

Abstract
All-trans retinoic acid (atRA), one of the active ingredients of vitamin A, exerts canonical activities to regulate gene expression mediated by nuclear RA receptors (RARs). AtRA could also elicit certain non-canonical activities including, mostly, rapid activation of extracellular signal regulated kinase 1/2 (ERK1/2); but the mechanism was unclear. In this study, we have found that cellular retinoic acid binding protein I (CRABPI) mediates the non-canonical, RAR- and membrane signal-independent activation of ERK1/2 by atRA in various cellular backgrounds. In the context of embryonic stem cells (ESCs), atRA/CRABPI-dependent ERK1/2 activation rapidly affects ESC cell cycle, specifically to expand the G1 phase. This is mediated by ERK stimulation resulting in dephosphorylation of nuclear p27, which elevates nuclear p27 protein levels to block G1 progression to S phase. This is the first study to identify CRABPI as the mediator for non-canonical activation of ERK1/2 by atRA, and demonstrate a new functional role for CRABPI in modulating ESC cell cycle progression.
Keywords: CRABPI, Retinoic Acid, ERK, Cell Cycle, p27
1. Introduction
All-trans retinoic acid (atRA), the principal active ingredient of vitamin A, exerts a wide spectrum of effects manifested in numerous aspects of biological processes such as growth, development, cell differentiation and proliferation, reproduction, neuron function and vision, etc [1]. AtRA is also used for certain therapeutic purposes and exerts its canonical activity by binding to nuclear RA receptors (RARs) to regulate gene expression [2–4]. However, before it enters the nucleus to act on RARs, in the cytoplasm atRA is bound by two types of cytosolic binding proteins named cellular retinoic acid binding protein I (CRABPI) and II (CRABPII). It is believed that CRABPI functions primarily to sequester atRA and to regulate its metabolism, whereas CRABPII binds atRA and delivers it to RARs [5, 6]. Increasingly, recent reports have revealed non-canonical activities of atRA in various biological contexts, including binding to PKCα to affect PKC activity [7], binding to membrane RAR to affect spine formation in hippocampal neurons [8], and rapid activation of extracellular signal regulated kinase 1/2 (ERK1/2). Among these, ERK activation by atRA has been most widely detected in different experimental model systems [9–11]. In most biological contexts, atRA-activated ERK1/2 could rapidly trigger several events without involving RARs, primarily to augment post-translational modification programs for proteins such as transcription factors and for chromatin conformation of specific gene loci. In the stem cell context, this activity modifies the conformation of the gene locus controlling the stemness feature, Oct4 [11, 12]. However, it remained unclear how atRA elicited such a non-canonical activity. This current study was designed to identify the key player that mediates the non-canonical activity of atRA to activate ERK1/2 using the embryonic stem cell (ESC) as the model system.
The ESC system has an enormous potential in both clinical applications and for addressing basic biological questions [13, 14]. However, to realize their full potential it is most critical to understand the mechanisms that control the unique feature of ESC, i.e. relatively fast proliferation without commitment or differentiation. The impetus for cell proliferation versus cell fate commitment is tightly regulated; one critical control is executed in regulating cell-cycle progression [13]. The cell cycle is comprised of four phases, G1, S, G2, and M; but ESC cell cycle is uniquely coordinated for a much shorter G1 phase by avoiding or shortening early G1. One principal mechanism mediating such a short G1 phase is the timely degradation of cyclin dependent kinase (CDK) interacting protein/kinase inhibitor protein (CIP/KIP), also known as p27 [15, 16]. Degrading p27 in early G1 abolishes its inhibition of CDKs, particularly the cyclin E-CDK2 complex, allowing cycle entry into the S phase and cell proliferation [13]. Thus, the level of p27, particularly nuclear p27, is critical to cell cycle control in ESC. P27 levels are modulated by a variety of posttranslational modifications at several key residues. Among these, one site, ser-10 phosphorylation, by Kinase-Interacting Stathmin (KIS) protein, Mini-Brain-Related Kinase 1 (MIRK) or Dual Tyrosine Phosphorylation Related Kinase 1, (DYRK1), is particularly important for regulating nuclear p27. Upon ser-10 phosphorylation, nuclear p27 is recruited to the nuclear export machinery and translocates to the cytoplasm where it is then ubiquitinated and subsequently degraded by the Kip1 ubiquitination-Promoting Complex (KPC). Depleting nuclear p27 relieves inhibition of cyclin E-CDK2 complex and allows progression into S phase in healthy, proliferating cells[16, 17]. Therefore, for ESC to commit to a specific fate, or to undergo differentiation, nuclear ser-10 phosphorylated p27 must be dephosphorylated in order to retain and accumulate p27 in the nuclei to lengthen G1. The control to dephosphorylate nuclear phosphorylated p27 is elusive.
