

Abstract
Cripto-1 is implicated in multiple cellular events, including cell proliferation, motility and angiogenesis, through the activation of an intricate network of signaling pathways. A crosstalk between Cripto-1 and the canonical Wnt/β-catenin signaling pathway has been previously described. In fact, Cripto-1 is a downstream target gene of the canonical Wnt/β-catenin signaling pathway in the embryo and in colon cancer cells and T-cell factor (Tcf)/lymphoid enhancer factor binding sites have been identified in the promoter and the first intronic region of the mouse and human Cripto-1 genes. We now demonstrate that Cripto-1 modulates signaling through the canonical Wnt/β-catenin/Tcf pathway by binding to the Wnt co-receptors low-density lipoprotein receptor-related protein (LRP) 5 and LRP6, which facilitates Wnt3a binding to LRP5 and LRP6. Cripto-1 functionally enhances Wnt3a signaling through cytoplasmic stabilization of β-catenin and elevated β-catenin/Tcf transcriptional activation. Conversely, Wnt3a further increases Cripto-1 stimulation of migration, invasion and colony formation in soft agar of HC11 mouse mammary epithelial cells, indicating that Cripto-1 and the canonical Wnt/β-catenin signaling co-operate in regulating motility and in vitro transformation of mammary epithelial cells.
Keywords: Cripto-1, Wnt signaling, β-catenin, LRP5, LRP6
1. Introduction
Cripto-1, also known as teratocarcinoma-derived growth factor-1 (TDGF-1), was initially isolated from human NTERA2 and mouse F9 embryonal carcinoma cells [1, 2]. Human and mouse Cripto-1 belong to the EGF-Cripto-1/FRL1/Cryptic (CFC) gene family and are localized in lipid rafts regions of the plasma membrane via a glycosilphosphatidylinositol (GPI) anchor [3]. Cripto-1 is expressed during early embryogenesis, functioning as a co-receptor for the transforming growth factor-β (TGF-β) family members Nodal, growth differentiation factor (GDF) 1 and 3, and enabling them to bind to serine/threonine kinase activin type I receptors Alk4 and Alk7 and activin type II receptor complex [4, 5]. Binding to the Alk4/7 and type II receptor complex in turn triggers activation and phosphorylation of the cytoplasmic receptor-activated Smad proteins, Smad2 and Smad3, which can then heterodimerize with Smad4 and activate gene transcription. Cripto-1 is expressed in human and mouse ES cells and regulates ES cell self-renewal and pluripotency [4]. In the adult mouse and human tissues, Cripto-1 expression is absent and/or very low. However, Cripto-1 is re-expressed in several different types of human carcinomas including gastric, breast, colon, lung, pancreatic, bladder, cervical, skin and ovarian cancers [4]. Furthermore, Cripto-1 functions as an oncogene in vitro, promoting cell proliferation, migration, invasion and epithelial to mesenchymal transition in a variety of epithelial cells [4-6].
The canonical Wnt/β-catenin/T-cell factor (Tcf) signaling pathway, similarly to Cripto-1, also performs essential functions in the maintenance of ES cell self-renewal and pluripotency, during different stages of embryonic development and in the etiology of several types of cancers in the colon, liver, skin, lung, breast, pancreas, ovary and prostate [7]. The Wnt/β-catenin signaling pathway is activated by binding of Wnt ligands, which are secreted palmitoylated glycoproteins, to a family of seven-pass transmembrane Frizzled (Fz) receptors and to single-pass transmembrane low-density lipoprotein receptor-related protein (LRP) 5 and LRP6 co-receptors [7]. In the absence of Wnt ligands, cytosolic β-catenin is phosphorylated by an Axin/adenomatous polyposis coli (APC)/glycogen synthase kinase 3β (GSK3β) complex and targeted for proteosomal degradation [7]. Conversely, activation of the Wnt/β-catenin signaling by secreted Wnts leads to inactivation of the Axin/APC/GSK3β complex and stabilization of cytosolic β-catenin. This facilitates β-catenin translocation into the nucleus and interaction with the Tcf/lymphoid enhancer factor (Lef) transcription factor family leading to the transcription of target genes [8].
A cross-talk between Cripto-1 and the canonical Wnt/β-catenin signaling pathway has been previously reported. For instance, mouse and human Cripto-1 have been identified as target genes in the Wnt/β-catenin signaling pathway during mouse embryogenesis and in mouse and human colon cancer cells [9, 10]. Moreover, FRL-1, an ortholog of Cripto-1 in Xenopus, can function as a co-receptor for Wnt 11, a non-canonical Wnt ligand, together with Xenopus Fz7 and the heparan sulphate proteoglycan Glypican-4, leading to the activation and stabilization of β-catenin [11].
In the present study, we demonstrate that mouse and human Cripto-1 enhance signaling through the canonical Wnt/β-catenin signaling pathway because of their ability to bind to LRP5 and LRP6 co-receptors. Conversely, Wnt3a increases Cripto-1 stimulation of migration, invasion and colony formation in soft agar of HC11 mouse mammary epithelial cells. These findings define novel activities of Cripto-1 and the canonical Wnt/β-catenin signaling pathway, revealing a new interaction between these two major signaling pathways that might be important in development and disease.
2. Materials and methods
2.1. Cell culture
293T cells (ATCC, Manassas, VA), 293T/Cripto-1 overexpressing cells [12], and NCCIT cells (ATCC) were cultured in DMEM containing 10% FBS. Mouse teratocarcinoma F9 cells were purchased from ATCC while F9 Cripto-1-/- cells were kindly provided by Dr. Michele Sanicola (Biogen-Idec, Cambridge, MA). F9 and F9 Cripto-1-/- cells were maintained in high glucose DMEM containing 10% FBS on gelatin coated cell culture plates. HC11 and HC11/Cripto-1 overexpressing cells [13] were grown in RPMI medium supplemented with 10%FBS.
2.2. Dual-luciferase assay
293T, F9 or F9 Cripto-1-/- cells were seeded in 24 well plates (5×104 cells/well) and incubated at 37°C, 5% CO2 overnight. Cells were then transfected with SuperTOPFLASH luciferase vector (50 ng for 293T cells and 500 ng for F9 or F9 Cripto-1-/- cells), 50 ng SuperFOPFLASH luciferase control vector or with 250 ng of p(n2)7-luciferase reporter construct together with pTK-Renilla (5 ng for 293T cells and 50 ng for F9 or F9 Cripto-1-/- cells) using LipoD293 transfection reagent (SignaGen Laboratories, Rockville, MD). After 5 h incubation, medium was changed to DMEM containing 0.5% FBS and cells were incubated for additional 16-20 h in the presence or absence of different concentrations of recombinant human (rh) Wnt3a (R&D Systems, Minneapolis, MN) alone or in combination with recombinant mouse (rm) Dkk1 (R&D Systems) for 24 h. The Alk4 inhibitor SB431542 (10 μM) (Ascent Scientific, Bristol, UK) was added to the cells after transfection for 16–20 h and together with rhWnt3a, during stimulation. The luciferase activity was measured using the Dual-luciferase reporter assay kit (Promega, Madison, WI) according to the manifacturer's instructions. These experiments were repeated three times with triplicate samples.
2.3. Co-immunoprecipitation assays
LRP6-GFP (kindly provided by Dr. Akira Kikuchi, Osaka University, Osaka, Japan), LRP5-turbo GFP (tGFP) (Origene, Rockville, MD), Wnt3a-HA (Millipore, Bedford, MA), Fz8 cysteine rich domain (CRD)-Fc (Addgene Inc., Cambridge, MA) [14], LRP5ΔC-myc (gift from Dr. Matthew Warman, Boston Children's Hospital, Boston, MA) [15], 3×FLAG-CR-1 WT, 3×-FLAG-CR-1 ΔEGF, 3×-FLAG-CR-1 ΔCFC, or 3×-FLAG-CR-1 ΔEGF ΔCFC plasmids were co-transfected into 293T cells, as previously described [16]. After 24 h, cells were harvested and lysed using radio immunoprecipitation assay (RIPA) buffer (50 mM Tris-Cl [pH 8.0], 150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholic acid, 0.1% SDS, 10 mg/ml Aprotinin, 10 mg/ml Leupeptin, 1 mM PMSF). For anti-FLAG immunoprecipitation, anti-FLAG M2 affinity gel (Sigma, St Louis, MO) was added to the cell lysates and incubated with rotation for 1 h at 4 °C. For anti-tGFP and anti-GFP immunoprecipitation, anti-tGFP (2 μg/ml) or anti-GFP (6 μg/ml) antibodies were added into the lysate and incubated with rotation for 3 h at 4 °C and then Protein G sepharose beads (GE Healthcare, Buckinghamshire, UK) were added and incubated for an additional hour. Beads were washed four times with high salt RIPA buffer containing 300 mM NaCl. Immunoprecipitated proteins were eluted using 2× Laemmli SDS sample buffer and boiled for 5 min. In the biotinylation assay, 293T cells, transiently expressing 3×FLAG-CR-1, LRP5-tGFP and LRP6-GFP, were washed with ice cold PBS (pH 8.0) and incubated with 2 mM NHS-PEG4-Biotin (Pierce, Rockford, IL) for 30 min on ice to avoid the internalization of the cell surface proteins. The cells were then washed twice with PBS containing 100 mM glycine to quench the reaction and to remove the excess biotin reagent and byproducts. Cells were then lysed using RIPA buffer and immunoprecipitation was performed as described above. Beads were then incubated with 1% SDS for 80 min at 37 °C and supernatant was collected. Streptoavidin agarose beads (Pierce) were added to the supernatant and mixed on a shaker for 3 h at 4 °C. The beads were washed three times with RIPA buffer and proteins eluted with 2× Laemmli SDS sample buffer. For the endogenous co-immunoprecipitation assay, NCCIT cells were lysed using a modified RIPA buffer (20 mM Tris-Cl [pH 8.0], 137 mM NaCl, 10% Glycerol, 1% Nonidet P-40, 1% deoxy-cholate, 10 mM NaF, 2 mM EDTA, 2 mM EGTA, 10 mg/ml Aprotinin, 10 mg/ml Leupeptin, 1 mM PMSF). 3 mg of proteins were incubated with anti-Cripto-1 antibody (1:50, V-17, Santa Cruz Biotechnology, Santa Cruz, CA) or anti-LRP6 antibody (1:50, C-10, Santa Cruz Biotechnology) and gently rotated for 16 h at 4 °C. Protein G magnetic sepharose beads (GE Healthcare) were then added for 2 h at 4 °C, Beads were washed with modified RIPA buffer four times and proteins were eluted with 80 μl of 2× Laemmli SDS sample buffer.
