

Abstract
The phosphatidylinositol 3 kinase (Pi3K)/Akt pathway is a major regulator of cell growth, proliferation, metabolism, survival, and angiogenesis. Despite extensive study, a thorough understanding of the modulation and regulation of this pathway has remained elusive. We have previously demonstrated that syndecan 4 (S4) regulates the intracellular localization of mTORC2, thus altering phosphorylation of Akt at serine473 (Ser473), one of two critical phosphorylation sites essential for the full activation of Akt[1]. Here we report that S4 also regulates the phosphorylation of Akt at threonine308 (Thr308), the second phosphorylation site required for the full Akt activation. A deletion of S4 resulted in lower levels of Thr308 phosphorylation both in vitro and in vivo. Furthermore, a deletion or knockdown of the S4 effector molecule PKCα led to a similar reduction in phosphorylation of Thr308 while overexpression of myristoylated PKCα rescued AktThr308 phosphorylation in endothelial cells lacking S4. Finally, PAK1/2 is also recruited to the rafts by the S4-PKCα complex and is required for AKT activation.
Introduction
The phosphatidylinositol 3 kinase (Pi3K) /Akt pathway regulates numerous cellular functions including proliferation, migration, and adhesion [2]. Consequently, it plays a key role in a number of processes among them cancer, metabolism and development. Akt is also a key regulator of angiogenesis due to its activity in endothelial cells [2, 3] In endothelial cells Akt is activated by multiple stimuli including growth factors, such as fibroblast growth factors (FGFs), vascular endothelial growth factors (VEGFs) and insulin-like growth factors (IGFs) among the others, as well by cell-cell and cell-matrix interactions [4]. Akt activation involves two phosphorylation events- phosphorylation of Thr308, by PDK1 and phosphorylation of Ser473 by mTORC2. The activation of Pi3K is thought to lead to the generation of 3, 4, 5-phosphoinositiol phosphate (PIP3) which, in turn, recruits Akt to the plasma cell membrane where it is phosphorylated by PDK1 and mTORC2. [5, 6]. Once activated, Akt phosphorylates a wide variety of downstream effector molecules, including eNOS, TSC2, GSK3, and FoxO [2].
Several lines of evidence have indicated the importance of Pi3K/Akt in angiogenesis. Thus, Akt1 is critical for the ischemic and cancer-mediated angiogenesis [7, 8] while deletion of the p110a isoform of Pi3K leads to vascular defects during embryonic development. PDK1, a downstream Pi3K pathway kinase, has also been reported to be vital in VEGF-mediated endothelial cell migration [9].
We have previously observed [1] that syndecan 4 (S4) regulates mTOR-dependent Akt activation by regulating the assembly and plasma membrane recruitment of the mTOR complex 2 (mTORC2) . Syndecan 4 belongs to a four member family of transmembrane proteoglycans [10]. All syndecans possess an extracellular domain that carries both heparan sulfate and chondroitin sulfate chains and a highly homologous intracellular domain. Syndecan-4 is unique among the rest of the Syndecan family in having a phosphatidylinositol-4,5-bisphosphate (PIP2) binding site that allows it to activate PKCα in the absence of changes in intracellular calcium levels [11-15]. This activation of PKCα is critical to several S4 functions, including its ability to activate RhoG and Rac1 and regulate cell migration [16-18].
Although S4 is rather ubiquitously expressed, levels of its expression vary and it is highly inducible in certain situations. For example, it is found at high levels in the ischemic myocardium [19], vascular tissues after injury [20] and in a variety of tumors [21]. S4 null mice display a delayed dermal wound healing reflecting impaired angiogenesis and a cell migration defect [22]. The ability of S4 to regulate angiogenesis is also suggested by the observation of enhanced neovascularization in the ischemic muscle following a combined FGF2/S4 delivery [23].
Since the process of angiogenesis is very much Akt-dependent, we undertook the current study to further understand S4’s role in the regulation of Akt activation in endothelial cells. We find that S4-dependent activation of PKCα is required for PAK-mediated recruitment of PDK1 and PAK1/2 to the membrane and phosphorylation of Akt Thr308.
