

Abstract
Myotonia congenita-inducing mutations in the muscle chloride channel CLC-1 normally result in reduced open probability (Po) of this channel. One well-accepted mechanism of the dominant inheritance of this disease involves a dominant-negative effect of the mutation on the function of the common gate of this homodimeric, double-barreled molecule. We report here a family with myotonia congenita characterized by muscle stiffness and clinical and electrophysiologic myotonic phenomena transmitted in an autosomal dominant pattern. DNA sequencing of DMPK and ZNF9 genes for myotonic muscular dystrophy types I and II was normal, whereas sequencing of CLC-1 encoding gene, CLCN1, identified a single heterozygous missense mutation, G233S. Patch-clamp analyses of this mutant CLC-1 channel in Xenopus oocytes revealed an increased Po of the channel’s fast gate, from ~ 0.4 in the wild type to > 0.9 in the mutant at −90 mV. In contrast, the mutant exhibits a minimal effect on the Po of the common gate. These results are consistent with the structural prediction that the mutation site is adjacent to the fast gate of the channel. Overall, the mutant could lead to a significantly reduced dynamic response of CLC-1 to membrane depolarization, from a 5-fold increase in chloride conductance in the wild type to a 2-fold increase in the mutant—this might result in slower membrane repolarization during an action potential. Since expression levels of the mutant and wild-type subunits in artificial model cell systems were unable to explain the disease symptoms, the mechanism leading to dominant inheritance in this family remains to be determined.
Keywords: myotonia congenita, muscle, chloride channel, CLCN1, dominant, gain of function
Introduction
Myotonia is a form of decreased muscle relaxation resulting from abnormal sarcolemnal membrane excitability. This symptom is associated with repeated firing of muscle action potentials that induce continued muscle contraction. The continuous contraction manifests clinically as muscle stiffness and aching, sometimes accompanied by diminished ability to terminate voluntary muscle contraction. Myotonia is seen in dystrophic disease, i.e. myotonic dystrophy, but also occurs in isolation from other muscle dysfunction(Heatwole and Moxley, 2007). At first approximation, the dysfunction can be the result of over-activity of the muscle sodium channels, which render the membrane excitable, or under-activity of the muscle chloride channels (CLC-1), which normally function to dampen excitability and stabilize the resting potential. Hence, the sodium channel mutations that have been identified have generally been of the dominantly-inherited, gain-of-function type, whereas the CLC-1 disorders have resulted from either dominantly (Thomsen type; MIM 160800) or recessively inherited (Becker type myotonia; MIM 255700) loss-of-function mutations. We report here a family with pure myotonia resulting from a dominantly inherited G233S mutation that has a complex effect on CLC-1 function. In Xenopus oocytes and HEK cells total Cl− conductance is reduced in mutant dimers and membrane expression of the mutant subunits is decreased. At the same time, biophysical studies reveal increase in the open probability of mutant CLC-1. Myotonia mutations at this codon have not been reported previously (Lossin and George, 2008; Moon et al., 2009; Pusch, 2002).
The CLC-1 chloride channel is a homodimer with each subunit comprising an anion pore. Each pore is controlled by an independent voltage-sensing fast gate (Chen, 2005; Dutzler, 2004; Dutzler, 2006; Jentsch, 2008). In addition, there appears to be a more slowly responding “common gate” that controls both pores simultaneously. It has been suggested that the common gate may involve the interaction of the two subunits. Therefore, in a “heterodimer” of a normally functioning wild-type (WT) subunit with an abnormally functioning mutant subunit affecting the common gate, current through both pores would be affected (Kubisch et al., 1998; Pusch et al., 1995; Saviane et al., 1999). Such a “dominant-negative” effect is thought to account for some of the dominant-type mutations located at or near the dimer interface of the channel. On the other hand, the recessive forms of CLC-1 mutations generally affect only the function within subunits, or, in some cases, affect the expression level of the CLC-1 protein in the sarcolemma (Papponen et al., 2008; Pusch, 2002; Wu et al., 2002). Whether the mutations are recessive or dominant, the majority of the CLCN1 mutations that cause myotonia are loss-of-function mutations because the chloride conductance contributed by CLC-1 is the dominant stabilizing force of the sarcolemma membrane potential.
In contrast, our electrophysiological analysis of the G233S mutant demonstrates that the mutation results in a near constitutively opened channel by rendering the fast-gate of the channel mostly opened while the common gating appears to be relatively normal. The functional characterizations of the mutant appear to be consistent with the structural role of the G233 residue according to the high-resolution structures of the homologous bacterial CLC molecules. This “gain-of-function” mutation for the molecular function of CLC-1 paradoxically appears to result in a reduction in membrane stability leading to myotonia.