2.4. Western blot analysis
Western blot analysis was performed as previously described [17]. Membranes were incubated with the following antibodies: anti-FLAG M2-HRP (1:2000, Sigma), anti-tGFP (1:1000, Origene), anti-GFP (1:1000, Roche, Indianapolis, IN), anti-human Cripto-1 (1:1000, Epitomics, Burlingame, CA), anti-β-catenin (1:2000, BD Biosciences, Franklin Lakes, NJ), anti-mouse Cripto-1 (1:1000, Cell Signaling Technology, Danvers, MA), anti-phospho LRP6 (1:1000, Cell Signaling Technology), anti-LRP5 (C-20, 1:1000, Santa Cruz Biotechnology), anti-LRP6 (C-10, 1:500, Santa Cruz Biotechnology), anti-β-actin-HRP (1:50000, Sigma), anti-phospho Smad2 S465/467 (1:1000, Cell Signaling Technology), and anti-Smad2 (L16D3, 1:1000, Cell Signaling Technology). Primary antibodies bound to their specific antigens were detected with anti-mouse IgG-HRP (1:2000, GE Healthcare) or anti-rabbit IgG-HRP (1:5000, Jackson Immuno Research, West Grove, PA) secondary antibodies, followed by ECL plus Western blot detection system. To detect the Cripto-1 protein co-immunoprecipitated together with LRP6 in NCCIT cells, anti-rabbit IgG Fc specific-HRP (1:10000, Jackson Immuno Research) was used as the secondary antibody. Densitometric analysis of the bands on the Western blots was performed using the NIH image program Image J available at http://rsbweb.nih.gov/nih-image/.
2.5. Immunocytochemistry
Immunofluorescence studies were conducted according to standard methods [18]. 293T or 293T/Cripto-1 cells transiently transfected with LRP6-GFP expression plasmid in 8-chamber coverglass slides were fixed in 4% paraformaldehyde for 10 mins, permeabilized in 0.1% Triton-X100 for 10 mins, and blocked in non-mammalian blocking buffer (Li-Cor Biosciences, Lincoln, NE) for 1 hr. The fixed cells were probed at 4° C overnight with primary antibodies in blocking buffer. The Cripto-1 rabbit monoclonal antibody (Epitomics) was diluted to a concentration of 85 μg/ml. Monoclonal mouse LRP6 antibody (R&D Systems) was diluted to 20 μg/ml. Cells were then washed three times with PBS. Alexa-fluor 488 goat anti-mouse (Invitrogen) and Alexa-fluor 594 goat anti-rabbit (Invitrogen) were diluted to a concentration of 10 μg/ml in blocking buffer. Cells were incubated with the secondary antibodies for 1 hr, and then washed three times with PBS. Cells were stained for 10 mins with the nuclear stain Hoechst 33258 (Invitrogen), which was diluted to a concentration of 2 μg/ml in PBS, and then rinsed three times with PBS. To control for background signal, control chambers were incubated with blocking buffer without the primary antibodies, and then followed by the secondary antibodies and Hoechst staining (data not shown). The cells were imaged on a LSM510 confocal microscope (Carl Zeiss, Inc., Thornwood, NY) using a 63× oil objective.
2.6. Wnt3a stimulation and GST-E-cadherin pull-down assay
F9 cells or F9 Cripto-1-/- cells (5×105 cells/plate) were seeded into 60 mm plates and incubated in serum containing 10% FBS DMEM medium for 24 h. Plates were then washed in PBS and incubated in DMEM 0.5% FBS for 16–20 h. Cells were then stimulated with various concentrations of rhWnt3a protein and incubated for various times. Cells were harvested, washed three times with ice cold PBS and lysed with lysis buffer (50 mM HEPES [pH 7.5], 10 mM sodium pyrophosphate, 50 mM NaF, 50 mM NaCl, 1 mM Na3VO4, 1% Triton X-100, 10 mg/ml Aprotinin, 10 mg/ml Leupeptin, 1 mM EDTA, 1 mM PMSF). 400 mg of proteins were incubated with 3 mg of recombinant GST-E-cadherin protein at 4 °C for 16 h. Then 20 μl of Glutathione-agarose beads (Santa Cruz Biotechnology) were added and incubated at 4 °C for 1 h. Glutathione-agarose beads were washed three times with lysis buffer and resuspended into 2× Laemmli SDS sample buffer and boiled for 5 min.
2.7. Real time PCR array and semi-quantitative PCR
Total RNA was extracted from F9 or F9 Cripto-1-/- cells stimulated for 18 hours with rhWnt3a (100ng/ml), using RNeasy Mini kit (QIAGEN, Hilden, Germany) according to the manufacturer's protocol.Two micrograms of total RNA was used for cDNA synthesis using the RETROscript kit (Ambion,Austin, TX) following the manufacturer's instructions. cDNA was used for qPCR with RT2 SYBR greenMaster Mix (QIAGEN) using RT2 Profiler PCR array (96 well plate) mouse Wnt signaling genes(QIAGEN, PAMM-243A). qPCR reaction was performed for 40 cycles (95 °C for 15 s, 60 °C for 60 s)using the Mx3005P QPCR System (Stratagene, La Jolla, CA). qPCR results were analyzed using the PCRarray data analysis software program (QIAGEN). For the semi-quantitative PCR, two microliters ofcDNA were amplified using Taq PCR master mix (QIAGEN) and PCR reaction was performed for 30cycles consisting of 94 °C for 30 s, 60 °C for 30 s and 72 °C for 30s. Primers used in the PCR reactionare the following: mouse Dkk1 (Forward: 5′-GCGGCAGCTGTCCGGTTCTT-3′, Reverse: 5′-AGCGCAAGGGTAGGGCTGGT-3′), mouse Cripto-1 (Forward: 5′TGTTCGCAAAGAGCACTGTGG-3′, Reverse: 5′-TGAGGTCCTGGTCCATCACTTGAC-3′) andmouse GAPDH (Forward: 5′-CCCTTCATTGACCTCAACTAC-′, Reverse: 5′-CCACCTTCTTGATGTCATCAT-3′). pSPORT1-Dkk1 (10 ng) (Open Biosystems, Huntsville, AL) was used as a positive control in the PCR reaction.
2.8. Cripto-1 re-expression in F9 Cripto-1-/- cells
F9 and F9 Cripto-1-/- cells were transfected according to manufacturer instructions with mPol2 empty vector or mPol2-Cr-1 lentiviral vectors (SAIC-Frederick, MD) and subsequently incubated with culture medium containing 5 μg/ml puromycin for 2 weeks. Stably transfected cells were then assessed in SuperTOPFLASH luciferase assays and Western blotting assays, as described above.
2.9. Migration and invasion assays
Migration and invasion assays were performed on fibronectin-coated (Chemicon, Temecula, CA) or Matrigel-coated Boyden chambers (Chemicon) as previously described [19, 20]. Briefly, serum starved HC11 and HC11/Cripto-1 cells were seeded in Boyden chambers at 3×105 cells/well in 24-well plates in the presence or absence of 50 ng/ml rhWnt3a and were incubated overnight at 37° C. Cells on the top side of the filter were removed. Migrated or invaded cells on the bottom side of the filter were fixed in methanol and stained with Hema 3 Stat Pack kit (Fisher Scientific, Hampton, NH). Ten random fields in each well were counted. In all the experiments, 2% fetal bovine serum was used as chemoattractant in the lower chamber. These experiments were repeated three times with duplicate samples.
2.10. Soft-agar assay
Anchorage-independent cell growth in soft-agar was performed as previously described [13]. Briefly, HC11 and HC11/Cripto-1 cells (5×104 cells/well) were seeded in 12-multiwell plates in the absence or presence of rhWnt3a recombinant protein (50 ng/ml). Treatment with rhWnt3a was repeated every three days for the entire length of the experiment. After 12 days, colonies were stained with nitroblue tetrazolium (Sigma) and counted. Soft-agar assay was performed in triplicate and repeated two times.