We now report studies to identify the direct mediator for atRA that can rapidly and non-canonically activate ERK1/2 in ESC, which is the cytosolic binding protein CRABPI. We then present data demonstrating that atRA-activated ERK1/2 acting through CRABPI stimulates events resulting in dephosphorylation of ser-10-phosphorylated p27, allowing rapid accumulation of p27 in the nucleus. This results in the inhibition of G1 progression in ESC, and consequently prolonged G1, which slows down proliferation and prepares cells for commitment to differentiation as triggered by RAR-mediated transcriptional programs.
2. Materials and Methods
2.1 Cell culture methods, plasmids and siRNAs
Cos-1 cells and CJ7 ESCs were maintained as described [12, 18] in medium containing dextran-coated charcoal-treated fetal bovine serum. Plasmid transfection and siRNA transfection was each conducted using Lipofectamine 2000 (Invitrogen) and Hiperfect transfection (Qiagen), respectively. Mouse CRABPI cDNA was cloned into pCMX-PL1. CRABPI and CRABPII siRNAs were from Qiagen. Mouse CRABPI siRNAs are 5′-CACGTGGGAGAATGAGAACAA-3′ and 5′-CAGCTTGTTCCTGCTTCATGA-3′. Mouse CRABPII siRNAs are 5′-CTGTGTGATTTAGAATATTTA-3′ and 5′-AAGGATCTGTTCTGCAAAGGA-3′.
2.2 Western blotting, cellular fractionation and chemicals
Whole cell lysate and nuclear/cytosolic fractions were prepared as described [19, 20]. Antibodies for β-actin (SC-47778), lamin (SC-7293), a-tubulin (SC-5286), ERK1 (SC-93), and ERK2 (SC-153) were from Santa Cruz. Antibodies for CRABPI (C1608), flag (F3165) were from Sigma. Anti-CRABP2 (10225-1-AP) was from Proteintech Group. Anti-phospho-p27-serine-10 (GTX61772) was from GeneTex. Anti-phospho-ERK1/2 (9101) and p27 (2552) were from Cell Signaling. Anti-GST (05–311) was from Upstate. AtRA (100nM), 9-cis RA, VEGF, and methyl-beta-cyclodextrin, MBCD (100uM) were from Sigma. AGN193109 (RAR antagonist, 100nM) was from Santa Cruz. EGF was from BD Biosciences. Retinol, LE540, 4-oxo-RA, 4-hydroxy-RA and FV80 were as described [21]. 5-(2-phenyl-pyrazolo[1,5-a][16]pyridin-3-yl)-1H-pyrazolo[3,4-c]pyridazin-3-ylamine (ERK1/2 inhibitor, 1 uM) was from EMD. 3H-RA was from Perkin Elmer.
2.3 In vitro ligand binding Assay
His-CRABP1 was purified from bacteria. In 300 μl of binding buffer (50 mm HEPES, 100 mm NaCl, 1 mm EDTA, 10% glycerol, 0.1% Nonidet P-40, pH 8.0), an equimolecular concentration (10 nM) of CRABP1 and [3H]RA was incubated for 40 min at room temperature in the presence of excess cold ligand. His-tagged CRABP1 was affinity-captured on nickel-nitrilotriacetic acid-agarose affinity beads (Qiagen) for 2 hrs at 4 °C, and washed twice with binding buffer. Ligand-bound CRABP1 was dispersed in scintillation mixtures, and the radioactivity was measured in a liquid scintillation counter (Beckman).
2.4 In vitro Kinase assay
Cells expressing flag-CRABPI were lysed and immunoprecipated [22] with anti-flag antibody. The precipitated complex was incubated with pure 0.4 μg GST-ERK2 (14–198, upstate) in 40 μl kinase buffer (20 mM MOPS pH7.2, 25 mM β-glycerolphosphate, 5mM EGTA, 1mM Sodium orthovanadate, 1mM DTT, 120uM ATP, 18mM MgCl2, 1X Protease inhibitor) at 30°C for 30min. Samples separated on SDS-PAGE gel were detected with anti-phospho-ERK1/2.
2.5 Flow Cytometry
After treatment, cells were harvested and fixed with absolute ethanol. Cells were then stained with staining buffer (10 ml PBS with 2 mg DNAse-free RNAse A and 0.4 ml of 500 μg/ml propidium iodine) at 37°C for 15min. Cells were analyzed with fluorescence-activated cell sorting (FACS, BD Biosciences).
2.6 Data Analysis
Analyses of data were performed using appropriate analysis of variance. Significant effects were followed with appropriate post hoc tests. In all cases, P values were two-tailed, and the comparison was considered statistically significant when P < 0.05. Data were presented as means ± SEM.