Materials and Methods
Electrodiagnostic Studies
Nerve conduction studies and needle electromyography were performed with surface electrodes and a monopolar needle using a Viking IV electromyographic system (CareFusion, San Diego, CA), following guidelines provided by the American Association of Neuromuscular and Electrodiagnostic Medicine. Temperature was monitored throughout the test with a skin thermistor probe, and maintained at 33° C. However, in intrinsic muscles of the hand, needle electromyography was recorded before and after applying an ice pack to the skin surface of the hand for 5 minutes.
Molecular Biology and Channel expressions in Xenopus Oocyte and HEK 293T Cells
The cDNA of the human WT CLCN1 gene was constructed in pTLN vector (Tseng et al., 2007), and the corresponding mutation of G233S was made using Quick Change II site-directed mutagenesis kit (Agilent Technologies).(Tseng et al., 2011) For oocyte injections the mRNAs of the WT channel and the G233S mutant were synthesized using SP6 polymerase (mMessage mMachine kit, Ambion Inc.). Approximately 3–5 ng of RNA was injected into one Xenopus oocyte, and patch-clamp recordings were performed ~1–5 days after oocyte injection.(Tseng et al., 2007; Tseng et al., 2011; Zhang et al., 2008). For HEK 293T cell transfections the mutant and wild type cDNA was inserted into the transfection vector, pcDNA3, which added either a C-terminal myc tag or a flag tag for protein identification.
Electrophysiological Recordings of the WT and mutant CLC-1
Analysis of WT and mutant CLC-1 channels made use of two-electrode voltage clamp (TEVC) techniques and inside-out patch-clamp recording methods in Xenopus oocytes injected with channel RNAs (Tseng et al., 2007; Tseng et al., 2011; Zhang et al., 2008). TEVC was used to examine the total whole oocyte current, and thus serves as an experimental method to compare the expression level of the WT CLC-1 channel and the G233S mutant in the cell membrane of Xenopus oocytes. For this set of experiments equal amounts of total RNA from WT, G233S or WT + G233S (1:1 ratio) were injected into each oocyte. Total current of each oocyte was measured using a TEVC amplifier (Warner Instruments/Harvard Apparatus). The current was then digitized by the Digidata 1320 A/D converter controlled by the pClamp9 software (Axon Instruments/Molecular Devices). More detailed biophysical studies were conducted in channels expressed in oocytes using patch-clamp methods on the excised membrane patches. For patch-clamp experiments, two voltage protocols (A and B) were used to elicit the channel current (see Fig. 1A). In protocol A (Fig. 1A, left), the membrane potential was stepped from the 0 mV holding voltage to various test voltages from +120 mV to −140 mV (in −20-mV steps) for 400 ms. Each test voltage is followed by a tail-voltage at −100 mV for 400 ms. The initial value of the tail-current was determined by fitting the induced tail-current with a double-exponential function, normalized to the maximal value. The normalized tail-current is the overall open probability (Po) of the channel at the preceding test-voltage. Because the channel contains two types of gating mechanisms, the fast-gating and the common gating, Po is the product of the open probability of the fast-gate (Pof) and that of the common-gate (Poc), namely Po = Pof × Poc. Protocol B (Fig. 1A, right) was exactly the same as protocol A, except that a 400-μs voltage step to +170 mV was inserted between the test-voltage and the tail-voltage. Because a short, but very positive, voltage step is enough to fully open the fast-gate (but does not alter the common-gate which has slower kinetics), the normalized initial tail-current (see Fig. 1A) represents Poc of the test voltage. Dividing the value of Po (from protocol A) by Poc (from protocol B) gave an estimate of Pof.
Figure 1. Comparison of the gating properties of the WT CLC-1 and the point mutant G233S.

(A) Voltage protocols used to evaluate the overall open probability (Po) of the channel (left) and the common-gate open probability (Poc, right), respectively. (B and C) Original recording traces of the WT (B) and G233S mutant (C) CLC-1 in responses to both voltage protocols.
Protein Expression in HEK 293T Cells
To assess the efficiency of expression of the G233S mutant, in vitro analysis was carried out with both wild-type CLC-1 and the G233S mutant cDNAs which were subcloned to pcDNA3 vector and genetically tagged with a myc epitope at the C-terminus of the protein. Human embryonic kidney (HEK) 293T cells were transfected, using Lipofectamine 2000 (Invitrogen) with the vector containing either WT or G233S cDNA. (Li et al., 2011) After culture for 48 hours, cells were lysed and the entire lysate or the biotinylated portion of the lysate were subjected to Western blot analysis.