Materials and Methods
Reagents and antibodies
Rabbit antibodies against AktThr308, total Akt, Akt1, Akt2, PKCα, PDK1, PDK1Ser241, PAK1, PAK2 were purchased from Cell Signaling Technology. Mouse antibodies against PKCα and flotillin were from BD Bioscience. Mouse antibodies against actin, and the Flag-tag were from Sigma. HUVEC cells were purchased from the tissue core laboratory at Yale University. Medium199 and DMEM as well as lipofectamine 2000 were from Invitrogen. SiRNAs against human Akt1, Akt2, S4 were obtained from OriGene. ShRNA plasmids against mouse and human Akt1, S4 were from Sigma. The lentiviral packaging plasmids pMDLg/pRRE, pRSV-Rev, pVCMV-VSV-G were from Addgene. Endothelial cell growth supplement was purchased from Biomedical Technologies.
Cell culture, transfection and lentivirus transduction
Mouse primary endothelial cells were isolated from the heart and lung as previously described [1] and maintained in DMEM supplemented with 20% FBS, non-essential amino acid, sodium pyruvate, penicillin, streptomycin at standard concentrations. HUVEC cells were cultured in Medium 199 with 20 %FBS. Both types of cells were cultured at 37 °C in 5% CO2.
SiRNAs were transfected into HUVEC with lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s protocols. The transfected cells were cultured for forty eight hours before used for the experiments.
ShRNA plasmids were transfected into HEK 293 cells according to the protocol from Lentiweb.com with modification. In brief, 10 μg shRNA plasmid together with 5 μg pMDLg/pRRE, 2.5 μg pRSV-Rev and 2.5 μg pVCMV-VSV-G were mixed with lipofectamine 2000 (Invitrogen) and transfected into 100 mm plate of HEK293 cells according to the transfection protocol of lipofectamine 2000 (Invitrogen). The viral supernatant was harvested forty eight to seventy two hours after transfection and used to infect mouse heart or lung primary endothelial cells for two days. The infected cells were selected by purimycin at 1 μg /ml for 3 days.
Adenovirus carrying PKCα with a myristoylation site at its N-terminal end (Ad-myr PKCα) was transduced into the mouse primary endothelial cells as previously described[1].
Immunoprecipitation and Western blot
Cells were lysed in PIPES lysis buffer (25 mM PIPES, pH7.0, 150 mM NaCl, 5 mM EDTA and 1% NP-40) or RIPA buffer (50 mM Tris-HCl, 150 mMNaCl, 1%NP-40, 0.5% Sodium deoxycholate, and 0.1% SDS together with protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (Roche). The protein concentration was determined by BCA Protein Assay kit (Thermo Scientific). To immuoprecipitate FLAG-tagged PKCα, cell lysates were incubated with Anti-FLAG M2 magnetic beads overnight at 4 °C, then the beads was washed six time with the lysis buffer, suspended in the reducing SDS-Sample buffer (Boston BioProducts) and subjected to SDS-PAGE (4-15% Criterion TGX Precast Gel, Bio-Rad Laboratories). The proteins were transferred to PVDF membrane, blocked with 5% milk, blotted with primary and HRP-conjugated secondary antibodies. The Western signals were visualized with G:BOX (Syngene).
Isolation of detergent-insoluble lipid raft
Isolation of lipid raft was described previously[1]. In brief, four 150 mm plates of cells were harvested and suspended in TNE buffer (25 mM Tris-HCl, pH7.4, 150 mM NaCl, 5 mM EDTA) in addition to the protease inhibitors as above. The cells were lysed by passage through a 22 g × 3″ needle 20 times to lyse the cells. Then Triton X-100 was added to the lysate to the final concentration at 1%. The lysate was adjusted to 40% (w/v) sucrose and laid at the bottom of an ultracentrifuge tube with a 5-30% sucrose discontinuous gradient on the top. The samples were centrifuged at 100,000g for 16 hours. Fractions were collected using a density fractionator. Western blotting with anti-flotillin Ab was used to identify the lipid raft fraction.