3. Results
3.1 Biphasic ERK1/2 activation by atRA
We previously reported rapid activation of ERK1/2 by atRA in a mouse embryonal carcinoma cell line P19 [23]. To investigate whether this was a general phenomenon common to various cellular contexts, we conducted a systematic kinetic study using ESC and COS-1 cells. As shown in Figure 1, atRA at a physiological concentration (100nM) rapidly, within 30 minutes, activated ERK1/2, which was followed by a second phase ERK1/2 activation approximately 8–12 hrs later. Interestingly, ERK2 activation appeared to be more profound as indicated by the stronger intensity of the lower band. Thus, atRA appears to elicit two phases of ERK1/2 activation in various cellular contexts including ESC and COS-1, as well as the previously reported P19 embryonal carcinoma cell line [11].
Fig. 1. AtRA induces a bi-phasic ERK1/2 activation.
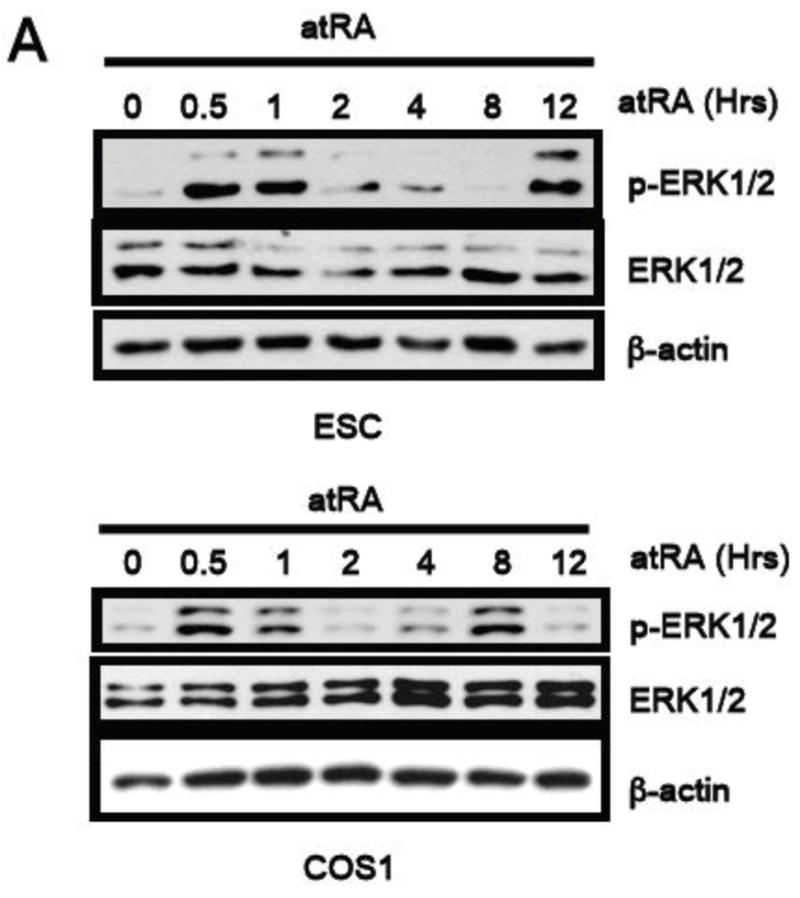
The kinetics of pERK1/2 activation in CJ7 mouse ESC and COS-1 cells triggered by a100 nM atRA treatment was determined by western blot analyses.
3.2 The first phase atRA-activation of ERK1/2 is independent of RAR and membrane signal
To determine the mechanisms responsible for the two phases of ERK1/2 activation elicited by atRA, we first tested whether this activation depended on the typical genomic action of atRA via RARs. Since ESC expresses multiple RARs, we used a pan-RAR antagonist AGN193109 to block the canonical action of atRA. As shown in Figure 2a, this pan-RAR antagonist blocked the second phase atRA-triggered ERK1/2 activation, leaving the first phase ERK1/2 activation unaffected, in both ESC and COS-1. These data suggest that RAR mediates the second phase atRA-elicited ERK1/2 activation. To further validate that the canonical, RAR-mediated, activity of atRA elicits only the second phase ERK1/2 activation, we employed a specific RAR agonist, FV80, to treat the cells and monitored their ERK1/2 activation profiles. As shown in Figure 2b, FV80 only induced the second phase ERK1/2 activation, which occurred approximately 8 to 16hrs later in ESC and COS-1 cells. This result further supports the notion that phase 1 ERK1/2 activation by atRA is RAR-independent.
Fig. 2. AtRA-induced first phase ERK1/2 activation is independent of RAR and membrane signaling.