2.11. Statistical analysis
Two-tailed Student's t test was used to determine the statistical significance of the quantitative results. Results with a P value <0.05 were considered statistically significant.
3. Results
3.1. Cripto-1 enhances the canonical Wnt/β-catenin/Tcf signaling pathway in 293T cells
To determine if Cripto-1 expression might alter the sensitivity of cells to Wnt/β-catenin signaling, we performed Tcf/Lef responsive SuperTOPFLASH luciferase assays in human kidney 293T cells, which do not express Cripto-1 [21]. We constructed a full-length Cripto-1 expression vector containing a 3×-FLAG tag (3×-FLAG-CR-1 WT) and three deletion mutant expression vectors, including 3×-FLAG-CR-1 ΔEGF plasmid lacking the EGF-like domain of Cripto-1, 3×-FLAG-CR-1 ΔCFC plasmid lacking the CFC domain of Cripto-1, and 3×-FLAG-CR-1 ΔEGF ΔCFC plasmid lacking both EGF and CFC domains (Fig. 1A). The full-length or deletion mutant constructs were then co-transfected into 293T cells together with a SuperTOPFLASH luciferase reporter vector and subsequently treated with recombinant human Wnt3a protein. Wnt3a strongly increased SuperTOPFLASH luciferase activity in 293T cells and co-expression of a full-length 3×-FLAG-CR-1 WT plasmid further enhanced in a dose-dependent manner Wnt3a stimulation of SuperTOPFLASH luciferase activity in 293T cells (Fig. 1B). In contrast, 3×-FLAG-CR-1 ΔEGF and 3×-FLAG-CR-1 ΔCFC plasmids induced only a modest not statistically significant increase in SuperTOPFLASH luciferase activity after Wnt3a treatment, as compared to Wnt3a only treated 293T cells (Fig. 1B). Finally, the 3×-FLAG-CR-1 ΔEGF ΔCFC plasmid was unable to potentiate the ability of Wnt3a to activate SuperTOPFLASH luciferase activity in 293T cells, indicating that both the EGF and CFC domains of Cripto-1 are required to enhance a canonical Wnt/β-catenin signaling pathway in 293T cells. No luciferase activity was detected following transfection of 293T cells with the control SuperFOPFLASH reporter luciferase vector either in the presence of Wnt3a alone or Wnt3a together with a 3×-FLAG CR-1 WT plasmid (Fig. 1B). A soluble form of Cripto-1, lacking the GPI anchor, which tethers Cripto-1 to the cell membrane (Fig. 2A), was unable to potentiate Wnt3a induction of a SuperTOPFLASH luciferase reporter construct in 293T cells, indicating that Cripto-1 attachment to the cell membrane is required for its modulation of a canonical Wnt/β-catenin signaling pathway in 293T cells (Fig. 2B).
Fig. 1.

Cripto-1 enhances Wnt3a induced SuperTOPFLASH luciferase activity in 293T cells. (A) Full-length (CR-1 WT) and deletion constructs (CR-1 ΔEGF lacking the EGF-like domain, CR-1 ΔCFC lacking the CFC domain, CR-1 ΔEGF ΔCFC lacking both EGF and CFC domains) of human Cripto-1 in a 3×-FLAG expression vector. (B) SuperTOPFLASH luciferase reporter assay in 293T cells transiently transfected with different concentrations of full-length 3×-FLAG CR-1 WT plasmid (1, 10 and 100 ng) or with 3×-FLAG CR-1 ΔEGF, 3×-FLAG CR-1 ΔCFC, 3×-FLAG CR-1 ΔEGF ΔCFC deletion constructs (10 and 100 ng) together with 50 ng of SuperTOPFLASH luciferase vector. Cells were subsequently treated with rhWnt3a protein (50 ng/ml) for 16-20 hr. Cells were also transfected with 50 ng of SuperFOPFLASH luciferase vector and subsequently stimulated with rhWnt3a. These results are the mean ±SD of triplicates from one of three separate experiments.
Fig. 2.

Cripto-1 lacking GPI-anchor does not enhance the canonical Wnt/β-catenin signaling pathway stimulated by Wnt3a in 293T cells. (A) Full-length (CR-1 WT) and GPI-anchor signal sequence deletion mutant (CR-1 ΔC) of human Cripto-1 in a 3×FLAG expression vector. (B) SuperTOPFLASH luciferase assay in 293T cells transiently transfected with different concentrations of 3×FLAG-CR-1 WT or 3×FLAG-CR-1 ΔC (10 and 100 ng) together with 50 ng of SuperTOPFLASH luciferase vector. Cells were subsequently treated with 50 ng/ml of rhWnt3a protein for 16-20 hr. These results are the mean ±SD of triplicates from one of three separate experiments.
3.2. Cripto-1 binds to LRP5 and LRP6 co-receptors on the cell surface of 293T and 293T/Cripto-1 cells
To elucidate the mechanism by which Cripto-1 enhances the canonical Wnt/β-catenin signaling pathway in response to Wnt3a stimulation, we evaluated if components of the Wnt/β-catenin signaling pathway might interact with Cripto-1 in a co-immunoprecipitation assay in 293T cells. The full-length 3×-FLAG-CR-1 WT plasmid was transiently expressed in 293T cells with the following expression vectors: Wnt3a-HA, the Wnt receptor Fz8 CRD-Fc and/or the Wnt co-receptor LRP5ΔC-myc. LRP5ΔC-myc was co-immunoprecipitated with 3×-FLAG-CR-1 WT in transiently transfected 293T cells (Fig. 3). Wnt3a-HA weakly co-immunoprecipitated with 3×FLAG-CR-1 WT, while Fz8 CRD-Fc co-immunoprecipitated with 3×-FLAG-CR-1 WT only in the presence of LRP5ΔC-myc and Wnt3a-HA (Fig. 3). Therefore, we evaluated whether Cripto-1 might directly interact with LRP5 and/or LRP6 in 293T cells. The full-length 3×FLAG-CR-1 WT plasmid or the deletion mutants 3×-FLAG-CR-1 ΔEGF, 3×-FLAG-CR-1 ΔCFC or 3×-FLAG-CR-1 ΔEGF ΔCFC constructs were transiently expressed in 293T cells together with an LRP5-tGFP expression plasmid and binding of these membrane proteins was evaluated by co-immunoprecipitation assays. LRP5-tGFP co-immunoprecipitated by the anti-FLAG antibody with both the full-length 3×FLAG-CR-1 WT and 3×-FLAG-CR-1 ΔEGF, although LRP5-tGFP co-immunoprecipitated to a lesser extent with the 3×-FLAG-CR-1 ΔCFC and 3×-FLAG-CR-1 ΔEGF ΔCFC deletion mutants (Fig. 4A). Reciprocal co-immunoprecipitation assays were also performed with an anti-tGFP antibody and full-length 3×-FLAG-CR-1 WT co-immunoprecipitated with LRP5-tGFP, while the 3×-FLAG-CR-1 ΔEGF or 3×-FLAG-CR-1 ΔCFC were less effective in binding to LRP5-tGFP in the co-immunopreciptation assays in 293T cells (Fig. 4B). In contrast, 3×-FLAG-CR-1 ΔEGF ΔCFC deletion mutant plasmid failed to bind to LRP5-tGFP in this pull-down assay (Fig. 4B). To verify proper transfection of different plasmids into 293T cells, we also performed Western blot analysis for the transfected proteins in the cell lysates, as shown in Figure 4A-B. Similar to LRP5-tGFP, LRP6-GFP also co-immunoprecipitated by the anti-FLAG antibody with 3×-FLAG-CR-1 WT and to a lesser extent with 3×-FLAG-CR-1 ΔEGF and 3×-FLAG-CR-1 ΔCFC in 293T cells, while no binding of LRP6-GFP to 3×-FLAG-CR-1 ΔEGF ΔCFC was detected (Fig. 4C). Similar binding data were obtained in reciprocal co-immunoprecipitation assays using the anti-GFP-antibody in 293T cells to pull down LRP6 with the different FLAG-tagged Cripto-1 proteins (Fig. 4D). Since Cripto-1, LRP5 and LRP6 reside on the cell membrane, we evaluated whether binding of these proteins occurs on the cell surface by biotinylation of membrane proteins. 3×-FLAG-CR-1 WT and LRP5-tGFP or LRP6-GFP co-transfected 293T cells were treated with plasma membrane impermeable NHS-PEG4-Biotin to biotinylate cell surface proteins. After co-immunoprecipitation with specific antibodies, a streptavidin affinity precipitation was performed in order to isolate biotinylated surface proteins [22]. As shown in Figure 5A-D, interaction between 3×-FLAG-CR-1 WT and LRP5-tGFP or LRP6-GFP was detected after streptavidin affinity purification of immunoprecipitated biotynilated membrane proteins, indicating that binding between these molecules occurs on the surface of the cells. We then evaluated whether interaction between Cripto-1 and LRP6 occurs between endogenously expressed proteins. Co-immunoprecipitation assays were therefore performed in NCCIT human embryonal carcinoma cells, which express high levels of endogenous Cripto-1 and LRP6 proteins [23]. As shown in Figure 5E, LRP6 was co-immunoprecipitated with Cripto-1 in NCCIT cells. Reciprocally, Cripto-1 was also found to bind to LRP6 in a co-immunoprecipitation assay in NCCIT cells (Fig. 5F). Furthermore, immunofluorescence studies in 293T/Cripto-1 cells transiently transfected with LRP6-GFP expression vector revealed that Cripto-1 was strongly co-localized with LRP6 on the cell membrane (Fig. 6). No specific immunofluorescence staining for Cripto-1 was detected in 293T cells lacking Cripto-1 expression (data not shown). Taken together, these results indicate that Cripto-1 binding to LRP5 and LRP6 occurs mainly on the cell surface.