Results
Endothelial cells (EC) isolated from S4-/- mice show decreased phosphorylation of Akt at the PDK1 site Thr308 in response to FGF2 compared to S4+/+ EC (Fig 1A). Furthermore, while stimulation of wild type EC with increasing amounts of FGF2 results in a dose-dependent increase in Akt Thr308 phosphorylation, no similar effect is seen in S4-/- EC (Fig 1B). In order to determine if the impairment of Akt Thr308 phosphorylation seen in S4-/- EC is FGF specific, we examined this in response to an unrelated growth factor IGF-1. Similarly to FGF2, IGF- induced Akt Thr308 phosphorylation was markedly reduced in S4-/- compared to S4+/+ EC (Fig 1C).
Figure 1. Decreased Akt Thr308 phosphorylation in syndecan 4 KO mice and primary endothelial cells.
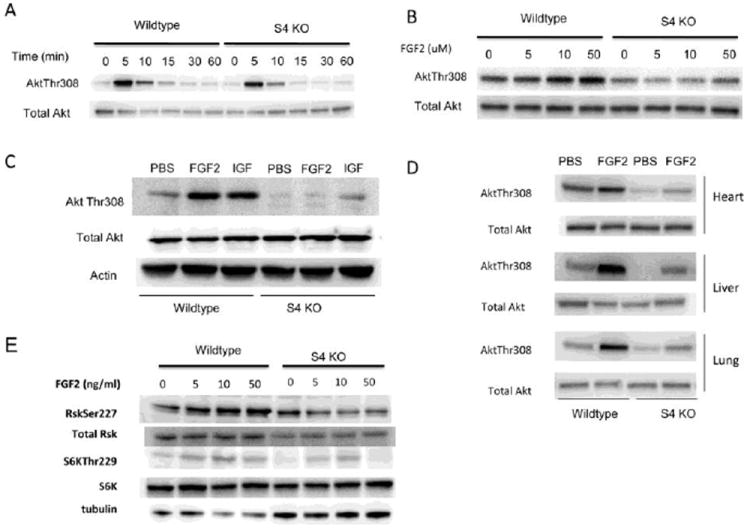
(A) Immunoblotting of total cell lysates from S4-/- and wildtype subconfluent primary heart EC that were serum-starved and then stimulated with 50ng/ml of FGF2 for the times indicated. Phosphorylation of Akt on Thr308 is reduced in S4-/- ECs relative to wildtype.
(B) Immunoblotting of total cell lysates from S4-/- and wildtype subconfluent primary heart EC that were serum-starved and then stimulated with the indicated dose of FGF2 for 5 minutes. Phosphorylation of Akt on Thr308 is reduced in S4-/- ECs relative to wildtype at all doses of FGF2.
(C) Immunoblotting of total cell lysates from S4-/- and wildtype subconfluent primary heart EC that were serum-starved and then stimulated with 50ng/ml FGF2 or 50ng/ml IGF for 5 minutes. Phosphorylation of Akt on Thr308 is reduced in S4-/- ECs relative to wildtype in response to IGF and FGF2.
(D) Western blot analysis of total heart, liver and lung lysates harvested 10 minutes following intravenous injection of FGF2 (50ng/gm body weight). Phosphorylation of Akt on Thr308 is reduced in S4-/- mice relative to wildtype in all tissues.
(E) Immunoblotting of total cell lysates from S4-/- and wildtype subconfluent primary heart EC that were serum-starved and then stimulated with 50ng/ml of FGF2 for the times indicated. Phosphorylation of PDK1 substrates Akt, Rsk and S6K are all reduced in S4-/- ECs relative to wildtype.
Next, in order to demonstrate that the reduction in S4-dependent Akt Thr308 phosphorylation is not strictly an in vitro phenomenon, we injected wild type and S4-/- mice with FGF2 and examined Akt activation in several organs 15 min later. Similar to the in vitro findings, there was a significant reduction in Akt phosphorylation in heart, liver and lung tissues isolated from syndecan-4-/- mice relative to control mice (Fig 1D).