(2a) Only the second phase of atRA ERK1/2 activation was blocked by RAR antagonist, AGN193109 (100 nM), in both ESC and COS-1 cells. (2b) The RAR agonist, FV80 (100 nM), activated only the second phase ERK1/2 activation in both ESC and COS-1 cells. (2c) Lipid raft inhibitor, methyl-beta-cyclodextrin (MBCD), effectively abolished EGF-activated ERK1/2 in ESC and VEGF-activated ERK1/2 in COS-1, but failed to inhibit atRA-triggered rapid phase 1 ERK1/2 activation in both cellular backgrounds.
We then compared atRA-activated phase 1 ERK1/2 to membrane signal activated ERK1/2 by employing epidermal growth factor (EGF), a well-established growth factor that activates mitogen activated protein kinase (MAPK) activity through membrane EGF receptor and is relevant to ESC growth/differentiation [24]. As shown in Figure 2c above, both atRA (100nM 1hr) and EGF rapidly activated ERK1/2 in ESC; however, a lipid raft inhibitor, methyl-beta-cyclodextrin (MBCD), effectively abolished EGF-activated ERK1/2 but failed to inhibit atRA-triggered rapid ERK1/2 activation. In the COS-1 system (2c, below), the relevant growth factor for membrane receptor tyrosine kinases is vascular endothelial growth factor (VEGF). MBCD appeared to also block the VEGF-activated ERK1/2, leaving atRA-triggered rapid ERK1/2 activation unaffected in the COS-1 context. These results indicate that atRA-elicited phase 1 ERK1/2 activation occurs in a membrane-independent manner, and is different from the typical membrane receptor tyrosine kinase activation of ERK1/2.
3.3 CRABPI as the mediator for phase 1 ERK1/2 activation
Given that atRA-elicited rapid ERK1/2 activation occurred independently of RAR and membrane signaling, we then examined other potential mediators for RA in the cytoplasm, such as the two specific atRA binding proteins, CRABPI and CRABPII. We first carried out loss-of-function assays by silencing CRABPI or CRABPII prior to atRA stimulation. As shown in Figure 3a left, silencing CRABPI effectively abrogated atRA-elicited rapid ERK1/2 activation. On the contrary, as shown in Figure 3a right, silencing CRABPII failed to block rapid ERK1/2 activation by atRA. This result indicates that CRABPI is probably responsible for the first phase atRA-activation of ERK1/2. Similarly, in the COS-1 background, silencing CRABPI (Figure 3b left) also resulted in a loss of RA-elicited rapid ERK1/2 activation. More importantly, a gain-of-function assay further validated this notion because elevating CRABPI triggered a dose-dependent enhancement in atRA-elicited rapid ERK1/2 activation (Figure 3b right).
Fig. 3. The first phase of atRA-induced biphasic ERK1/2 activation is CRABPI dependent.

(a) CRABPI, but not CRABPII, knockdown in ESC abrogated atRA-induced phase 1 ERK1/2 activation. (b) In COS-1, CRABPI knockdown also abrogated phase 1 activation of ERK1/2 induced by atRA, and elevating CRABPI levels enhanced atRA-induced ERK1/2 activation in a dose dependent manner. (c) Left, in vitro competitive binding assays of various retinoid compounds were conducted using 3H-atRA bound CRABPI. Right, 4-oxo-RA (100 nM), a CRABPI ligand, readily activated the first phase ERK1/2 activation in COS-1 cells. *indicates P< 0.05. (d) In vitro kinase assay using partially immuno-purified atRA/CRABPI complex to activate purified ERK1/2.
We then postulated that ligand binding to CRABPI was critical to atRA-elicited ERK1/2 activation, and compared CRABPI-binding ability of a spectrum of potential CRABPI ligands and several negative control compounds. We performed competitive in vitro binding assays using purified CRABPI protein and 3H-atRA in the presence of an excess amount of each cold ligand. As shown in Figure 3c left, the positive control, cold atRA, effectively competed with 3H-atRA for binding to CRABPI. Two atRA metabolites, 4-oxo-RA and 4-hydroxy-RA, that have been previously shown as potential CRABPI ligands [25], could also effectively compete with 3H-RA for binding to CRABPI. The RAR agonist FV80 failed to compete for CRABPI binding (Figure 3c). Interestingly, FV80 could elicit only the second, RAR-mediated, phase of ERK1/2 activation as shown earlier (Figure 2a). Since CRABPI could bind atRA metabolites including 4-oxo-RA and 4-hydroxy-RA with a high affinity, we then examined if these atRA metabolites could elicit phase 1 ERK1/2 activation. As shown in Figure 3c right, 4-oxo-RA very effectively elicited phase 1 ERK1/2 activation; it also activated ERK1/2 at later time points, 8 to 16 hours, likely through RARs [26]. These data support that ligand binding to CRABPI contributes to rapid ERK1/2 activation.