Western blotting
Transfected HEK293T cells were washed with ice-cold PBS [(in mM) 136 NaCl, 2.5 KCl, 1.5 KH2PO4, 6.5 Na2HPO4, pH 7.4] and resuspended in a hypotonic buffer (10 mM Tris, pH 8.0). To assay the total protein expression level, cell lysates were directly employed for immunoblotting analyses. Alternatively, cell lysates were biotinylated and pulled down with streptavidin agarose beads before loading into the gel. After adding Laemmli buffer, samples were sonicated on ice and heated at 70 C for 5 min. The proteins were then separated by 7.5% SDS-PAGE, electrophoretically transferred to nitrocellulose membranes, and detected using mouse anti-myc (clone 9E10) or mouse anti-β-actin (1:5000; Sigma) antibodies. Blots were then exposed to horseradish-peroxidase conjugated anti-mouse IgG (1:5000; Thermo Scientific), and revealed by an enhanced chemiluminescence detection system (Thermo Scientific). To quantify the protein density, densitometric scans of anti-myc density with respect to the cognate β-actin density was first determined. The normalized expression level of G233S mutant with respect to β-actin expression was then further normalized to the normalized expression value of WT.
Biotinylation
Intact transfected cells were washed extensively with PBS supplemented with 0.5 mM CaCl2, 2 mM MgCl2 (CM-PBS), followed by incubation in 1 mg/ml sulfo-NHS-LC-biotin (Thermo Scientific) in CM-PBS at 4 C for 30 min. Biotinylation was terminated by removing the biotin reagents and rinsing with CM-PBS and Tris-buffered saline (TBS) (20 mM Tris, 150 mM NaCl, pH 7.6) for three times. Cells were solubilized in RIPA buffer [(in mM) 20 Tris-HCl, 150 NaCl, 1% Triton X-100, 0.5% Na-deoxycholate, 0.1% SDS, 1 EDTA, 1 phenylmethylsulfonyl fluoride, pH 7.4] supplemented with protease inhibitor cocktail. Solubilized cell lysates were incubated for 16 hours at 4 C with streptavidin-agarose beads (Thermo Scientific). Beads were washed three times in lysis buffer and twice with TBS. The biotin-streptavidin complexes were eluted from the beads by boiling for 5 min in Laemmli sample buffer and were subject to SDS-PAGE and immunoblotting. To compare the efficiency of cell membrane protein expression to total cell membrane expression, a surface expression ratio was first calculated by dividing the biotinylated protein density by the corresponding standardized total protein density. The surface expression ratio of G233S was then normalized to that of WT CLC-1.
Results
Case Reports
Case 1
A 31 year-old male developed “hand cramping” during his job entering data on a computer keyboard, described as “sudden paralysis” of his fingers with the third and fourth fingers flexed and the second finger extended. Each episode lasted less than twenty seconds and was sometime relieved by manually moving the fingers with the opposite hand. In response to questioning, he described occasional mild difficulty, since childhood, in releasing his grip when grasping a doorknob or shaking hands – although, he never was certain that the symptom was abnormal. On the other hand, he was athletic as a child and did well in sports. The symptoms have gradually progressed over the subsequent seven years, occurring with increased frequency and duration. In addition, during this time, there has also been the slow progression of occasional muscle fasciculations and diffuse muscle aching relieved by moving about. Mental status, cranial nerve, motor, sensory and coordination examinations have been normal during this time period except for difficulty releasing his hand and foot grip and the presence of asymmetrical thenar eminence percussion myotonia.
Case 2
The 57 year-old father of case 1 had diffuse muscle stiffness and aching with frequent muscle cramps beginning in his mid-twenties and progressing slowly up to the present. His examination revealed slow release of grip and percussion myotonia of the thenar eminence.
Electrodiagnostic and Histologic Studies
For both cases, electrodiagnostic testing was normal except for myotonic discharges, unaffected by warming or cooling, in all muscle groups tested (Fig. 2.). Interestingly, even though myotonic discharges were readily elicited, even by minimal movements of the electromyography recording needle, no after-discharges potentials were observed in any of the muscle group tested. No repetitive compound muscle action potentials after single nerve stimulation were noted. Deltoid muscle biopsy of case 1 was normal by light microscopy and histochemical analysis.
Figure 2. Electrodiagnostic findings.

Needle EMG recording performed in the first dorsal interosseous muscle of the proband showing an example of classical waxing and waning myotonic discharges triggered by movement of the needle electrode.
Family History
The proband’s father (Case 2) has had slowly progressive life-long muscle aching and stiffness, with frequent “cramps” as noted above. Case 1’s mother and only sibling are asymptomatic (Fig. 3 and Table 1). There is no consanguinity. The proband’s eight-year old son complains of aching shoulders and neck. All family members have undergone neurological examinations (DPR) and only the proband’s father has muscle stiffness or myotonic phenomena.
Figure 3. Pedigree of myotonia phenotype.