The reduced PDK1-dependent Akt phosphorylation in response to both FGF2 and IGF1 in S4-/- cells suggests that this may not be just an Akt defect and that other PDK1-dependent kinases may be impaired as well. In addition to Akt, PDK1 also phosphorylates other members of the AGC kinase family including Rsk and S6K. We find that FGF2 activation of both Rsk and S6K is also decreased in S4-/- EC relative to WT cells (Fig 1E), thus demonstrating a global reduction in PDK1 activity in the absence of S4.
Since a major element of syndecan-4 dependent signaling is the membrane recruitment and activation of PKCα, we next examined the role of PKCα in PDK1-dependent signaling. A knockdown of PKCα expression in wild type endothelial cells using two different siRNA sequences significantly reduced FGF2-dependent Akt Thr308 phosphorylation (Fig 2A). This result was independently confirmed by isolating primary endothelial cells from wild type and PKCα-/- mice and stimulating them with FGF2. PKCα-/- EC demonstrated a similar reduction in Akt phosphorylation in response to FGF2 (Fig. 2B).
Figure 2. AktThr308 phosphorylation is dependent upon PKCα.
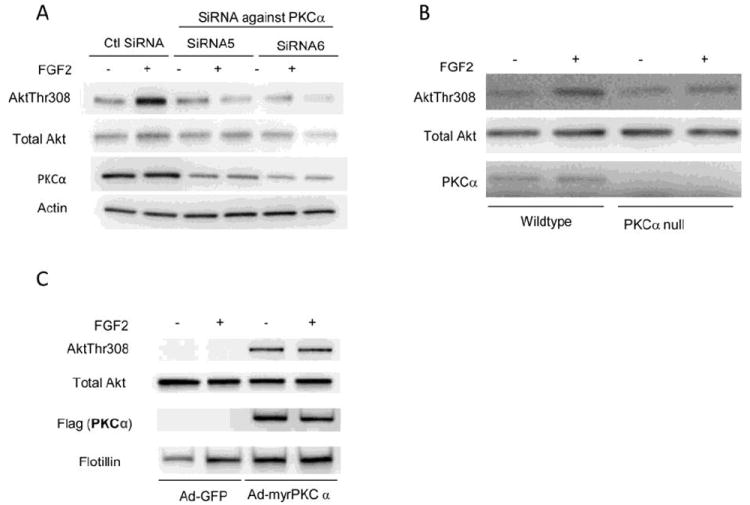
(A) Western blotting of HUVEC cells transfected with control and PKCα siRNAs for forty-eight hours, serum-starved and stimulated for 5 minutes with FGF2(50ng/ml). FGF2 induced phosphorylation of Akt on T hr308 is reduced in PKCα knockdown HUVEC.
(B) Western blotting of wildtype and PKCα knockout primary heart endothelial cells, serum-starved and stimulated for 5 minutes with FGF2(50ng/ml). FGF2 induced phosphorylation of Akt on Thr308 is reduced in PKCα knockout ECs relative to wildtype.
(C) Western blotting of S4-/- ECs transduced with either Ad-GFP (control) or Ad-myr PKCα for two days, then serum-starved, stimulated for 5 minutes with FGF2(50ng/ml) and lipid raft fractions isolated. Transduction of S4-/- EC with myrPKCα fully restores AktThr308 phosphorylation that is not FGF dependent.
Given that Akt activation is PKCα dependent, we next examined whether the expression of a membrane-targeted form of PKCα (myrPKCα) could rescue Akt activation in S4-/- endothelial cells. Transduction of S4-/- EC with an adenoviral myrPKCα construct (Ad-myrPKCα) resulted in the robust expression of PKCα and its localization to the plasma membrane rafts. Furthermore, this resulted in the complete restoration of Akt Thr308 phosphorylation (Fig 2C). Of note, expression of myrPKCα by itself was sufficient to induce Akt1 Thr308 phosphorylation, suggesting that the key role of FGF stimulation is to localize PKCα to the cell membrane via S4.