To validate the functional role of atRA/CRABPI in mediating rapid ERK1/2 activation, we performed an in vitro kinase assay that detected enzymatic activation of ERK1/2 using partially purified holo-CRABPI complexes. ERK1/2 activity was assayed by western blot analysis with a phospho-specific ERK1/2 antibody. As shown in Figure 3d, only the CRABPI complex purified from atRA-treated cells activated ERK1/2. On the contrary, CRABPI complexes purified from control cells failed to activate ERK1/2 in this in vitro reaction. Together, these data show that the holo-CRABPI complex can activate ERK1/2 both in-vivo and in-vitro.
3.4 AtRA/CRABPI-dependent ERK1/2 rapidly stimulates events leading to p27 stability
It is well established that upon exposure to atRA, ESCs lose their stemness property and usually undergo differentiation. This is attributed to the action of atRA binding to RARs that regulate gene transcription, which is required for numerous cellular processes in the committed cells. The RAR-mediated canonical activity of atRA typically alters numerous cellular processes including elevating ERK1/2 level and activity, as demonstrated in the second phase ERK1/2 activation that can be suppressed by RAR antagonists (Figures 1 & 2). Since atRA can also rapidly elicit RAR-independent ERK1/2 activation, through binding to the cytosolic protein CRABPI, it was interesting to examine the physiological relevance of this type of rapid ERK1/2 activation elicited by atRA in ESC cultures. The most unique property of ESC cultures is attributed to their unlimited proliferation with a relatively much shorter cell cycle (~8 hrs) due to, primarily, a much shorter G1 phase. Control of G1 phase is critical to ESC cell fate, either to continue the cycle progression or to exit the cycle and commit to a certain lineage. We postulated that atRA/CRABPI-dependent rapid ERK1/2 activation in ESC may contribute to a rapid alteration in ESC cell cycle control; therefore we monitored the cell cycle profile of ESC cultures exposed to atRA. In this experiment, we found most significant immediate changes in G1 phase (for detail, see later Figure 5). We then examined potential regulators for G1, and found a significant and rapid effect of atRA on the level of p27, a critical CDK inhibitor that prevents S entry. Considering the delicate nature of ESC, in this series of experiments, we have examined endogenous components without any ectopic expression in order to ensure we capture the true biological phenomena.
Fig. 5. AtRA interferes with cell cycle progression in G1.

(a) FACS analyses of atRA treatment and CRABPI expression in ESC cell cycle regulation. Upper panels show the FACS results with G1 and S gating indicated on each representative chart, panels at the bottom show the quantified results. AtRA expanded, primarily, the G1 phase population, which was abolished by CRABPI knockdown. (b) A proposed model. In the cytoplasm, atRA binds to CRABPI to activate ERK1/2, which is then translocated to the nucleus to stimulates events leading dephosphorylation of ser-10 phosphorylated p27, allowing p27 to be accumulated in the nucleus to negatively regulate cell cycle progression through G1.
As shown in Figure 4a left group, atRA rapidly (1 hr) activated ERK1/2 (1st panel, pERK), without affecting its protein level (2nd panel). Following atRA treatment, Ser-10 phosphorylated p27 (that will be degraded) level was rapidly reduced (3rd panel), and as a result, the p27 protein level was increased (4th panel). Protein levels of CRABPI, CRABPII and actin remained constant (5th–7th panels). In order to validate whether the holo-CRABPI-activated ERK1/2 translocated to the nucleus to act on nuclear p27, we performed cellular fractionation experiments to examine total and phosphoprotein levels of both ERK1/2 and p27. As shown in the middle group (nuclear fractions), atRA indeed elicited a rapid (within the first time point, 1 hr) increase in nuclear ERK1/2 activation (1st panel, pERK1/2) without altering ERK1/2 protein levels (2nd panel). Importantly, there was also a rapid reduction in the nuclear ser-10-phosphorylated p27 level (3rd panel, p-p27-s10) with a concurrent increase in the nuclear p27 protein level (4th panel, p27). Interestingly, both CRABPI and CRABPII were barely detectable in the nuclear fraction, and were mostly distributed in the cytosolic fraction (see right group), under this exposure condition (5th & 6th panels). Lamin shows the control for nuclear fractions (7th panel). The cytosolic fractions were evaluated as shown in the right group. Upon atRA treatment, cytosolic pERK1/2 rapidly accumulated but then decreased (1st panel) without detectable changes in its protein level (2nd panel). Levels of cytosolic p27, CRABPI, α-tubulin remained unchanged. Based upon all these data, we observed a continuous increase in nuclear pERK1/2 (the middle group) and a decrease in cytosolic pERK1/2 (the right group at the bottom), with total ERK1/2 protein levels unaltered. These fractionation data would suggest that atRA activation of ERK1/2 occurs first in the cytoplasm and pERK1/2 is then translocated to the nucleus and accumulated there to, plausibly, act on its nuclear target. This would then modulate (suppress) p27 phosphorylation, and correspondingly causes an increase in nuclear p27 protein level. This is because phosphorylation prevents p27’s nuclear export [15]. Furthermore, silencing CRABPI blocked RA-triggered dephoshorylation of nuclear p-S10-p27, and as a result, CRABPI deficient cells failed to accumulate nuclear p27 (Fig. 4B).