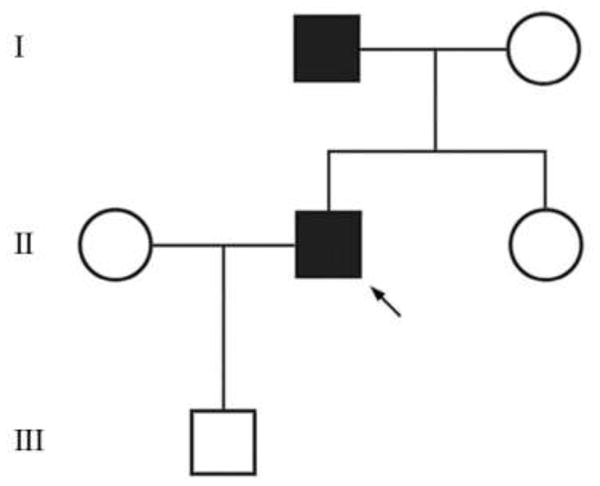
See Table 1 for phenotypic details and mutation analysis.
Table 1.
Summary of Clinical and Laboratory Results
| Subject | Clinical Myotonia | Myotonic Discharges | Muscle Biopsy | Heterozygous for G233S Mutation |
|---|---|---|---|---|
| Mother | No | No | N.D. | − |
| Father | Yes | Yes | N.D. | + |
| Sister | No | No | N.D. | − |
| Proband | Yes | Yes | Normal | + |
| Son | Unknown | ND | ND | ND |
DNA Analysis
Genomic DNA sequencing was carried out commercially (Athena Diagnostics, Worcester, MA). For the proband, sequencing of DMPK and ZNF9 for myotonic dystrophy (DM1 and DM2 respectively) was normal. Chloride channel CLCN1 sequencing of 23 exons and each surrounding intron revealed a single heterozygous 697G>A (NM_000083.2) missense mutation resulting in a glycine to serine amino acid change at codon 233 in exon 5. No other mutations were identified. For CLCN1, the proband’s father (Case 2) had the same heterozygous mutation, whereas the proband’s mother and sister had normal sequences (Fig. 3 and Table 1).
Electrophysiological Analysis of the Mutant CLC-1 Channel, G233S
The mRNAs of the WT human CLCN1 gene and the G233S mutant gene were injected into Xenopus oocytes, and the oocytes expressing the WT and mutant channels were subjected to patch clamp recording using the excised inside-out patch recording mode, using the two voltage protocols described above (Fig. 1A). Fig. 1B & C show the original patch-clamp recording traces of the WT and mutant channels, respectively. The overall open probabilities (Po) of these two channels are shown in Fig. 4A, while the estimated open probability of fast-gate (Pof) and that of the common gate (Poc) are shown in Fig. 4B (Po = Pof × Poc). It can be seen from the original recording traces that the inward current of the mutant at negative voltage is large compared to that the WT channel (compare original traces shown in Fig. 1B & C and the calculated Po shown in Fig. 4A). This effect is mostly due to a large open probability of the fast gate of the mutant channel—even at the very negative membrane potential of −140 mV. The fast-gate Pof of the G233S mutant is more than 0.8 while that of the WT channel is only ~0.2 (Fig. 4B). On the other hand, the effect of the G233S mutation on the common-gate Po is small—the common gate Po is increased by ~ 0.1 at the negative voltage range by the mutation (Fig. 4B).
Figure 4. Open probability (Po) of the WT and the G233S mutant channel.

(A) Overall open probability (Po = Pof × Poc) of the WT and mutant channels calculated from the tail-current from the recordings obtained with protocol A. (B) The open probability of the fast-gate (Pof, left panel) and of the common gate (Poc) as a function of the voltage for the WT channel (black squares) and G233S mutant (red squares).
Protein Expression Studies
To compare the total protein expression efficiency of myc-tagged WT CLC-1 and myc-tagged G233S mutant in mammalian cells, we performed Western blot analyses of transfected HEK293T cells. To this end, we probed Western blots of whole cell homogenates of the transfected cells for the C-terminal myc epitope. We also probed for β-actin as an internal control. As shown in Fig. 5A, the amounts of the expressed WT or the mutant protein, in comparison with the amount of endogenous β-actin, are qualitatively similar to each other. However, G233S surface expression was reduced to about half of the WT expression (Fig. 5A), suggesting that, in addition to the observed abnormal voltage-dependent activation curves, the mutant is abnormal in its trafficking to the membrane, This experimental system was also used to search for a dominant negative effect of the mutant. In cells transfected with a 1:1 ratio of WT to G233S mutant, the surface expression of the myc-labeled WT was not reduced in the presence of the mutant (Fig. 5B). This latter result from the co-expression of WT and mutant suggests that G233S mutant does not generate a dominant negative effect on the expression of the WT subunit.