In a previous study Higuchi et al reported that PAK serves as a scaffold protein mediating AktThr308 phosphorylation by PDK1 [24]. In order to examine the role of PAK in the PKCα-dependent Akt phosphorylation by PDK1, we first set to determine whether PAK1 and PKCα are present in the same protein complex. The analysis of an immunoprecipitate generated by a pull-down with an antibody against a myrPKCα tag in Ad-myrPKCα transduced EC revealed the presence of PAK1, while no co-immunoprecipitation was detected in GFP-transduced cells (Fig 3A). Since syndecan-4 recruits PKCα to the membrane, we next examined whether transduction of S4-/- endothelial cells with Ad-myrPKCα results in PDK1 membrane recruitment. Isolation of lipid raft fractions from S4-/- cells following Ad-myrPKCα transduction resulted in a significant increase in both PDK1 and PAK at the membrane that was not further increased by FGF2 stimulation (Fig 3B).
Figure 3. PAK1 and PAK2 are components of the S4-PKCα complex in lipid rafts.
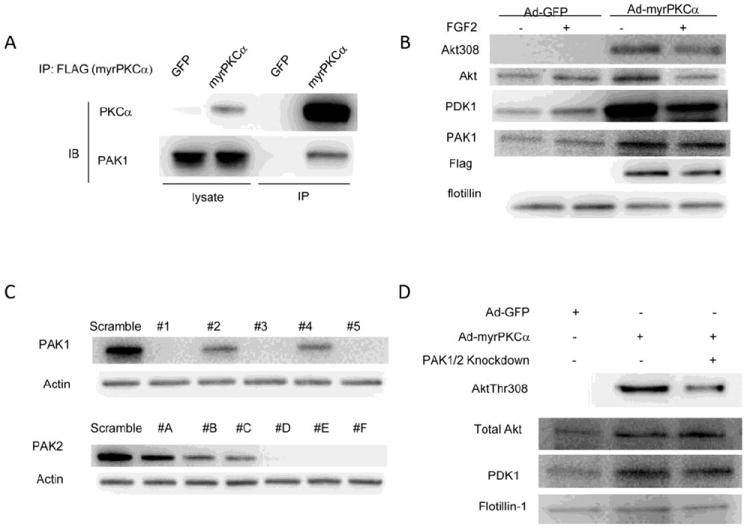
(A) Western blotting of ECs transduced with either Ad-GFP (control) or Ad-myr PKCα for two days and immunoprecipitated for PKCα. Transduction of ECs with Ad- PKCα results in co-immunoprecipitation of PAK1 with FLAG tagged myrPKCα PAK1.
(B) Western blotting of S4-/- ECs transduced with either Ad-GFP (control) or Ad-myr PKCα for two days, then serum-starved, stimulated for 5 minutes with FGF2(50ng/ml) and lipid raft fractions isolated. Expression of myrPKCα results in increased membrane localization of both PDK1 and PAK.
(C) Western blotting of cells transduced for two days with lentiviruses carrying various shRNAs against mouse PAK1 or PAK2. Efficient knockdown of PAK1 and PAK2 is seen with several shRNAs. use PAK1shRNA3 in combination with PAK2shRNAD to knockdown PAK1 and 2.
(D) Western blotting of S4-/- ECs transduced with PAK1/PAK2 shRNAs in combination with either Ad-GFP (control) or Ad-myrPKCα for two days, then serum-starved, stimulated for 5 minutes with FGF2(50ng/ml) and lipid raft fractions isolated. Knockdown of PAK1/2 reduces myr PKCα-mediated rescue of AktThr308 phosphorylation in S4-/- ECs.
