D-gal-treated mice, which may be associated with an oxidative stress-related increase in the expression of p66Shc and Ser36-P-p66Shc and the translocation of p66Shc to the mitochondria. Considering that oxidative stress has been associated with numerous human aging processes in addition to presbycusis, the role of p66Shc as a signaling agent may have important therapeutic implications.
Funding Statement
This work was supported by grants from the Major State Basic Research Development Program of China (973 Program) (2011CB504504) and the National Nature Science Foundation of China (30730094 and 81000408). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Someya S, Prolla TA (2010) Mitochondrial oxidative damage and apoptosis in age-related hearing loss. Mech Ageing Dev 131: 480–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yamasoba T, Someya S, Yamada C, Weindruch R, Prolla TA, et al. (2007) Role of mitochondrial dysfunction and mitochondrial DNA mutations in age-related hearing loss. Hear Res 226: 185–193. [DOI] [PubMed] [Google Scholar]
- 3. Seidman MD, Khan MJ, Bai U, Shirwany N, Quirk WS (2000) Biologic activity of mitochondrial metabolites on aging and age-related hearing loss. Am J Otol 21: 161–167. [DOI] [PubMed] [Google Scholar]
- 4. Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408: 239–247. [DOI] [PubMed] [Google Scholar]
- 5. Beckman KB, Ames BN (1998) The free radical theory of aging matures. Physiol Rev 78: 547–581. [DOI] [PubMed] [Google Scholar]
- 6. Jang YC, Remmen VH (2009) The mitochondrial theory of aging: insight from transgenic and knockout mouse models. Exp Gerontol 44: 256–260. [DOI] [PubMed] [Google Scholar]
- 7. Mohamed SA, Hanke T, Erasmi AW, Bechtel MJ, Scharfschwerdt M, et al. (2006) Mitochondrial DNA deletions and the aging heart. Exp Gerontol 41: 508–517. [DOI] [PubMed] [Google Scholar]
- 8. Nicklas JA, Brooks EM, Hunter TC, Single R, Branda RF (2004) Development of a quantitative PCR (TaqMan) assay for relative mitochondrial DNA copy number and the common mitochondrial DNA deletion in the rat. Environ Mol Mutagen 44: 313–320. [DOI] [PubMed] [Google Scholar]
- 9. Zeng Z, Zhang Z, Yu H, Corbley MJ, Tang Z, et al. (1999) Mitochondrial DNA deletions are associated with ischemia and aging in Balb/c mouse brain. J Cell Biochem 73: 545–553. [PubMed] [Google Scholar]
- 10. Seidman MD, Bai U, Khan MJ, Murphy MJ, Quirk WS, et al. (1996) Association of mitochondrial DNA deletions and cochlear pathology: a molecular biologic tool. Laryngoscope 106: 777–783. [DOI] [PubMed] [Google Scholar]
- 11. Seidman MD, Bai U, Khan MJ, Quirk WS (1997) Mitochondrial DNA deletions associated with aging and presbyacusis. Arch Otolaryngol Head Neck Surg 123: 1039–1045. [DOI] [PubMed] [Google Scholar]
- 12. Zhang X, Han D, Ding D, Dai P, Yang W, et al. (2002) Deletions are easy detectable in cochlear mitochondrial DNA of Cu/Zn superoxide dismutase gene knockout mice. Chin Med J (Engl) 115: 258–263. [PubMed] [Google Scholar]
- 13. Zhang X, Han D, Ding D, Dai P, Yang W, et al. (2002) Cochlear mitochondrial DNA3867bp deletion in aged mice. Chin Med J (Engl) 115: 1390–1393. [PubMed] [Google Scholar]
- 14. Chen B, Zhong Y, Peng W, Sun Y, Kong WJ (2010) Age-related changes in the central auditory system: comparison of D-galactose-induced aging rats and naturally aging rats. Brain Res 1344: 43–53. [DOI] [PubMed] [Google Scholar]
- 15. Chen B, Zhong Y, Peng W, Sun Y, Hu YJ, et al. (2011) Increased mitochondrial DNA damage and decreased base excision repair in the auditory cortex of D-galactose-induced aging rats. Mol Biol Rep 38: 3635–3642. [DOI] [PubMed] [Google Scholar]
- 16. Kong WJ, Wang Y, Wang Q, Hu YJ, Han YC, et al. (2006) The relation between D-galactose injection and mitochondrial DNA 4834 bp deletion mutation. Exp Gerontol 41: 628–634. [DOI] [PubMed] [Google Scholar]
- 17. Kong WJ, Hu YJ, Wang Q, Wang Y, Han YC, et al. (2006) The effect of the mtDNA4834 deletion on hearing. Biochem Biophys Res Commun 344: 425–430. [DOI] [PubMed] [Google Scholar]
- 18. Zhong Y, Hu YJ, Chen B, Peng W, Sun Y, et al. (2011) Mitochondrial transcription factor A overexpression and base excision repair deficiency in the inner ear of rats with D-galactose-induced aging. FEBS J 278: 2500–2510. [DOI] [PubMed] [Google Scholar]