Figure 5. Comparison of the protein expression efficiency for the wild-type CLC-1 and the G233S mutant.

(A) Total and surface expression levels of Myc-tagged WT CLC-1 and the G233S mutant in HEK293T cells. Left panel: Western blot of total proteins (left two lanes) and the biotinylated proteins (right two lanes) for WT and G233S. The amount of proteins loaded into each of the left two lanes was ~1/10 of that of the two right lanes. Membranes were immunoblotted with the anti-myc (Top) or the anti-β-actin (Bottom) antibody. Middle & Right panels: densitometric scans of anti-Myc density with respect to the cognate β-actin signal for the total-protein and the biotinylated-protein experiments, respectively. * indicates a statistically significant difference (p < 0.05, n = 4). (B) Co-expression with the G233S mutant fails to affect the surface expression of the WT CLC-1 channel. (Left panel) Western blot of the Myc-tagged CLC-1-WT co-expressed with Flag-tagged WT or Flag-tagged G233S (1:1 molar ratio) in HEK293T cells. Cell lysates were either directly immunoblotted (Total) or subject to biotinylation and streptavidin pull-down prior to immunoblotting analyses (Surface). Quantification of the total protein density (Middle panel) and surface expression efficiency (Right panel) revealed no significant difference between the two co-expression conditions (n = 3). (C) Comparison of the amplitude of the whole oocyte current generated from the expression of WT, G233S and WT+G233S (1:1 ratio). The current at +60 mV was used for the comparison (n = 32).
Whole Oocyte Currents
To examine the (surface) expression of functional channels, we injected the same amounts of RNA of WT, mutant, or both (1:1 mutant/wild-type ratio) into Xenopus oocytes, and then followed the growth of the whole oocyte current (at +60 mV) for three days. For oocytes expressing the G233S mutant CLC-1 channels, the conductance of oocytes expressing G233S was reduced (mean ± SEM on day 3 after RNA injection, 1.9 ± 0.2 μA) compared to 23.1 ± 1.8 μA for WT (Fig. 5C). To address possible dominant negative effects of the G233S mutant, oocytes were also injected with a 1:1 mixture of mutant and WT RNA (with the same amount of total injected RNA). Conductance in these cells was midway between the WT and the mutant, 15.1 ± 1.5 μA, demonstrating the absence of a dominant negative effect.
Discussion
More than 130 distinct mutations of CLCN1 have been reported in patients with pure myotonia. (Colding-Jorgensen, 2005; Gao et al., 2010; Kumar et al., 2010; Lossin and George, 2008; Lyons et al., 2010; Moon et al., 2009; Morales et al., 2008; Papponen et al., 2008; Pusch, 2002; Ryan et al., 2002; Shalata et al., 2010) Analysis of many of these “naturally occurring” mutations has contributed to the understanding of the molecular function of this molecule as well as providing data on the pathogenic mechanisms involved in the individual patients/families. Most mutations have been single nucleotide missense mutations, which have been distributed diffusely throughout the coding regions of the gene without apparent hotspots(Lossin and George, 2008). The vast majority are recessively inherited, with a few of these exhibiting a semidominant (Mailander et al., 1996; Pusch, 2002) inheritance pattern, i.e., occurrence of both recessive and dominant patterns. In none of the latter subgroup have the differences between the mechanisms involved in the dominantly inherited vs. the recessively inherited forms been delineated. In addition, pure dominantly inherited mutations occur but make up less than ten percent of total CLCN1 mutations. In this group, the location of the mutations is topologically relatively more restricted, primarily to the transmembrane regions of the protein (Duffield et al., 2003; Lossin and George, 2008; Pusch, 2002), and most of these mutations that have been studied physiologically have exhibited a shift in the channel activation curve toward positive membrane potentials, resulting in significantly reduced resting membrane stabilizing effects (Grunnet et al., 2003; Lin et al., 2006; Pusch, 2002).
The molecular mechanism involved in the increased open probability in the G233S mutant homodimers at near resting membrane potentials is expected. The mutation G233A was artificially made in previous experiments by others, and the mutation was found to disrupt the closing of the channel upon membrane hyperpolarization (Fahlke et al., 1997). The residue G233 of CLC-1 is within a conserved GKEGP motif present in CLC family members (Fig. 6A). The negatively charged side-chain of the glutamate residue E232 in this conserved motif is thought to be the fast-gate of mammalian CLC channels. Fig. 6B shows the high resolution crystal structure of bacterial CLC proteins. It can be seen that the homologous E148 residue projects its negatively charged side chain into the pore, obstructing the ion transport pathway. Because G233 is next to the corresponding glutamate residue (E232) in CLC-1, it is reasonable to conclude that the G233S mutation disrupts the fast-gating mechanism of CLC-1. On the other hand, the G233S mutation only slightly affects the common gating of CLC-1 (Fig. 4).