In order to examine whether PAK is required for the PKCα-dependent phosphorylation of Akt Thr308 by PDK1, we knocked down both PAK1 and PAK2 isoforms in S4-/- endothelial cells and then determined the effect of this knockdown upon PDK1/Akt membrane localization and Akt phosphorylation. The efficient knockdown of both PAK isoforms was obtained with 3 different shRNAs for each isoform. A combined knockdown of both PAK1 and PAK2 was used to study PAK role in S4/PKCα/Akt activation pathway. In agreement with the above data, Akt and PDK1 levels were low in S4-/- EC and Akt Thr308 phosphorylation was essentially absent (Fig 3D, left lane). Transduction of these cells with Ad-myrPKCα increased Akt and PDK levels in the lipid rafts and dramatically increased Akt Thr308 phosphorylation (Fig 3D, middle lane). However, the knockdown of PAK1/PAK2 markedly reduced Akt1Thr308 phosphorylation while having no substantial effect on AKT or PDK1 rafts presence (Fig 3D, right lane).
Discussion
The proteoglycan syndecan-4 regulates signaling pathways and biological functions through its association with growth factor receptors and integrins [25-27] In this study and in our previous work [1], we have focused on the role of S4 in the PI3K/Akt signaling pathway. Here we report that S4 regulates PDK1-dependent Akt activation via the PKCα/PDK1/PAK complex. The absence of S4 results in decreased PDK1 presence in the membrane rafts leading to decrease activation of not only Akt, but also other PDK1-dependent kinases including Rsk (Ser227 site) and S6K (Thr229 site). While S4 and PKCα are required for PDK1 recruitment to the rafts, PAK1 and PAK2 proteins are needed for the efficient PDK1-dependent Akt phosphorylation. Together, S4/PKCα/PAK complex emerges as a key regulator of Akt activation (Fig 4).
Figure 4. The working model of the role of syndecan-4 in the PDK1/Akt pathway.
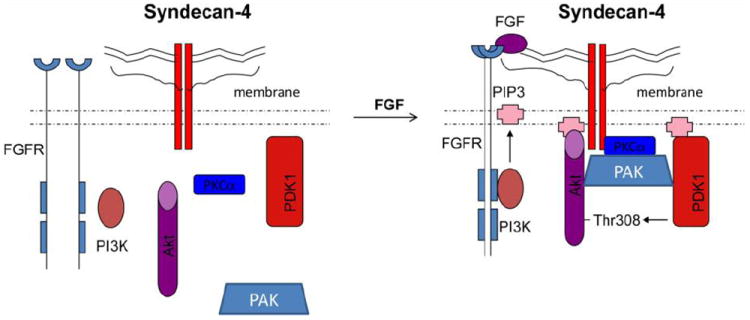
Treament of FGF induces dimerization of syndecan 4 which, in turn, recruits PKCα, PAK, PDK1 and Akt to form a complex in the membrane raft. Formation of the complex facilitates Akt phosphorylation at Thr308 by PDK1.
Syndecan-4 has traditionally been considered a FGF2 receptor [28] and for that reason in the current study we focused on the role of S4 in FGF2-dependent Akt activation. However, the effect is clearly not FGF2-limited as IGF stimulation also fails to activate Akt in S4-/- endothelial cells. More likely, any soluble growth factor or extracellular matrix protein that is able to interact with and cluster S4 is activating Akt in a S4-dependent manner. A recent report demonstrated that S4 binds tissue transglutaminase (TG2) [29]. TG2, in addition to its protein cross-linking enzymatic activity, can associates with a number of ECM proteins including fibronectin and integrins that interact with S4 [26, 30] and with receptor tyrosine kinases such as PDGF receptor[29]. Potentially S4 may modulate IGF-mediated Akt activation via its interaction with a TG2-growth factor receptor complex.
Previously we have shown that S4 modulates Akt phosphorylation at Ser473, the mTORC2 phosphorylation site [1], via recruitment of PKCα . In this report, we have focused on the role of S4 in regulating phosphorylation of Akt on Thr308, a site phosphorylated by PDK1 that is essential for the full Akt activation. We found that S4-dependent activation of PKCα is essential for PDK1-dependent Akt activation. Since PDK1 is constitutively active due to its intermolecular phosphorylation at Ser241 [31], PKCα’s primary role is to localize PDK1 to the lipid rafts where it can then phosphorylate Akt1 and other kinases. This conclusion is supported by the finding that overexpression of PKCα increases PDK1 levels in the lipid rafts of S4-/- EC that in turn dramatically increases AktThr308 phosphorylation. Importantly, this increase in the Akt phosphorylation is not due the increased presence of Akt itself in the raft or to any other activity of FGF2, but solely to the increased PDK1 presence.