Fig. 3.

Interaction between CR-1 and Wnt ligand or Wnt receptors. Mouse Wnt3a-HA, human Fz8 CRD-Fc, human LRP5ΔC-myc and full-length 3×FLAG-CR-1 WT expression plasmids were co-transfected into 293T cells. After 24 h, cells were harvested and lysed in RIPA buffer. The cell lysates were treated with 10 μl of anti-FLAG M2 affinity gel for 2 h at 4 °C. The Affinity gels were washed with RIPA buffer for 4 times. Proteins were then eluted by 2× Laemmli SDS sample buffer and separated by SDS-PAGE. Western blotting was carried out using following antibodies: anti-FLAG, anti-myc, anti-HA and anti-human IgG-HRP.
Fig. 4.

Cripto-1 binds to LRP5 and LRP6 in 293T cells. 293T cells were co-transfected with LRP-5-tGFP or LRP6-GFP expression vectors together with Cripto-1 full-length or deleted expression plasmids. 3×-FLAG Cripto-1 was immunoprecipated using an anti-FLAG mouse monoclonal antibody and the immunoprecipitated proteins were analyzed by Western blot with anti-tGFP, anti-GFP and anti-FLAG mouse monoclonal antibodies (A and C). LRP5-tGFP or LRP6-GFP were immunoprecipitated with anti-tGFP or anti-GFP mouse monoclonal antibody and immunoprecipitated proteins were analyzed by Western blot with anti-tGFP, anti-GFP and anti-FLAG mouse monoclonal antibodies (B and D). Cell lysates of 293T cells transiently transfected with various expression vectors as described above were analyzed by Western blot for Cripto-1 using an anti-FLAG antibody, for LRP5 using anti-tGFP antibody and for LRP6 using anti-GFP antibody (A through D).
Fig. 5.

Cripto-1 binds to LRP5 and LRP6 co-receptors on the cell surface. 293T cells transiently transfected with Cripto-1, LRP5 and/or LRP6 expression plasmids were incubated with 2mM NHS-PEG4-Biotin and then lysed using RIPA buffer. Following immunoprecipitation with anti-FLAG or anti-GFP monoclonal antibodies, cell surface biotinylated proteins were isolated by adding streptavidin agarose beads to the immunoprecipitated proteins for 3 h at 4 °C. A and C show co-immunoprecipitation of LRP5-tGFP (A) or LRP6-GFP (C) with 3×-FLAG CR-1 WT only in the presence of biotin. B and D show co-immunoprecipitation of 3×-FLAG CR-1 WT with LRP5-tGFP (B) or LRP6-GFP (D) only in the presence of biotin. Cell lysates of immunoprecipitated samples were also analyzed by Western blot to ensure expression of transfected plasmids. (E) Endogenous Cripto-1 and LRP6 co-immunoprecipitate in NCCIT human embryonal carcinoma cells. NCCIT protein lysates were immunoprecipitated with an anti-Cripto-1 (V-17) rabbit polyclonal antibody and probed with an LRP6 monoclonal antibody (E) or immunoprecipitated with an anti-LRP6 monoclonal antibody and probed with and anti-Cripto-1 rabbit polyclonal antibody (F). * indicates the immunoglobulin light chain.
Fig. 6.

Cripto-1 and LRP6 co-localize on the cell membrane. 293T/Cripto-1 cells transiently transfected with LRP6-GFP expression vector were stained with anti-LRP6 (green) and anti-Cripto-1 (red) antibodies. Nuclei were visualized by Hoechst 33258 (blue). The merged image shows significant co-localization of Cripto-1 and LRP6 on the cell membrane. The arrows indicate clear examples of co-localization.
3.3. Cripto-1 potentiates Wnt3a induced β-catenin signaling in F9 mouse embryonal carcinoma cells
We next evaluated the biological significance of the interaction between Cripto-1 with the LRP5 and LRP6 co-receptors in F9 and F9 Cripto-1-/- cells. F9 cells are mouse embryonal carcinoma cells that express high levels of endogenous Cripto-1, while F9 Cripto-1-/- cells carry a deletion of the Cripto-1 gene and are therefore null for Cripto-1 expression [24]. We therefore assessed if there are differences in the levels of stabilized active β-catenin in F9 and F9 Cripto-1-/- cells using an E-cadherin pull-down assay [25]. β-catenin can bind to the intracellular domain of E-cadherin. After Wnt3a stimulation of the β-catenin/Tcf βathway, β-catenin can then be captured by a pull-down assay with GST tagged E-cadherin. F9 and F9 Cripto-1-/- cells were treated with recombinant human Wnt3a protein (20 and 50 ng/ml) for 6 hours and active stabilized β-catenin was precipitated from the cell lysates with GST-E-cadherin followed by binding to glutathione-agarose beads. Western blot analysis for β-catenin revealed that a greater amount of β-catenin co-precipitated with E-cadherin in F9 cells as compared to F9 Cripto-1-/- cells, as assessed by densitometric analysis of the Western blots (Fig. 7A). Equal levels of β-catenin were expressed in F9 and F9 Cripto-1-/- cells, while undetectable levels of Cripto-1 protein expression was seen in F9 Cripto-1-/- cells (Fig. 7A). We also evaluated phosphorylation of LRP5 and LRP6 co-receptors following Wnt3a stimulations in F9 and F9 Cripto-1-/- cells using an anti-phospho LRP6 antibody. This antibody recognizes a common phosphorylation site in both LRP5 and LRP6 co-receptors, corresponding to phosphorylated Ser1490 in LRP6 and phosphorylated Ser1502 in LRP5, and therefore can detect phoshorylated forms of both LRP5 and LRP6 co-receptors. As shown in Figure 7B, Wnt3a (50 ng/ml) induced a robust phosphorylation of both LRP5 and LRP6 in F9 cells in a time dependent manner, while F9 Cripto-1-/- cells showed a less pronounced phosphorylation of LRP5 and LRP6 in response to Wnt3a treatment. Equal amounts of LRP5 and LRP6 co-receptors were detected in both cell lines (Fig. 7B). To ascertain the transcriptional activity of the canonical Wnt/β-catenin pathway, we also performed a SuperTOPFLASH luciferase reporter assay in F9 and F9 Cripto-1 -/- cells following Wnt3a treatment. Treatment with different concentrations of Wnt3a (2-200 ng/ml) of F9 cells induced a dose-dependent increase in SuperTOPFLASH luciferase activity with detectable reporter activation observed even at 2 ng/ml of Wnt3a (Fig. 8A). In contrast, F9 Cripto-1-/- cells showed a significant increase in SuperTOPFLASH luciferase activity only when higher concentrations of Wnt3a protein were used (50, 100 and 200 ng/ml), while no significant increase in SuperTOPFLASH luciferase activity was detected after stimulation with low concentrations of Wnt3a protein (2 and 10 ng/ml), as compared to unstimulated F9 Cripto-1-/- cells (Fig. 8A). In addition, the levels of activation of SuperTOPFLASH luciferase activity in response to Wnt3a (10 and 50 ng/ml) were significantly lower in F9 Cripto-1-/- cells as compared to F9 wild type cells (Fig. 8A). We also assessed whether Cripto-1 expression in F9 cells might modulate expression of genes that are responsive to the canonical and non canonical Wnt signaling pathway, using a real time PCR array of 84 Wnt signaling and target genes [26, 27]. F9 and F9 Cripto-1-/- cells were treated with Wnt3a protein (100 ng/ml) for 18 h and mRNA was isolated followed by real time PCR array analysis. F9 Cripto-1-/- cells treated with Wnt3a protein showed a significant upregulation of several genes involved in the Wnt signaling pathway as compared to F9 control cells, including the non canonical Wnt5a, the Wnt inhibitors secreted frizzled-related protein 2 (sFRP2) and disabled homolog 2 (Dab2), and ectonucleotidepyrophosphatase/phosphodiesterase 2 (Enpp2), suggesting that Cripto-1 might function as a negative regulators of these genes (Fig. 8B). In contrast, a significant downregulation of Calcium channel, voltage-dependent, alpha2/delta subunit 3 (Cacna2d3), c-Myc and Prostaglandin-enodoperoxide synthase 2 (Ptgs2) was detected in F9 Cripto-1-/- cells as compared to F9 wild type cells, indicating that these genes require Cripto-1 expression and are therefore positively regulated by Cripto-1 (Fig. 8B). To further confirm that Cripto-1 expression is indeed important for activation of the Wnt/β-catenin signaling pathway in F9 cells, we reconstituted Cripto-1 expression in F9 Cripto-1-/- cells using a lentiviral vector that drives Cripto-1 gene expression under the control of a mouse RNA polymerase II promoter (mPol2). As shown in Figure 8C, transfection of the Cripto-1 lentiviral vector in F9 Cripto-1-/- cells was able to reconstitute Cripto-1 expression in these cells, although at lower levels than the one found in F9 wild type cells. SuperTOPFLASH luciferase assays in F9 wild type, F9 Cripto-1-/- cells transfected with a control lentiviral empty vector (F9 Cripto-1-/-/EV) and F9 Cripto-1-/- cells expressing exogenous lentiviral Cripto-1 (F9 Cripto-1-/-/Cripto-1) revealed that Cripto-1 re-expression in F9 Cripto-1-/- cells was not able to fully rescue the response of F9 Cripto-1-/-/Cripto-1 cells to Wnt3a stimulation (10 ng/ml) of SuperTOPFLASH luciferase activation, as compared to the levels of SuperTOPFLASH activation in F9 wild type cells (Fig. 8D). However, SuperTOPFLASH luciferase activity in response to Wnt3a stimulation was slightly but significantly higher in F9 Cripto-1-/-/Cripto-1 cells as compared with F9 Cripto-1-/-/EV cells (Fig. 8D). These results suggest that Cripto-1 expression is critical for sensitization of a canonical Wnt/β-catenin pathway in F9 cells to suboptimal concentrations of Wnt3a. Furthermore, Cripto-1 re-expression in F9 cells lacking Cripto-1 expression partially rescues Wnt3a activation of the canonical Wnt/β-catenin pathway.