- 19. Zhong Y, Hu YJ, Yang Y, Peng W, Sun Y, et al. (2011) Contribution of common deletion to total deletion burden in mitochondrial DNA from inner ear of d-galactose-induced aging rats. Mutat Res 712: 11–19. [DOI] [PubMed] [Google Scholar]
- 20. Boguski MS (2002) Comparative genomics: the mouse that roared. Nature 420: 515–516. [DOI] [PubMed] [Google Scholar]
- 21. Rajendran M, Thomes P, Zhang L, Veeramani S, Lin MF (2010) p66Shc–a longevity redox protein in human prostate cancer progression and metastasis: p66Shc in cancer progression and metastasis. Cancer Metastasis Rev 29: 207–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, et al. (1992) A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell 70: 93–104. [DOI] [PubMed] [Google Scholar]
- 23. Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, et al. (2005) Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122: 221–233. [DOI] [PubMed] [Google Scholar]
- 24. Migliaccio E, Mele S, Salcini AE, Pelicci G, Lai KM, et al. (1997) Opposite effects of the p52shc/p46shc and p66shc splicing isoforms on the EGF receptor-MAP kinase-fos signalling pathway. EMBO J 16: 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, et al. (1999) The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 402: 309–313. [DOI] [PubMed] [Google Scholar]
- 26. Berry A, Capone F, Giorgio M, Pelicci PG, de Kloet ER, et al. (2007) Deletion of the life span determinant p66Shc prevents age-dependent increases in emotionality and pain sensitivity in mice. Exp Gerontol 42: 37–45. [DOI] [PubMed] [Google Scholar]
- 27. Percy CJ, Brown L, Power DA, Johnson DW, Gobe GC (2009) Obesity and hypertension have differing oxidant handling molecular pathways in age-related chronic kidney disease. Mech Ageing Dev 130: 129–138. [DOI] [PubMed] [Google Scholar]
- 28. Rota M, LeCapitaine N, Hosoda T, Boni A, De Angelis A, et al. (2006) Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66shc gene. Circ Res 99: 42–52. [DOI] [PubMed] [Google Scholar]
- 29. Ljubicic V, Menzies KJ, Hood DA (2010) Mitochondrial dysfunction is associated with a pro-apoptotic cellular environment in senescent cardiac muscle. Mech Ageing Dev 131: 79–88. [DOI] [PubMed] [Google Scholar]
- 30. Yao X, Rarey KE (1996) Detection and regulation of Cu/Zn-SOD and Mn-SOD in rat cochlear tissues. Hear Res 96: 199–203. [DOI] [PubMed] [Google Scholar]
- 31. VOSTEEN KH (1960) The histochemistry of the enzymes of oxygen metabolism in the inner ear. Trans Southeast Sect Am Urol Assoc 1960: 230–241. [PubMed] [Google Scholar]
- 32. Lu J, Wu DM, Hu B, Zheng YL, Zhang ZF, et al. (2010) NGF-Dependent activation of TrkA pathway: A mechanism for the neuroprotective effect of troxerutin in D-galactose-treated mice. Brain Pathol 20: 952–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lu J, Wu DM, Zheng YL, Hu B, Zhang ZF, et al. (2010) Ursolic acid attenuates D-galactose-induced inflammatory response in mouse prefrontal cortex through inhibiting AGEs/RAGE/NF-kappaB pathway activation. Cereb Cortex 20: 2540–2548. [DOI] [PubMed] [Google Scholar]
- 34. Zhang ZF, Lu J, Zheng YL, Hu B, Fan SH, et al. (2010) Purple sweet potato color protects mouse liver against d-galactose-induced apoptosis via inhibiting caspase-3 activation and enhancing PI3K/Akt pathway. Food Chem Toxicol 48: 2500–2507. [DOI] [PubMed] [Google Scholar]
- 35. Shan Q, Lu J, Zheng Y, Li J, Zhou Z, et al. (2009) Purple sweet potato color ameliorates cognition deficits and attenuates oxidative damage and inflammation in aging mouse brain induced by d-galactose. J Biomed Biotechnol 2009: 564737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu CM, Ma JQ, Lou Y (2010) Chronic administration of troxerutin protects mouse kidney against D-galactose-induced oxidative DNA damage. Food Chem Toxicol 48: 2809–2817. [DOI] [PubMed] [Google Scholar]
- 37. Sun Y, Tang W, Chang Q, Wang Y, Kong W, et al. (2009) Connexin30 null and conditional connexin26 null mice display distinct pattern and time course of cellular degeneration in the cochlea. J Comp Neurol 516: 569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brossas JY, Barreau E, Courtois Y, Treton J (1994) Multiple deletions in mitochondrial DNA are present in senescent mouse brain. Biochem Biophys Res Commun 202: 654–659. [DOI] [PubMed] [Google Scholar]