In addition to PKCα-dependent recruitment of PDK to the rafts, the full activation of Akt Thr308 phosphorylation also requires the presence of PAK1 and PAK2 proteins. In agreement with a prior publication [24], PAKs presence did not affect PDK or AKT recruitment but was necessary for AKT phosphorylation. Our data shows that PAK interacts with PKCα either directly or as a part of the S4/PKCα complex. This conclusion is based on the demonstration of a co-immunoprecipitation between PKCα and PAK1, increased PAK1 levels in the lipid rafts after myrPKCα overexpression in S4-/- EC, and finally, on the observation that a PAK1/2 knockdown reduces PKCα-mediated Akt-Thr308 phosphorylation in lipid raft. Taken together, these three lines of evidence indicate that PAK is a key component of the S4/PKCα/Akt signaling complex.
Together with the prior demonstration of the critical role played by the S4/PKCα complex in regulation of mTORC2 assembly and mTOR-dependent Akt phosphorylation and activation [32], these results identify S4 as a key player in the regulation of Akt activity.
Highlights.
Syndecan-4 dependent PKCα activation is necessary for PDK1 recruitment to the rafts
Syndecan-4/PKC-alpha also recruit PAKL1/PAK2
The presence of PDK1/PAK1/PAK2 is required for Akt activation
These results place syndecan-4a t the center of Akt signaling regulation
Acknowledgments
Supported by an NIH grant HL62289 (MS)
The authors thank Dr. Anthony Lanahan for his help in the preparation of the manuscript and Dr. Zhen Zhuang for the assistance in isolation of mouse primary endothelial cells.
Abbreviations
- S4
syndecan 4
- PI3K
phosphatidylinositol 3 kinase
- myrPKCα
myristoylated protein kinase C
- PAK
p21 protein (CDC42/Rac)-activated kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Partovian C, Ju R, Zhuang ZW, Martin K, Simons Michael. Molecular Cell. 2008;32:140–149. doi: 10.1016/j.molcel.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fayard E, Xue G, Parcellier A, Bozulic L, Hemmings BA. Curr Top Microbiol Immunol. 2010;346:31–56. doi: 10.1007/82_2010_58. [DOI] [PubMed] [Google Scholar]
- 3.Karar J, Maity A. Front Mol Neurosci. 2011;4:1–7. doi: 10.3389/fnmol.2011.00051. Article 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shiojima I, Walsh K. Circulation research. 2002;90:1243–1250. doi: 10.1161/01.res.0000022200.71892.9f. [DOI] [PubMed] [Google Scholar]
- 5.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 6.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Current biology. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 7.Ackah E, Yu J, Zoellner S, Iwakiri Y, Skurk C, Shibata R, Ouchi N, Easton RM, Galasso G, Birnbaum MJ, Walsh K, Sessa WC. The Journal of clinical investigation. 2005;115:2119–2127. doi: 10.1172/JCI24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J, Somanath PR, Razorenova O, Chen WS, Hay N, Bornstein P, Byzova TV. Nat Med. 2005;11:1188–1196. doi: 10.1038/nm1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bekhite MM, Finkensieper A, Binas S, Muller J, Wetzker R, Figulla HR, Sauer H, Wartenberg M. Journal of cell science. 2011;124:1819–1830. doi: 10.1242/jcs.077594. [DOI] [PubMed] [Google Scholar]
- 10.Choi Y, Chung H, Jung H, Couchman JR, Oh ES. Journal of the International Society for Matrix Biology. 2011;30:93–99. doi: 10.1016/j.matbio.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Horowitz A, Murakami M, Gao Y, Simons M. Biochemistry. 1999;38:15871–15877. doi: 10.1021/bi991363i. [DOI] [PubMed] [Google Scholar]