Figure 6. Molecular basis of the G233S channelopathy.

Shown on top of the figure is the conserved GKEGP motif of CLC molecules, in which the side-chain of the glutamate residue (labeled with grey star) is currently thought to be the fast gate. The X-ray structure of the CLC-ec1 molecule is depicted at the bottom, with the extracellular side of the molecule being on top. The critical glutamate (E148 in CLC-ec1 or E232 in CLC-1) was colored in CPK code while G149 of CLC-ec1 (G233 of CLC-1) is colored in orange. The two green spheres are the Cl− ions seen in the crystal structure of the bacterial CLC molecule. Curved arrows represent the anion transport pathways (one in each subunit).
Besides an increase in the fast-gate open probability for the G233S mutant, our analysis has identified another abnormality of the mutant—channel the surface expression of the G233S mutant appears to be reduced. The total chloride current from oocytes expressing G233S alone is reduced (Fig. 5C), which in homozygotes might result in myotonia. The cause of this reduction is likely related to the reduced insertion of the mutant channels into the cell membrane as biochemically shown in the reduction of the biotinylated portion of the expressed channels (Fig. 5A). However, since myotonia does not occur until the total CLC-1 conductance falls below ~ 20 % – 40 % of normal (Furman and Barchi, 1978), for a heterozygous channel even the complete absence of conductance through the mutant subunit alone would not be adequate to produce the clinical syndrome in the face of a normally functioning WT subunit. Therefore, many dominant mutants of myotonia exert a dominant negative effect in which the mutation in one subunit can reduce the function of the WT subunit, through interfering with the interaction of the two subunits. (Grunnet et al., 2003; Kubisch et al., 1998; Saviane et al., 1999; Wu et al., 2002). Also a number of previously reported dominantly inherited CLCN1 mutations that result in changes in voltage dependence have been identified in exon 8, which encodes the portion of the protein that forms the interface between the two subunits of the molecule (Duffield et al., 2003; Lossin and George, 2008; Pusch, 2002). Functionally, many of these mutations affect the common (or slow) gating mechanism that controls current flow through both pores (Macias et al., 2007; Simpson et al., 2004; Warnstedt et al., 2002).
A dominant negative effect can also occur at the protein expression level. It is conceivable that the expression of the mutant subunit in the cell membrane is defective (as a result of either a defect in protein synthesis or in membrane trafficking or both), and this defect may then render the surface expression of heterodimeric channels difficult. Our analysis of G233S channel expression indicates that this type of dominant negative effect appears not to underlie the dominant hereditary pattern in this family. The surface expression level of the G233S mutant is indeed lower than that of the WT subunit in both Xenopus oocytes and HEK293T cells (Fig. 5A & C). However, experiments of WT/mutant co-expression in transfected HEK293T cells have demonstrated no statistical difference in the amount of surface WT channel subunit whether the WT channel is co-expressed with the G233S mutant or not. (Fig. 5B). In Xenopus oocytes (Fig. 5C), the G233S mutant generates much smaller current but the oocytes that were co-transfected with WT and G233S (in 1:1 ratio) still produced total whole oocyte current larger than 50 % of that from oocytes expressing WT subunit alone. These results suggest that the G233S mutant does not exert a dominant negative effect on the surface expression level or the functional current of the WT channel, at least not in Xenopus oocytes or in mammalian HEK293T cells.
It should be noted that our observations for channel expressions in the HEK293T or Xenopus oocyte model system may not mimic the expression patterns in native muscle fibers in vivo. If the expression of the mutant greatly exceeds that of the WT subunit in native muscle membrane, myotonia may occur even if G233S appears to be a gain-of-function mutation with respect to the molecular function of the CLC-1 molecule (the mutation increases CLC-1’s open probability). However, with respect to the repolarization of the membrane voltage after the firing of an action potential, the G233S mutant appears, in fact, to be a loss-of-function mutation. From the voltage-dependent activation curve of the WT channel, the overall Po (Pof × Poc) of the WT channel is increased by 5 fold from a resting potential of −90 mV (Po ≈ 0.2) to the peak of the action potential of > 40 mV (Po ≈ 1). On the other hand, the increase of the overall Po for the G233S mutant is less than 2 fold (from Po ≈ 0.6 at −90 mV to Po ≈1 at > 40 mV) (Fig. 4A). Because an increase of Cl− conductance in response to membrane depolarization is critical to repolarize the membrane potential from the peak of an action potential back to the resting state, a defect in the dynamics of the response to a voltage change as occurs in the G233S mutant may contribute to the over excitability of the membrane and the clinical myotonia. In this regard, the effect of the G233S mutation can be considered to be similar to those caused by the loss-of-function mutations of CLCN1 that result in reduced conductance at positive membrane potentials.