Fig. 4. AtRA induces p27 nuclear accumulation by reducing p27 serine-10 phosphorylation.

(a) Kinetic analyses of relevant endogenous components, as well as loading and fractionation controls, in the whole cells (the upper group), the nuclear fractions (the middle group) and the cytosolic fractions (the lower group) of atRA-treated ESC. (b) Silencing CRABPI abolished atRA-triggered elevation in P27 levels, and, consistently, decreased the p-S10-P27 levels. Silencing and loading controls were shown at the two lower panels.
3.5 AtRA/CRABPI prolongs ESC G1 phase
Finally, to demonstrate the biological relevance of this signaling pathway to ESC, we performed a flow cytometry analysis to determine how CRABPI may play a role in cell cycle regulation of ESC transiently exposed to atRA. As shown in Figure 5b, under control silencing, atRA rapidly increased the fraction of G1 phases by approximately 18% (increase from approximately 32% to 38% of total cell population). This is consistent with the accumulation of p27 following atRA treatment. But when the endogenous CRABPI was silenced, the rapid effect of atRA to expand G1 population was abolished.
4. Discussion
In this study, we identified, for the first time, a functional role for CRABPI in mediating rapid activation of ERK1/2 by atRA in ESC, which is independent of RAR and membrane signals. We further demonstrated an important biological consequence of this non-canonical activity of atRA in ESC cycle regulation, primarily to elevate the level of a CDK inhibitor, p27, which expands the G1 population. Based upon these results, we summarized the signal transduction pathway that manifested the rapid non-canonical activity of atRA in the context of ESC cell cycle regulation as shown in Figure 5b. AtRA first binds to CRABPI in the cytoplasm, resulting in rapid ERK1/2 activation that is independent of membrane signal and RAR. The activated ERK1/2 translocates to the nucleus to stimulate dephosphorylation of p27 at serine 10. Dephosphorylation of p27 allows this protein to be retained and accumulated in the nucleus to inhibit G1-S progression. This current study elucidates such a non-canonical mechanism through which atRA rapidly disturbs ESC cell cycle homeostasis and prepares the cells for the execution of differentiation programs mediated by RARs. Presumably, CRABPI level could affect the sensitivity, or vulnerability, of the stem cells to extracellular stimuli or environmental insults. To this end, it is interesting that in another cell differentiation model, the fibroblast-adipocyte differentiation system, the crabp1 gene appears to undergo very specific epigenetic changes along the course of adipocyte differentiation [27].
ERK1/2 kinases play intricate roles in ES cell biology. ERK1/2 was first considered for self-renewal due to its constitutive basal activity in ESC cultures, but was surprisingly found to also promote ESC differentiation [28]. Further supporting this differentiation-promoting role of ERK1/2 came from the finding that genetic or pharmacological inhibition of this pathway blocks ES cell differentiation [29]. Therefore, a homeostatic control of ERK1/2 activity is crucial to the health of ESCs. To this end, it is thought that a prolonged MAPK/ERK signal leads to differentiation and/or growth arrest, while a short MAPK/ERK signal is mitogenic [30]. Our result suggests that not only the duration of MAPK/ERK activation, but also the context of the activation, contributes to the ultimate biological consequences. It would be of importance to understand the precise modes of ERK1/2 activation by atRA/CRABPI. Since chemicals disturbing membrane signals do not block the rapid effect of atRA on ERK1/2, it is unlikely that its mode of action adopts the canonical pathway of ERK1/2 activation.
In attempting to decipher how atRA/CRABPI might activate ERK1/2, we found a correlation between ligand binding of CRABPI and its ability to activate ERK1/2 (Figure 3). In addition to atRA, we found that two atRA metabolites, 4-oxo-RA and 4-hydroxy-RA, that could bind CRABPI, were also able to elicit the first phase ERK1/2 activation. 4-oxo-RA metabolite has been documented to have biological activities in a variety of contexts, but mostly attributable to its action through RARs. For instance, 4-oxo-RA could alter anteroposterial patterning in the Xenopus embryo, restore germ cell production in vitamin A–deficient mice, stimulate alveolar regeneration [26], and induce differentiation of promyelocytic leukemia cells [31]. Our finding that these atRA metabolites also elicited an activity through CRABPI would suggest other potential physiological or pathological actions of atRA metabolites. Additionally, it would be of potential therapeutic interest to profile the retinoid compounds that specifically bind CRABPI. While classical studies have shown the function of CRABPI primarily in regulating atRA metabolism, such as to sequester atRA [32], the exact physiological role for CRABPI remains unresolved. Our current study demonstrates a new functional role for CRABPI in cell cycle regulation. Interestingly, several genetic association studies have reported correlation of Crabp1 expression with cancers/tumors [33–35], mostly suggesting CRABPI as a tumor suppressor, which might be relevant to the presumptive role of CRABPI in modulating stem cells’ cell cycle progression as we detected in the ESC system. This current study challenges the classical dogma that CRABPI merely plays a seemingly more passive role as a binding protein, and indicates a necessity to re-evaluate the exact physiological function(s) of CRABPI.