- 12.Horowitz A, Simons M. The Journal of biological chemistry. 1998;273:25548–25551. doi: 10.1074/jbc.273.40.25548. [DOI] [PubMed] [Google Scholar]
- 13.Oh ES, Woods A, Couchman JR. The Journal of biological chemistry. 1997;272:11805–11811. doi: 10.1074/jbc.272.18.11805. [DOI] [PubMed] [Google Scholar]
- 14.Oh ES, Woods A, Couchman JR. The Journal of biological chemistry. 1997;272:8133–8136. doi: 10.1074/jbc.272.13.8133. [DOI] [PubMed] [Google Scholar]
- 15.Oh ES, Woods A, Lim ST, Theibert AW, Couchman JR. The Journal of biological chemistry. 1998;273:10624–10629. doi: 10.1074/jbc.273.17.10624. [DOI] [PubMed] [Google Scholar]
- 16.Bass MD, Roach KA, Morgan MR, Mostafavi-Pour Z, Schoen T, Muramatsu T, Mayer U, Ballestrem C, Spatz JP, Humphries MJ. The Journal of cell biology. 2007;177:527–538. doi: 10.1083/jcb.200610076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elfenbein A, Rhodes JR, Meller J, Schwartz MA, Matsuda M, Simons M. The Journal of cell biology. 2009;186:75–83. doi: 10.1083/jcb.200810179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tkachenko E, Elfenbein A, Tirziu D, Simons M. Circulation research. 2006;98:1398–1404. doi: 10.1161/01.RES.0000225283.71490.5a. [DOI] [PubMed] [Google Scholar]
- 19.Li J, Brown LF, Laham RJ, Volk R, Simons M. Circulation research. 1997;81:785–796. doi: 10.1161/01.res.81.5.785. [DOI] [PubMed] [Google Scholar]
- 20.Nikkari ST, Jarvelainen HT, Wight TN, Ferguson M, Clowes AW. Am J Pathol. 1994;144:1348–1356. [PMC free article] [PubMed] [Google Scholar]
- 21.Yoneda A, Lendorf ME, Couchman JR, Multhaupt HA. J Histochem Cytochem. 2012;60:9–21. doi: 10.1369/0022155411428469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Echtermeyer F, Streit M, Wilcox-Adelman S, Saoncella S, Denhez F, Detmar M, Goetinck P. The Journal of clinical investigation. 2001;107:R9–R14. doi: 10.1172/JCI10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jang E, Albadawi H, Watkins MT, Edelman ER, Baker AB. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:1679–1684. doi: 10.1073/pnas.1117885109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higuchi M, Onishi K, Kikuchi C, Gotoh Y. Nature cell biology. 2008;10:1356–1364. doi: 10.1038/ncb1795. [DOI] [PubMed] [Google Scholar]
- 25.Byron A, Morgan MR, Humphries MJ. Current biology. 2010;20:R1063–1067. doi: 10.1016/j.cub.2010.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morgan MR, Humphries MJ, Bass MD. Nature reviews Molecular cell biology. 2007;8:957–969. doi: 10.1038/nrm2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tkachenko E, Rhodes JR, Simons M. Circulation research. 2005;96:488–500. doi: 10.1161/01.RES.0000159708.71142.c8. [DOI] [PubMed] [Google Scholar]
- 28.Horowitz A, Tkachenko E, Simons M. The Journal of cell biology. 2002;157:715–725. doi: 10.1083/jcb.200112145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belkin AM. The FEBS journal. 2011;278:4704–4716. doi: 10.1111/j.1742-4658.2011.08346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woods A, Longley RL, Tumova S, Couchman JR. Archives of biochemistry and biophysics. 2000;374:66–72. doi: 10.1006/abbi.1999.1607. [DOI] [PubMed] [Google Scholar]
- 31.Mora A, Komander D, van Aalten DM, Alessi DR. Semin Cell Dev Biol. 2004;15:161–170. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 32.Partovian C, Ju R, Zhuang ZW, Martin KA, Simons M. Mol Cell. 2008;32:140–149. doi: 10.1016/j.molcel.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]