- 39. HARMAN D (1956) Aging: a theory based on free radical and radiation chemistry. J Gerontol 11: 298–300. [DOI] [PubMed] [Google Scholar]
- 40. Gong Y, Liu L, Xie B, Liao Y, Yang E, et al. (2008) Ameliorative effects of lotus seedpod proanthocyanidins on cognitive deficits and oxidative damage in senescence-accelerated mice. Behav Brain Res 194: 100–107. [DOI] [PubMed] [Google Scholar]
- 41. Lu J, Wu DM, Hu B, Cheng W, Zheng YL, et al. (2010) Chronic administration of troxerutin protects mouse brain against D-galactose-induced impairment of cholinergic system. Neurobiol Learn Mem 93: 157–164. [DOI] [PubMed] [Google Scholar]
- 42. Wu DM, Lu J, Zheng YL, Zhou Z, Shan Q, et al. (2008) Purple sweet potato color repairs d-galactose-induced spatial learning and memory impairment by regulating the expression of synaptic proteins. Neurobiol Learn Mem 90: 19–27. [DOI] [PubMed] [Google Scholar]
- 43. Ho SC, Liu JH, Wu RY (2003) Establishment of the mimetic aging effect in mice caused by D-galactose. Biogerontology 4: 15–18. [DOI] [PubMed] [Google Scholar]
- 44. Song X, Bao M, Li D, Li YM (1999) Advanced glycation in D-galactose induced mouse aging model. Mech Ageing Dev 108: 239–251. [DOI] [PubMed] [Google Scholar]
- 45. Inal ME, Kanbak G, Sunal E (2001) Antioxidant enzyme activities and malondialdehyde levels related to aging. Clin Chim Acta 305: 75–80. [DOI] [PubMed] [Google Scholar]
- 46. Eshaghian A, Vleugels RA, Canter JA, McDonald MA, Stasko T, et al. (2006) Mitochondrial DNA deletions serve as biomarkers of aging in the skin, but are typically absent in nonmelanoma skin cancers. J Invest Dermatol 126: 336–344. [DOI] [PubMed] [Google Scholar]
- 47. Meissner C, Bruse P, Mohamed SA, Schulz A, Warnk H, et al. (2008) The 4977 bp deletion of mitochondrial DNA in human skeletal muscle, heart and different areas of the brain: a useful biomarker or more? Exp Gerontol 43: 645–652. [DOI] [PubMed] [Google Scholar]
- 48. Dumont P, Burton M, Chen QM, Gonos ES, Frippiat C, et al. (2000) Induction of replicative senescence biomarkers by sublethal oxidative stresses in normal human fibroblast. Free Radic Biol Med 28: 361–373. [DOI] [PubMed] [Google Scholar]
- 49. Ventura A, Maccarana M, Raker VA, Pelicci PG (2004) A cryptic targeting signal induces isoform-specific localization of p46Shc to mitochondria. J Biol Chem 279: 2299–2306. [DOI] [PubMed] [Google Scholar]
- 50. Orsini F, Migliaccio E, Moroni M, Contursi C, Raker VA, et al. (2004) The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J Biol Chem 279: 25689–25695. [DOI] [PubMed] [Google Scholar]
- 51. Nemoto S, Combs CA, French S, Ahn BH, Fergusson MM, et al. (2006) The mammalian longevity-associated gene product p66shc regulates mitochondrial metabolism. J Biol Chem 281: 10555–10560. [DOI] [PubMed] [Google Scholar]
- 52. Gopalakrishna R, Jaken S (2000) Protein kinase C signaling and oxidative stress. Free Radic Biol Med 28: 1349–1361. [DOI] [PubMed] [Google Scholar]
- 53. Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, et al. (2007) Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 315: 659–663. [DOI] [PubMed] [Google Scholar]
- 54. Le S, Connors TJ, Maroney AC (2001) c-Jun N-terminal kinase specifically phosphorylates p66ShcA at serine 36 in response to ultraviolet irradiation. J Biol Chem 276: 48332–48336. [DOI] [PubMed] [Google Scholar]
- 55. Lebiedzinska M, Duszynski J, Rizzuto R, Pinton P, Wieckowski MR (2009) Age-related changes in levels of p66Shc and serine 36-phosphorylated p66Shc in organs and mouse tissues. Arch Biochem Biophys 486: 73–80. [DOI] [PubMed] [Google Scholar]
- 56. Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, et al. (2007) Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 315: 659–663. [DOI] [PubMed] [Google Scholar]
- 57. Wulf G, Finn G, Suizu F, Lu KP (2005) Phosphorylation-specific prolyl isomerization: is there an underlying theme? Nat Cell Biol 7: 435–441. [DOI] [PubMed] [Google Scholar]
- 58. Zhang W, Liu Y, An Z, Huang D, Qi Y, et al. (2011) Mediating effect of ROS on mtDNA damage and low ATP content induced by arsenic trioxide in mouse oocytes. Toxicol In Vitro 25: 979–984. [DOI] [PubMed] [Google Scholar]
- 59. Trinei M, Giorgio M, Cicalese A, Barozzi S, Ventura A, et al. (2002) A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene 21: 3872–3878. [DOI] [PubMed] [Google Scholar]