Our identification of CRABPI as the direct mediator of atRA to rapidly activate ERK1/2 in multiple cellular backgrounds expands the reservoir through which atRA can elicit specific biological activities. AtRA/CRABPI-elicited rapid ERK1/2 activation may provide a dynamic and timely mechanism that most likely facilitates a certain cellular environment favorable to a particular signaling pathway or sensitive to a specific growth factor or signal input. In the case of ESC cultures, this may be key to their homeostasis, or the modulation of their sensitivity (such as to differentiation signals). Previously, we reported that atRA, also through its non-canonical activation of ERK1/2, could inhibit ESC stemness feature by converting transcription activator TR2 into its suppressive form to rapidly regulate the key stem cell regulator, Oct4 [11]. By rapidly augmenting Oct4 chromatin/transcription factor complex, atRA could also very quickly lower Oct4 level, which may be a prelude to the loss of stemness. This current study not only identifies the direct mediator of the non-canonical activity of atRA, but also expands the scope of atRA’s non-canonical activities that also include cell cycle regulation.
Cell cycle is regulated by numerous inputs that act on G1, S, G2 or M phases. ESC is characterized by a much shorter G1, which is due to timely phosphorylation of p27 and its subsequent degradation. While phosphorylation of p27 in naturally cycling cells is well understood, which can occur in the nucleus or cytosol and in different types of cells [15], but how dephosphorylation of phosphorylated (on ser-10) p27 is executed and controlled has remained elusive. This remains an active line of investigation. Since we detected apparent nuclear translocation of phosphorylated ERK1/2 (Figure 4), presumably nuclear pERK1/2 can activate certain phosphatase(s) to target p27. Further studies are needed to determine whether there are specific phosphatases that can be activated by ERK1/2 in the nucleus of ESC.
Highlights.
AtRA activates ERK1/2 in a biphasic manner
CRABPI mediates phase 1, non-canonical activation of ERK1/2 by atRA
Phase 1 ERK1/2 activation by atRA is independent of RAR and membrane signaling events
Non-canonical activity of atRA/CRABPI negatively modulates G1 progression
Acknowledgments
This work was supported by NIH grants DK54733, DK60521 and K02-DA13926, and Distinguished McKnight Professorship of University of Minnesota (L.-N. Wei). We thank technical assistance from Y Tsui and K Chang.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Duester G. Cell. 2008;134:921–931. doi: 10.1016/j.cell.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei LN. Annu Rev Pharmacol Toxicol. 2003;43:47–72. doi: 10.1146/annurev.pharmtox.43.100901.140301. [DOI] [PubMed] [Google Scholar]
- 3.Samarut E, Rochette-Egly C. Mol Cell Endocrinol. 2012;348:348–360. doi: 10.1016/j.mce.2011.03.025. [DOI] [PubMed] [Google Scholar]
- 4.Soprano DR, Teets BW, Soprano KJ. Vitamins and hormones. 2007;75:69–95. doi: 10.1016/S0083-6729(06)75003-8. [DOI] [PubMed] [Google Scholar]
- 5.D’Ambrosio DN, Clugston RD, Blaner WS. Nutrients. 2011;3:63–103. doi: 10.3390/nu3010063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross AC. Faseb J. 1993;7:317–327. doi: 10.1096/fasebj.7.2.8440409. [DOI] [PubMed] [Google Scholar]
- 7.Ochoa WF, Torrecillas A, Fita I, Verdaguer N, Corbalan-Garcia S, Gomez-Fernandez JC. Biochemistry. 2003;42:8774–8779. doi: 10.1021/bi034713g. [DOI] [PubMed] [Google Scholar]
- 8.Chen N, Napoli JL. Faseb J. 2008;22:236–245. doi: 10.1096/fj.07-8739com. [DOI] [PubMed] [Google Scholar]