Fig. 7.

β-catenin stabilization and LRP5/6 phosphorylation in F9 and F9 Cripto-1-/- cells treated with rhWnt3a. (A) F9 and F9 Cripto-1-/- cells were stimulated with different concentrations of rhWnt3a and activated β-catenin was immunoprecipitated with GST-E-cadherin. Glutathione agarose beads were then added to immunoprecipitated proteins and eluted proteins were analyzed by Western blot using an anti-β-catenin antibody. Densitometric analysis of immunoprecipitated β-catenin was performed with Image J program. Protein lysates were analyzed by Western blot for β-catenin, Cripto-1 and β-actin expression (B) Western blot analysis of phosphorylated LRP5/6 in F9 and F9 Cripto-1-/- cells after rhWnt3a treatment. Density ratio (P/T) corresponds to phosphorylated LRP6 (P-LRP6) divided by total LRP6 (LRP6).
Fig. 8.

Wnt/ β-catenin signaling in F9 and F9 Cripto-1-/- cells. (A) Luciferase reporter assay in F9 and F9 Cripto-1-/- cells transiently transfected with Tcf/Lef responsive SuperTOPFLASH luciferase reporter vector. After transfection, cells were stimulated with different concentrations of rhWnt3a (B) Wnt signaling molecules real time PCR array in F9 and F9 Cripto-1-/- cells. Cells were treated with/without rhWnt3a (100 ng/ml) for 18 h, cDNA was synthesized from total RNA and real time PCR array was performed according to manufacturer's instructions. (C) and (D) Re-expression of Cripto-1 in F9 Cripto-1-/- cells partially rescues sensitivity of F9 Cripto-1-/- cells to rhWnt3a stimulation. (C) A lentiviral Cripto-1 expression vector or a control empty vector were infected into F9 Cripto-1-/- cells, stably expressing lentiviral vectors cells were established and Cripto-1 expression was assessed by Western blot analysis. (D) F9 Cripto-1-/- cells, stably expressing a lentiviral Cripto-1 expression vector or a control empty vector, were compared to F9 control cells in a SuperTOPFLASH luciferase assay after rhWnt3a stimulation (10 ng/ml). P value was calculated using Student's t test.
3.4.Cripto-1 enhances the canonical Wnt/β-catenin signaling pathway in response to Wnt3a independently of a Smad2/3 signaling pathway in F9 cells
Cripto-1, together with its ligand Nodal, induces activation and phosphorylation of Smad2/3 signaling proteins, that in turn bind to Smad4 and translocate into the nucleus to regulate transcription of specific target genes [17]. We have previously demonstrated that phosphorylated Smad2 can activate the Wnt/β-catenin signaling pathway independently of Smad4 [28]. We therefore evaluated whether activation of the Wnt/β-catenin signaling pathway by Cripto-1 might be due in part to activation and phosphorylation of Smad2/3 upon interaction with Alk4/Alk7 receptors. The Smad2/3 pathway is constitutively activated in F9 and F9 Cripto-1-/- cells, although the basal levels of phosphorylation of Smad2 are lower in F9 Cripto-1-/- cells as compared to wild type F9 cells (Fig. 9A). We therefore performed a SuperTOPFLASH luciferase reporter assay in F9 and F9 Cripto-1-/- cells stimulated with Wnt3a (100 ng/ml) in the presence or absence of the Alk4/5/7 inhibitor, SB431542. As expected, a significant increase in SuperTOPFLASH luciferase activity was observed in F9 cells following Wnt3a stimulation, while a weak response to Wnt3a was obtained in F9 Cripto-1-/- cells with respect to SuperTOPFLASH luciferase activation (Fig. 9B). Interestingly, treatment of the F9 wild type or F9 Cripto-1-/- cells with the SB431542 inhibitor did not have any significant effect on the ability of Wnt3a to enhance SuperTOPFLASH luciferase activity in either cell line, indicating that the Smad2/3 pathway is not required for activation of a canonical Wnt/β-catenin signaling pathway in this context (Fig. 9B). In contrast, the SB431542 inhibitor strongly inhibited Smad2 signaling in F9 cells as assessed by FAST responsive left side specific enhancer in Nodal derived (n2)7-luciferase reporter assay (Fig. 9C) and in a Smad2 phosphorylation assay (Fig. 9A) [29]. Furthermore, Wnt3a stimulation of F9 or F9 Cripto-1-/- cells did not exert any effect on (n2)7-luciferase reporter activity regardless of the presence or absence of SB431542 inhibitor (Fig. 9C).
Fig. 9.

Activation of the canonical Wnt/β-catenin signaling pathway is independent of a Smad2/3 signaling pathway in F9 cells. (A) Western blot analysis for phospho Smad2 (P-Smad2) and total Smad2 in F9 and F9 Cripto-1-/- cells stimulated with rhWnt3a (10 ng/ml) in the presence or absence of SB431542 inhibitor. Density ratio indicates densitometric values of phosphorylated Smad2/total Smad2. (B) SuperTOPFLASH luciferase reporter assay and (C) (n2)7-luc reporter assay in F9 and F9 Cripto-1-/- cells. Cells were incubated in the presence of the Alk4/5/7 inhibitor SB431542 to block activation of the Smad2/Smad3 signaling pathway.
3.5. F9 cells are more resistant to Dickkopf-1 (Dkk1) inhibition of Wnt/β-catenin signaling than F9Cripto-1-/- cells
Our results suggest that Cripto-1 may facilitate binding of Wnt3a to the co-receptors LRP5 and LRP6, thereby enhancing activation of a canonical Wnt/β-catenin signaling pathway in response to low or limiting concentrations of Wnt3a. Dkk1, a negative regulator of the canonical Wnt/β-catenin signaling pathway, binds to LRP5 and LRP6 co-receptors and blocks through steric hindrance Wnt bnding to LRP5 and LRP6 and simultaneously stimulates their internalization through the cell surface receptor Kremen, which is localized in clathrin-coated vescicles [30-32]. We therefore assessed the effects of Dkk1 treatment on Wnt3 stimulation of a SuperTOPFLASH luciferase activity in wild type and Cripto-1 null F9 cells. F9 and F9 Cripto-1-/- cells were transfected with a SuperTOPFLASH reporter plasmid and subsequently treated with 100 ng/ml of Wnt3a protein in the absence or presence of different concentrations of recombinant mouse Dkk1 protein. As shown in Figure 10A, Dkk1 inhibited SuperTOPFLASH luciferase activation by Wnt3a in a dose dependent manner in both F9 and F9 Cripto-1-/- cells. However, F9 Cript-1-/- cells were more sensitive to the inhibitory effect of Dkk1 treatment as compared to wild type o Cripto-1 expressing F9 cells with respect to Wnt3a stimulation of a SuperTOPFLASH luciferase reporter assay. To further exclude the possibility that endogenous expression levels of Dkk1 might contribute to the different sensitivity of the F9 and F9 Cripto-1-/- F9 cells to Dkk1 treatment, we performed semi-quantitative PCR for Dkk1 in either F9 or F9 Cripto-1-/- cells. No expression of Dkk1 mRNA was detected in F9 or F9 Cripto-1-/- cells, while a Dkk1 cDNA used as positive control was clearly amplified by the PCR reaction (Fig. 10B).
Fig. 10.

F9 cells are more resistant to Dkk1 inhibition of the Wnt/β-catenin signaling pathway than F9 Cripto-1-/- cells. (A) SuperTOPFLASH assay in F9 and F9 Cripto-1-/- cells treated with rhWnt3a (100 ng/ml) alone or in combination with different concentration of rmDkk1. (B) Dkk1 mRNA expression in F9 and F9 Cripto-1-/- cells as assessed by RT-PCR.