Because we were unable to detect a dominant negative effect of the G233S mutation, the mechanism involved in the dominant inheritance pattern of this family is not clear. In theory, there are other mechanisms that may explain the dominant hereditary pattern (Colding-Jorgensen, 2005; Raj et al., 2010) of the G233S mutation, including a differential allelic expression favoring mutant homodimers (Colding-Jorgensen, 2005; Duno et al., 2004; Mailander et al., 1996) or reduced transport of the heterodimer into the membrane from the endoplasmic reticulum (Papponen et al., 2008). Other mechanisms that have also be reported include founder effects (linkage disequilibrium with genes modifying CLC-1 expression or function) (Koty et al., 1996) and incomplete mutation detection, the presence of an as yet unidentified mutation in a modifying gene (Zhang et al., 1996). Studies of additional families with similar dominant mutations may provide additional information on the underlying mechanisms
Acknowledgments
Supported by research grants to Dr. Richman from National Institutes of Health (R21NS071325-01), Muscular Dystrophy Association (MDA114815), Myasthenia Gravis Foundation of California; grants to Dr. Maselli from National Institutes of Health (R01NS049117-01), Muscular Dystrophy Association, Myasthenia Gravis Foundation of America and Myasthenia Gravis Foundation of California; grants to Dr. Tang from National Science Council, Taiwan (NSC 96-2320-B-002-069-MY3) and grants to Dr. Chen from National Institutes of Health (R01GM065447).
References
- Chen TY. Structure and function of clc channels. Annu Rev Physiol. 2005;67:809–839. doi: 10.1146/annurev.physiol.67.032003.153012. [DOI] [PubMed] [Google Scholar]
- Colding-Jorgensen E. Phenotypic variability in myotonia congenita. Muscle Nerve. 2005;32:19–34. doi: 10.1002/mus.20295. [DOI] [PubMed] [Google Scholar]
- Duffield M, et al. Involvement of helices at the dimer interface in ClC-1 common gating. J Gen Physiol. 2003;121:149–161. doi: 10.1085/jgp.20028741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duno M, et al. Difference in allelic expression of the CLCN1 gene and the possible influence on the myotonia congenita phenotype. Eur J Hum Genet. 2004;12:738–743. doi: 10.1038/sj.ejhg.5201218. [DOI] [PubMed] [Google Scholar]
- Dutzler R. The structural basis of ClC chloride channel function. Trends Neurosci. 2004;27:315–320. doi: 10.1016/j.tins.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Dutzler R. The ClC family of chloride channels and transporters. Curr Opin Struct Biol. 2006;16:439–446. doi: 10.1016/j.sbi.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Fahlke C, et al. Pore-forming segments in voltage-gated chloride channels. Nature. 1997;390:529–32. doi: 10.1038/37391. [DOI] [PubMed] [Google Scholar]
- Furman RE, Barchi RL. The pathophysiology of myotonia produced by aromatic carboxylic acids. Ann Neurol. 1978;4:357–65. doi: 10.1002/ana.410040411. [DOI] [PubMed] [Google Scholar]
- Gao F, et al. Novel chloride channel gene mutations in two unrelated Chinese families with myotonia congenita. Neurol India. 2010;58:743–6. doi: 10.4103/0028-3886.72163. [DOI] [PubMed] [Google Scholar]
- Grunnet M, et al. Characterization of two new dominant ClC-1 channel mutations associated with myotonia. Muscle Nerve. 2003;28:722–732. doi: 10.1002/mus.10501. [DOI] [PubMed] [Google Scholar]
- Heatwole CR, Moxley RT., III The nondystrophic myotonias. Neurotherapeutics. 2007;4:238–251. doi: 10.1016/j.nurt.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ. CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Crit Rev Biochem Mol Biol. 2008;43:3–36. doi: 10.1080/10409230701829110. [DOI] [PubMed] [Google Scholar]
- Koty PP, et al. Myotonia and the muscle chloride channel: dominant mutations show variable penetrance and founder effect. Neurology. 1996;47:963–968. doi: 10.1212/wnl.47.4.963. [DOI] [PubMed] [Google Scholar]
- Kubisch C, et al. ClC-1 chloride channel mutations in myotonia congenita: variable penetrance of mutations shifting the voltage dependence. Hum Mol Genet. 1998;7:1753–1760. doi: 10.1093/hmg/7.11.1753. [DOI] [PubMed] [Google Scholar]
- Kumar KR, et al. A novel CLCN1 mutation (G1652A) causing a mild phenotype of thomsen disease. Muscle Nerve. 2010;41:412–5. doi: 10.1002/mus.21610. [DOI] [PubMed] [Google Scholar]