- 9.Nayak S, Shen M, Bunaciu RP, Bloom SE, Varner JD, Yen A. Leuk Lymphoma. 2010;51:1734–1747. doi: 10.3109/10428194.2010.501535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J, Zhou R, He Q, Li WI, Zhang T, Niu B, Zheng X, Xie J. Neurotoxicology. 2009;30:599–604. doi: 10.1016/j.neuro.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 11.Gupta P, Ho PC, Huq MM, Ha SG, Park SW, Khan AA, Tsai NP, Wei LN. Proc Natl Acad Sci U S A. 2008;105:11424–11429. doi: 10.1073/pnas.0710561105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chuang YS, Huang WH, Park SW, Persaud SD, Hung CH, Ho PC, Wei LN. Stem Cells. 2011;29:660–669. doi: 10.1002/stem.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orford KW, Scadden DT. Nat Rev Genet. 2008;9:115–128. doi: 10.1038/nrg2269. [DOI] [PubMed] [Google Scholar]
- 14.Guan K, Chang H, Rolletschek A, Wobus AM. Cell Tissue Res. 2001;305:171–176. doi: 10.1007/s004410100416. [DOI] [PubMed] [Google Scholar]
- 15.Starostina NG, Kipreos ET. Trends Cell Biol. 2012;22:33–41. doi: 10.1016/j.tcb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chu IM, Hengst L, Slingerland JM. Nat Rev Cancer. 2008;8:253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 17.Wander SA, Zhao D, Slingerland JM. Clin Cancer Res. 2011;17:12–18. doi: 10.1158/1078-0432.CCR-10-0752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho PC, Gupta P, Tsui YC, Ha SG, Huq M, Wei LN. Cellular signalling. 2008;20:1911–1919. doi: 10.1016/j.cellsig.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ho PC, Tsui YC, Lin YW, Persaud SD, Wei LN. Molecular and cellular endocrinology. 2012;351:176–183. doi: 10.1016/j.mce.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gupta P, Ho PC, Ha SG, Lin YW, Wei LN. PloS one. 2009;4:e4363. doi: 10.1371/journal.pone.0004363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kagechika H, Kawachi E, Hashimoto Y, Shudo K. J Med Chem. 1989;32:834–840. doi: 10.1021/jm00124a016. [DOI] [PubMed] [Google Scholar]
- 22.Ho PC, Lin YW, Tsui YC, Gupta P, Wei LN. Cell metabolism. 2009;10:516–523. doi: 10.1016/j.cmet.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta P, Ho PC, Huq MM, Ha SG, Park SW, Khan AA, Tsai NP, Wei LN. Proc Natl Acad Sci U S A. 2008;105:11424–11429. doi: 10.1073/pnas.0710561105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heo JS, Lee YJ, Han HJ. Am J Physiol Cell Physiol. 2006;290:C123–133. doi: 10.1152/ajpcell.00142.2005. [DOI] [PubMed] [Google Scholar]
- 25.Fogh K, Voorhees JJ, Astrom A. Arch Biochem Biophys. 1993;300:751–755. doi: 10.1006/abbi.1993.1104. [DOI] [PubMed] [Google Scholar]
- 26.Maden M. Am J Respir Cell Mol Biol. 2006;35:260–267. doi: 10.1165/rcmb.2006-0029OC. [DOI] [PubMed] [Google Scholar]
- 27.Wei LN. Biochim Biophys Acta. 2012;1821:206–212. doi: 10.1016/j.bbalip.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burdon T, Smith A, Savatier P. Trends Cell Biol. 2002;12:432–438. doi: 10.1016/s0962-8924(02)02352-8. [DOI] [PubMed] [Google Scholar]
- 29.Wang J, Yen A. J Biol Chem. 2008;283:4375–4386. doi: 10.1074/jbc.M708471200. [DOI] [PubMed] [Google Scholar]
- 30.Traverse S, Gomez N, Paterson H, Marshall C, Cohen P. Biochem J. 1992;288(Pt 2):351–355. doi: 10.1042/bj2880351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de The H, Chen Z. Nat Rev Cancer. 2010;10:775–783. doi: 10.1038/nrc2943. [DOI] [PubMed] [Google Scholar]
- 32.Fiorella PD, Napoli JL. J Biol Chem. 1991;266:16572–16579. [PubMed] [Google Scholar]
- 33.Miyake T, Ueda Y, Matsuzaki S, Miyatake T, Yoshino K, Fujita M, Nomura T, Enomoto T, Kimura T. J Cancer Res Clin Oncol. 2011;137:715–722. doi: 10.1007/s00432-010-0930-8. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka K, Imoto I, Inoue J, Kozaki K, Tsuda H, Shimada Y, Aiko S, Yoshizumi Y, Iwai T, Kawano T, Inazawa J. Oncogene. 2007;26:6456–6468. doi: 10.1038/sj.onc.1210459. [DOI] [PubMed] [Google Scholar]
- 35.Hawthorn L, Stein L, Varma R, Wiseman S, Loree T, Tan D. Head Neck. 2004;26:1069–1083. doi: 10.1002/hed.20099. [DOI] [PubMed] [Google Scholar]