3.6. Wnt3a enhances proliferation, motility and growth in soft agar of Cripto-1 overexpressing HC11mouse mammary epithelial cell
We next evaluated whether co-operation between Cripto-1 and the canonical Wnt/β-catenin signaling pathway might have any biological significance. HC11 are mouse mammary epithelial cells that lack any Cripto-1 expression (Fig. 11A). We have previously demonstrated that overexpression of Cripto-1 in HC11 cells increases their migratory potential in Boyden chambers and in wound healing assays [13, 33]. We therefore assessed whether Wnt3a treatment of HC11 control and HC11/Cripto-1 expressing cells had any significant effect on migration, invasion and growth in soft-agar of these cells. As expected, HC11/Cripto-1 cells showed enhanced migratory and invasive capabilities as compared to HC11 control cells (Fig. 11B-C). Wnt3a treatment of HC11 control cells slightly increased migration and invasion of these cells. In contrast, a statistically significant increase in cell migration and invasion was observed in HC11/Cripto-1 cells following treatment with Wnt3a recombinant protein as compared to untreated HC11/Cripto-1 cells (Fig. 11B-C). Furthermore, Wnt3a treatment (50 ng/ml) dramatically increased colony formation in soft agar of HC11/Cripto-1 cells but not of HC11 wild type cells (Fig. 11D), indicating that activation of a canonical Wnt/β-catenin signaling pathway in the context of Cripto-1 expression further enhances motility and in vitro transformation of mammary epithelial cells that is regulated by Cripto-1.
Fig. 11.

Wnt3a enhances Cripto-1 stimulated migration, invasion and growth in soft agar of HC11 mouse mammary epithelial cells. (A) Western blot analysis for Cripto-1 and β-actin in HC11 control cells (WT) and HC11 stably expressing Cripto-1 (HC11/Cripto-1). Migration (B) and invasion (C) assays of HC11 and HC11/Cripto-1 in the presence or absence of rhWnt3a protein (50 ng/ml). (D) Growth in soft agar of HC11 and HC11/Cripto-1 cells in the presence or absence of rhWnt3a (50 ng/ml). Colonies were counted after 2 weeks. * P <0.05 as compared to HC11 untreated cells; **P< 0.05 as compared to HC11/Cripto-1 untreated cells.
4. Discussion
A number of different extracellular soluble or matrix-associated proteins such as the R-Spondins (RSPOs), secreted frizzled-related proteins (Sfrps), Dkk1, Cerberus, connective-tissue growth factor (CTGF), Wnt inhibitory factor-1 (WIF-1) and Wnt-1 induced secreted protein (WISP-1) are known to function as co-modulators by either enhancing or inhibiting the canonical Wnt/β-catenin/Tcf signaling pathway [34, 35]. We have previously demonstrated that Cripto-1 can function as a chaperone protein by facilitating the intracellular proteolytic processing of the Notch 1 receptor in the endoplasmic reticulum (ER)-Golgi network through a furin-like protein convertase [16]. This S1 proteolytic cleavage, which is accelerated by Cripto-1, results in the sensitization to ligand-induced activation of Notch signaling. Likewise, cross-talk of the canonical Wnt/β-catenin signaling pathway with Cripto-1 has been previously reported in Xenopus. In fact, the non-canonical Wnt ligand, Wnt11, together with Xenopus Cripto-1, FRL1, can enhance the canonical Wnt/β-catenin signaling pathway, specifying dorsal axis formation in Xenopus embryos [11]. Co-immunoprecipitation assays demonstrated a direct interaction between FRL1 and Wnt11, and lack of the EGF-like domain in FRL1 severely reduced interaction between these two proteins.
In our study human Cripto-1 weakly interacts with Wnt3a in a co-immununoprecipitation assay in 293T cells, while this interaction is highly enhanced by co-expression of Fz8 and the LRP5 co-receptor. In fact, ectopic or endogenous Cripto-1 can directly bind to LRP5 and LRP6 co-receptors, facilitating Wnt3a binding to the Fz receptors (Fig. 12). Deletion of either the EGF or CFC domains of Cripto-1 significantly impaired binding of Cripto-1 to LRP5 or LRP6, while deletion of both EGF and CFC domains completely blocked interaction among these proteins. We also demonstrate that Cripto-1 can enhance the canonical Wnt/β-catenin signaling pathway when activated by Wnt3a in a Tcf/Lef responsive SuperTOPFLASH luciferase assay in 293T cells (Fig. 12). In this respect, deletion of the Cripto-1 gene in F9 mouse embryonal carcinoma cells leads to an attenuated response to Wnt3a-induced β-catenin stabilization and SuperTOPFLASH reporter assay activation. In particular, Cripto-1 expression in F9 cells is essential for sensitization of a canonical Wnt/β-catenin pathway to suboptimal concentrations of Wnt3a. The ability of Cripto-1 to enhance Wnt/β-catenin requires tethering of Cripto-1 to the plasma membrane as soluble Cripto-1, which lacks the GPI anchor, is ineffective in this capacity. Likewise, initial binding of Cripto-1 to LRP5/LRP6 occurs on the plasma membrane and not in an intracellular compartment.
Fig. 12.
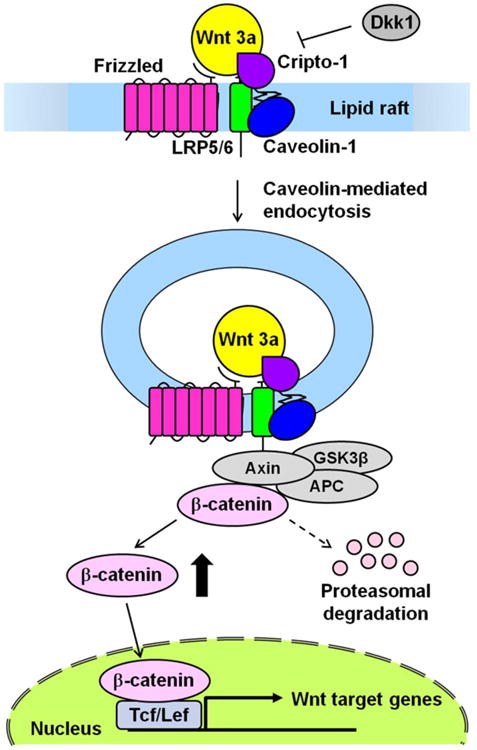
Proposed model of Cripto-1 modulation of the Wnt/β-catenin signaling pathway. Cripto-1 can regulate the Wnt/β-catenin signaling pathway through the binding of LRP5/6, which also bind to caveolin-1 in the lipid raft as well as Cripto-1. Cripto-1 facilitates the phosphorylation of LRP5/6 induced by Wnt3a. The phosphorylated LRP6 leads to recruitment of the Axin complex (Axin, APC and GSK3β) to the Wnt receptor complex. The receptor complex is then internalized via a caveolin-mediated route to induce the cytoplasmic accumulation of β-catenin. β-catenin can then form a complex with Tcf/Lef and activate the transcription of target genes after translocation to the nucleus. Cripto-1 also might interfere with the internalization of LRP5/6 that is facilitated by Dkk1, which is a potent inhibitor of the Wnt/β-catenin signaling pathway.
Analysis of a panel of genes that are in the canonical or non-canonical Wnt signaling pathway or that are directly or indirectly modulated by the Wnt/β-catenin signaling pathway using a PCR array showed a different gene expression profile in F9 and F9 Cripto-1-/- cells following Wnt3a stimulation. Twelve genes were found to be significantly upregulated in F9 Cripto-1-/- cells, indicating that Cripto-1 might function as a negative regulator for these genes. Among these genes we identified cell signaling molecules, such as FGF9 and Gdf5, Wnt5a, Sfrp2, and the tumor suppressor endocytic adaptor protein Dab-2. Notably, Cripto-1 blocked expression of Wnt/β-catenin target genes such as Sfrp2, Wnt5a and Dab-2, which are Wnt/β-catenin antagonists. In a similar context, Cripto-1 negatively regulated expression of the Notch inhibitor Dlk1. Enpp2 was upregulated over 7 fold in F9 Cripto-1-/- cells compared to F9 control cells. Enpp2 encodes for a catalytic protein known as Autotaxin, which is involved in cellular membrane lipid metabolism and remodeling [36]. Interestingly, Autotaxin, which is a phosphodiesterase and phospholipase, can either facilitate cell proliferation and migration or can possess antitumor activity by inhibiting cancer invasion and metastasis in a context dependent manner. Among the genes downregulated in F9 Cripto-1-/- cells and therefore positively regulated by Critpo-1 we identified c-myc, Ptgs2 (Cox-2), which is involved in inflammation, and Cacna2d3, a membrane protein involved in intracellular calcium transport [37]. Cripto-1 might therefore be directly involved in regulating signaling molecules modulated by the Wnt/β-catenin signaling pathway. Cripto-1 is also a direct downstream target gene of the canonical Wnt/β-catenin signaling pathway during embryogenesis and in colon cancer cells and Tcf/Lef specific binding sites have been identified in the first intronic region of the human Cripto-1 gene, suggesting the existence of a reciprocal regulation among these two signaling pathways [9, 10]. Previous studies have demonstrated that Cripto-1 can signal through the activation of a Nodal/Alk4/Alk7/Smad2/3/4 signaling pathway or a Nodal-independent signaling pathway that triggers activation of the p44 MAPK/AKT intracellular signaling cascade [17]. Modulation of the canonical Wnt/β-catenin signaling pathway by Cripto-1 in response to Wnt3a stimulation is independent of the Nodal/Alk4/Alk7 signaling pathway, since a specific inhibitor of the Alk4/Alk5/Alk7 serine threonine kinase receptors does not interfere with the ability of Cripto-1 to enhance Wnt3a stimulation of a SuperTOPFLASH luciferase activity in F9 cells. Reciprocally, Wnt3a does not affect Nodal and Cripto-1-dependent activation of a Smad2-responsive FAST reporter.
Interaction between Cripto-1 and the canonical Wnt/β-catenin signaling pathway might be important in the context of mammary tumor formation that can be initiated by aberrant Cripto-1 or Wnt/β-catenin signaling [38, 39]. We have previously demonstrated that Cripto-1 overexpression in mouse mammary epithelial cell lines enhances proliferation, migration and invasion and increases in vitro transformation as shown by the ability of these Cripto-1 expressing cells to form colonies in semi solid agar [13, 33]. Activation of the canonical Wnt/β-catenin signaling pathway by Wnt3a stimulation in mouse mammary epithelial cells stably expressing Cripto-1 further enhances the transforming activities of Cripto-1 in vitro Wnt3a treatment significantly enhances migration, invasion and colony formation in soft-agar of HC11/Cripto-1 cells as compared to HC11 wild type cells, which lack Cripto-1 expression. This might be significant in the context of mammary tumorigenesis, since activated β-catenin is found in mammary tumors of transgenic mice overexpressing human Cripto-1 in the mammary gland and since mouse Cripto-1 is upregulated in Wnt1 mammary tumors [13, 39].
Internalization of the LRP5 and LRP6 co-receptors might also be important in the ability of Cripto-1 to modulate the Wnt/β-catenin signaling pathway. For example, RSPO1, a potent activator of the canonical Wnt/β-catenin signaling pathway, enhances Wnt signaling by interfering with Dkk1/Kremen mediated internalization of LRP6 and by simultaneously binding to Fz receptors and the the G-protein-coupled receptors LGR4 and LGR5 via clathrin endocytosis [40, 41]. In this respect, F9 wild type cells expressing Cripto-1 are more resistant to Dkk1 inhibition of the Wnt/β-catenin signaling pathway as compared to F9 Cripto-1 null cells, suggesting that Cripto-1, similarly to RSPO1, might also inhibit the internalization of LRP5/LRP6 that is facilitated by Dkk1 and which requires clathrin [42]. In fact, several studies have shown that endosomal vescicles can control Wnt signaling specificity and diversity. For example, Wnt3a stimulation induces the caveolin-1 dependent internalization of LRP6 ultimately leading to β-catenin accumulation [43]. In contrast, the adaptor protein Dab-2 facilitates internalization of LRP6 in clathrin-coated vescicles such that the β-catenin disruption complex remains intact thereby inhibiting Wnt/β-catenin signaling [44]. We have previously shown that Cripto-1 is localized in lipid rafts and directly binds to Caveolin-1 [45], suggesting that Cripto-1 may also enhance the canonical Wnt/β-catenin signaling pathway by potentially sequestering LRP5/6 in lipid rafts and facilitating LRP5 or LRP6 internalization into early endosomes or caveosomes through interaction with Caveolin-1. Finally, Cripto-1 can bind to the heat shock protein, ER stress response protein GRP78/BIP on the surface of tumor cells and within the ER [46]. GRP78 is normally a chaperone protein that assists in the correct tertiary folding of proteins in the ER [47]. GRP78 binds to Wnt1 and Wnt3a and facilitates the folding, post-translational processing and secretion of Wnt3a [48]. This interaction between GRP78 and different Wnt proteins may possibly be expedited by formation of a terniary complex with Cripto-1 on the plasma membrane, which would then lead to enhanced Wnt/β-catenin signaling through LRP5 and LRP6.
5. Conclusion
In conclusion, we have demonstrated that Cripto-1 modulates the canonical Wnt/β-catenin signaling pathway through direct interaction with the LRP5 and LRP6 co-receptors. Cross-talk between Cripto-1 and the Wnt/β-catenin signaling pathway might play a role in mammary transformation leading to a more aggressive behavior of mammary cancer cells.
Highlights.
Cripto-1 enhances the canonical Wnt/β-catenin signaling pathway.
Cripto-1 binds to the Wnt co-receptors LRP5 and LRP6 on the cell surface.
Cripto-1 induces cytoplasmic stabilization of β-catenin.
Wnt3a enhances Cripto-1 stimulation of mammary epithelial cell motility and growth.
Acknowledgments
We thank Drs. Michele Sanicola, Xi He, Matthew Warman, Roel Nusse and Akira Kikuchi for providing materials. This work was supported by the National Institutes of Health intramural funding. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Abbreviations
- TDGF-1
teratocarcinoma-derived growth factor-1
- CFC
Cripto-1/FRL1/Cryptic
- GPI
glycosilphosphatidylinositol
- TGF-β
transforming growth factor-β
- GDF
growth differentiation factor
- Tcf
T-cell factor
- Fz
Frizzled
- LRP
low-density lipoprotein receptor-related protein
- APC
adenomatous polyposis coli
- GSK3β
glycogen synthase kinase 3β
- Lef
lymphoid enhancer factor
- rh
recombinant human
- rm
recombinant mouse
- CRD
cysteine rich domain
- RIPA
radio immunoprecipitation assay
- Cacna2d3
Calcium channel, voltage-dependent, alpha2/delta subunit 3
- Ptgs2
Prostaglandin-enodoperoxide synthase 2
- mPol2
mouse RNA polymerase II promoter
- Dkk1
Dickkopf-1
- RSPO
R-Spondin
- SFRP
secreted frizzled-related protein
- CTGF
connective-tissue growth factor
- WIF-1
Wnt inhibitory factor-1
- WISP-1
Wnt-1 induced secreted protein
- ER
endoplasmic reticulum
- Dab-2
disbled-2
Footnotes
Author contribution: T.N., M.S., S.L., Y.E.G., J.S.R., D.S.S., and C.B. designed the research. T.N., H.K., T.T., and C.B. performed experiments. T.N., M.C.R., N.P.C., M.G., and A.B. analyzed and interpreted data. T.N., H.K., T.T., D.S.S., and C.B. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
David S. Salomon, Email: salomond@mail.nih.gov.
Caterina Bianco, Email: biancoc@mail.nih.gov.
References
- 1.Dono R, Montuori N, Rocchi M, De Ponti-Zilli L, Ciccodicola A, Persico MG. Am J Hum Genet. 1991;49(3):555–565. [PMC free article] [PubMed] [Google Scholar]
- 2.Ciccodicola A, Dono R, Obici S, Simeone A, Zollo M, Persico MG. Embo J. 1989;8(7):1987–1991. doi: 10.1002/j.1460-2075.1989.tb03605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Minchiotti G, Parisi S, Liguori G, Signore M, Lania G, Adamson ED, Lago CT, Persico MG. Mech Dev. 2000;90(2):133–142. doi: 10.1016/s0925-4773(99)00235-x. [DOI] [PubMed] [Google Scholar]
- 4.de Castro NP, Rangel MC, Nagaoka T, Salomon DS, Bianco C. Future Oncol. 2010;6(7):1127–1142. doi: 10.2217/fon.10.68. [DOI] [PubMed] [Google Scholar]
- 5.Bianco C, Rangel MC, Castro NP, Nagaoka T, Rollman K, Gonzales M, Salomon DS. Am J Pathol. 2010;177(2):532–540. doi: 10.2353/ajpath.2010.100102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rangel MC, Karasawa H, Castro NP, Nagaoka T, Salomon DS, Bianco C. Am J Pathol. 2012;180(6):2188–2200. doi: 10.1016/j.ajpath.2012.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reya T, Clevers H. Nature. 2005;434(7035):843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 8.Nusse R. Cell Res. 2005;15(1):28–32. doi: 10.1038/sj.cr.7290260. [DOI] [PubMed] [Google Scholar]
- 9.Hamada S, Watanabe K, Hirota M, Bianco C, Strizzi L, Mancino M, Gonzales M, Salomon DS. Biochem Biophys Res Commun. 2007;355(1):240–244. doi: 10.1016/j.bbrc.2007.01.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morkel M, Huelsken J, Wakamiya M, Ding J, van de Wetering M, Clevers H, Taketo MM, Behringer RR, Shen MM, Birchmeier W. Development. 2003;130(25):6283–6294. doi: 10.1242/dev.00859. [DOI] [PubMed] [Google Scholar]
- 11.Tao Q, Yokota C, Puck H, Kofron M, Birsoy B, Yan D, Asashima M, Wylie CC, Lin X, Heasman J. Cell. 2005;120(6):857–871. doi: 10.1016/j.cell.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe K, Hamada S, Bianco C, Mancino M, Nagaoka T, Gonzales M, Bailly V, Strizzi L, Salomon DS. J Biol Chem. 2007;289(49):35772–35786. doi: 10.1074/jbc.M707351200. [DOI] [PubMed] [Google Scholar]
- 13.Strizzi L, Bianco C, Normanno N, Seno M, Wechselberger C, Wallace-Jones B, Khan NI, Hirota M, Sun Y, Sanicola M, Salomon DS. J Cell Physiol. 2004;201(2):266–276. doi: 10.1002/jcp.20062. [DOI] [PubMed] [Google Scholar]
- 14.Semenov MV, Tamai K, Brott BK, Kuhl M, Sokol S, He X. Curr Biol. 2001;11(12):951–961. doi: 10.1016/s0960-9822(01)00290-1. [DOI] [PubMed] [Google Scholar]
- 15.Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, Zacharin M, Oexle K, Marcelino J, Suwairi W, Heeger S, Sabatakos G, Apte S, Adkins WN, Allgrove J, Arslan-Kirchner M, Batch JA, Beighton P, Black GC, Boles RG, Boon LM, Borrone C, Brunner HG, Carle GF, Dallapiccola B, De Paepe A, Floege B, Halfhide ML, Hall B, Hennekam RC, Hirose T, Jans A, Juppner H, Kim CA, Keppler-Noreuil K, Kohlschuetter A, LaCombe D, Lambert M, Lemyre E, Letteboer T, Peltonen L, Ramesar RS, Romanengo M, Somer H, Steichen-Gersdorf E, Steinmann B, Sullivan B, Superti-Furga A, Swoboda W, van den Boogaard MJ, Van Hul W, Vikkula M, Votruba M, Zabel B, Garcia T, Baron R, Olsen BR, Warman ML. Cell. 2001;107(4):513–523. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 16.Watanabe K, Nagaoka T, Lee JM, Bianco C, Gonzales M, Castro NP, Rangel MC, Sakamoto K, Sun Y, Callahan R, Salomon DS. J Cell Biol. 2009;187(3):343–353. doi: 10.1083/jcb.200905105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bianco C, Adkins HB, Wechselberger C, Seno M, Normanno N, De Luca A, Sun Y, Khan N, Kenney N, Ebert A, Williams KP, Sanicola M, Salomon DS. Mol Cell Biol. 2002;22(8):2586–2597. doi: 10.1128/MCB.22.8.2586-2597.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woldemichael GM, Turbyville TJ, Linehan WM, McMahon JB. Cancer Res. 2011;71(1):134–142. doi: 10.1158/0008-5472.CAN-10-0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bianco C, Strizzi L, Rehman A, Normanno N, Wechselberger C, Sun Y, Khan N, Hirota M, Adkins H, Williams K, Margolis RU, Sanicola M, Salomon DS. Cancer Res. 2003;63(6):1192–1197. [PubMed] [Google Scholar]
- 20.Bianco C, Strizzi L, Ebert A, Chang C, Rehman A, Normanno N, Guedez L, Salloum R, Ginsburg E, Sun Y, Khan N, Hirota M, Wallace-Jones B, Wechselberger C, Vonderhaar BK, Tosato G, Stetler-Stevenson WG, Sanicola M, Salomon DS. J Natl Cancer Inst. 2005;97(2):132–141. doi: 10.1093/jnci/dji011. [DOI] [PubMed] [Google Scholar]
- 21.Watanabe K, Bianco C, Strizzi L, Hamada S, Mancino M, Bailly V, Mo W, Wen D, Miatkowski K, Gonzales M, Sanicola M, Seno M, Salomon DS. J Biol Chem. 2007;282(43):31643–31655. doi: 10.1074/jbc.M702713200. [DOI] [PubMed] [Google Scholar]
- 22.Holman D, Henley JM. J Neurosci Methods. 2007;160(2):302–308. doi: 10.1016/j.jneumeth.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mancino M, Esposito C, Watanabe K, Nagaoka T, Gonzales M, Bianco C, Normanno N, Salomon DS, Strizzi L. Cancer Res. 2009;69(5):1717–1721. doi: 10.1158/0008-5472.CAN-08-2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schiffer SG, Foley S, Kaffashan A, Hronowski X, Zichittella AE, Yeo CY, Miatkowski K, Adkins HB, Damon B, Whitman M, Salomon D, Sanicola M, Williams KP. J Biol Chem. 2001;276(41):37769–37778. doi: 10.1074/jbc.M104774200. [DOI] [PubMed] [Google Scholar]
- 25.Orsulic S, Huber O, Aberle H, Arnold S, Kemler R. J Cell Sci. 1999;112(Pt 8):1237–1245. doi: 10.1242/jcs.112.8.1237. [DOI] [PubMed] [Google Scholar]
- 26.Lickert H, Domon C, Huls G, Wehrle C, Duluc I, Clevers H, Meyer BI, Freund JN, Kemler R. Development. 2000;127(17):3805–3813. doi: 10.1242/dev.127.17.3805. [DOI] [PubMed] [Google Scholar]
- 27.Shibamoto S, Winer J, Williams M, Polakis P. Exp Cell Res. 2004;292(1):11–20. doi: 10.1016/j.yexcr.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 28.Hirota M, Watanabe K, Hamada S, Sun Y, Strizzi L, Mancino M, Nagaoka T, Gonzales M, Seno M, Bianco C, Salomon DS. Cell Signal. 2008;20(9):1632–1641. doi: 10.1016/j.cellsig.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saijoh Y, Adachi H, Sakuma R, Yeo CY, Yashiro K, Watanabe M, Hashiguchi H, Mochida K, Ohishi S, Kawabata M, Miyazono K, Whitman M, Hamada H. Mol Cell. 2000;5(1):35–47. doi: 10.1016/s1097-2765(00)80401-3. [DOI] [PubMed] [Google Scholar]
- 30.David Holman JMH. Journal of Neuroscience Methods. 2007;160:302–308. doi: 10.1016/j.jneumeth.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mao B, Wu W, Davidson G, Marhold J, Li M, Mechler BM, Delius H, Hoppe D, Stannek P, Walter C, Glinka A, Niehrs C. Nature. 2002;417(6889):664–667. doi: 10.1038/nature756. [DOI] [PubMed] [Google Scholar]
- 32.Mao B, Wu W, Li Y, Hoppe D, Stannek P, Glinka A, Niehrs C. Nature. 2001;411(6835):321–325. doi: 10.1038/35077108. [DOI] [PubMed] [Google Scholar]
- 33.Wechselberger C, Ebert AD, Bianco C, Khan NI, Sun Y, Wallace-Jones B, Montesano R, Salomon DS. Exp Cell Res. 2001;266(1):95–105. doi: 10.1006/excr.2001.5195. [DOI] [PubMed] [Google Scholar]
- 34.Rey JP, Ellies DL. Dev Dyn. 2010;239(1):102–114. doi: 10.1002/dvdy.22181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kikuchi A, Yamamoto H, Sato A, Matsumoto S. Int Rev Cell Mol Biol. 2011;291:21–71. doi: 10.1016/B978-0-12-386035-4.00002-1. [DOI] [PubMed] [Google Scholar]
- 36.Tania M, Khan MA, Zhang H, Li J, Song Y. Biochem Biophys Res Commun. 2010;401(4):493–497. doi: 10.1016/j.bbrc.2010.09.114. [DOI] [PubMed] [Google Scholar]
- 37.Chu PJ, Best PM. J Mol Cell Cardiol. 2003;35(2):207–215. doi: 10.1016/s0022-2828(02)00313-9. [DOI] [PubMed] [Google Scholar]
- 38.Incassati A, Chandramouli A, Eelkema R, Cowin P. Breast Cancer Res. 2010;12(6):213. doi: 10.1186/bcr2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wechselberger C, Strizzi L, Kenney N, Hirota M, Sun Y, Ebert A, Orozco O, Bianco C, Khan NI, Wallace-Jones B, Normanno N, Adkins H, Sanicola M, Salomon DS. Oncogene. 2005;24(25):4094–4105. doi: 10.1038/sj.onc.1208417. [DOI] [PubMed] [Google Scholar]
- 40.Binnerts ME, Kim KA, Bright JM, Patel SM, Tran K, Zhou M, Leung JM, Liu Y, Lomas WE, 3rd, Dixon M, Hazell SA, Wagle M, Nie WS, Tomasevic N, Williams J, Zhan X, Levy MD, Funk WD, Abo A. Proc Natl Acad Sci U S A. 2007;104(37):14700–14705. doi: 10.1073/pnas.0702305104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carmon KS, Gong X, Lin Q, Thomas A, Liu Q. Proc Natl Acad Sci U S A. 2012;108(28):11452–11457. doi: 10.1073/pnas.1106083108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kikuchi A, Yamamoto H, Sato A. Trends Cell Biol. 2009;19(3):119–129. doi: 10.1016/j.tcb.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 43.Yamamoto H, Sakane H, Michiue T, Kikuchi A. Dev Cell. 2008;15(1):37–48. doi: 10.1016/j.devcel.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 44.Jiang Y, He X, Howe PH. EMBO J. 2012;31(10):2336–2349. doi: 10.1038/emboj.2012.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bianco C, Strizzi L, Mancino M, Watanabe K, Gonzales M, Hamada S, Raafat A, Sahlah L, Chang C, Sotgia F, Normanno N, Lisanti M, Salomon DS. Am J Pathol. 2008;172(2):345–357. doi: 10.2353/ajpath.2008.070696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shani G, Fischer WH, Justice NJ, Kelber JA, Vale W, Gray PC. Mol Cell Biol. 2008;28(2):666–677. doi: 10.1128/MCB.01716-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang LH, Zhang X. J Cell Biochem. 2010;110(6):1299–1305. doi: 10.1002/jcb.22679. [DOI] [PubMed] [Google Scholar]
- 48.Verras M, Papandreou I, Lim AL, Denko NC. Mol Cell Biol. 2008;28(23):7212–7224. doi: 10.1128/MCB.00947-08. [DOI] [PMC free article] [PubMed] [Google Scholar]