- Li HY, et al. Hyperphosphorylation as a defense mechanism to reduce TDP-43 aggregation. PLoS One. 2011;6:e23075. doi: 10.1371/journal.pone.0023075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MJ, et al. Functional characterization of CLCN1 mutations in Taiwanese patients with myotonia congenita via heterologous expression. Biochem Biophys Res Commun. 2006;351:1043–1047. doi: 10.1016/j.bbrc.2006.10.158. [DOI] [PubMed] [Google Scholar]
- Lossin C, George AL., Jr Myotonia congenita. Adv Genet. 2008;63:25–55. doi: 10.1016/S0065-2660(08)01002-X. [DOI] [PubMed] [Google Scholar]
- Lyons MJ, et al. Novel CLCN1 mutation in carbamazepine-responsive myotonia congenita. Pediatr Neurol. 2010;42:365–8. doi: 10.1016/j.pediatrneurol.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Macias MJ, et al. Myotonia-related mutations in the distal C-terminus of ClC-1 and ClC-0 chloride channels affect the structure of a poly-proline helix. Biochem J. 2007;403:79–87. doi: 10.1042/BJ20061230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailander V, et al. Novel muscle chloride channel mutations and their effects on heterozygous carriers. Am J Hum Genet. 1996;58:317–324. [PMC free article] [PubMed] [Google Scholar]
- Moon IS, et al. Novel CLCN1 mutations and clinical features of Korean patients with myotonia congenita. J Korean Med Sci. 2009;24:1038–44. doi: 10.3346/jkms.2009.24.6.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales F, et al. Clinical and molecular diagnosis of a Costa Rican family with autosomal recessive myotonia congenita (Becker disease) carrying a new mutation in the CLCN1 gene. Rev Biol Trop. 2008;56:1–11. doi: 10.15517/rbt.v56i1.5505. [DOI] [PubMed] [Google Scholar]
- Papponen H, et al. F413C and A531V but not R894X myotonia congenita mutations cause defective endoplasmic reticulum export of the muscle-specific chloride channel CLC-1. Muscle Nerve. 2008;37:317–325. doi: 10.1002/mus.20922. [DOI] [PubMed] [Google Scholar]
- Pusch M. Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Hum Mutat. 2002;19:423–434. doi: 10.1002/humu.10063. [DOI] [PubMed] [Google Scholar]
- Pusch M, et al. Mutations in dominant human myotonia congenita drastically alter the voltage dependence of the CIC-1 chloride channel. Neuron. 1995;15:1455–1463. doi: 10.1016/0896-6273(95)90023-3. [DOI] [PubMed] [Google Scholar]
- Raj A, et al. Variability in gene expression underlies incomplete penetrance. Nature. 2010;463:913–8. doi: 10.1038/nature08781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan A, et al. A novel alteration of muscle chloride channel gating in myotonia levior. J Physiol. 2002;545:345–354. doi: 10.1113/jphysiol.2002.027037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saviane C, et al. The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. J Gen Physiol. 1999;113:457–68. doi: 10.1085/jgp.113.3.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalata A, et al. Myotonia congenita in a large consanguineous Arab family: insight into the clinical spectrum of carriers and double heterozygotes of a novel mutation in the chloride channel CLCN1 gene. Muscle Nerve. 2010;41:464–9. doi: 10.1002/mus.21525. [DOI] [PubMed] [Google Scholar]
- Simpson BJ, et al. Characterization of three myotonia-associated mutations of the CLCN1 chloride channel gene via heterologous expression. Hum Mutat. 2004;24:185. doi: 10.1002/humu.9260. [DOI] [PubMed] [Google Scholar]
- Tseng PY, et al. Cytoplasmic ATP inhibition of CLC-1 is enhanced by low pH. J Gen Physiol. 2007;130:217–21. doi: 10.1085/jgp.200709817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng PY, et al. Binding of ATP to the CBS domains in the C-terminal region of CLC-1. J Gen Physiol. 2011;137:357–68. doi: 10.1085/jgp.201010495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnstedt M, et al. The myotonia congenita mutation A331T confers a novel hyperpolarization-activated gate to the muscle chloride channel ClC-1. J Neurosci. 2002;22:7462–7470. doi: 10.1523/JNEUROSCI.22-17-07462.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu FF, et al. Novel CLCN1 mutations with unique clinical and electrophysiological consequences. Brain. 2002;125:2392–2407. doi: 10.1093/brain/awf246. [DOI] [PubMed] [Google Scholar]
- Zhang J, et al. Mutations in the human skeletal muscle chloride channel gene (CLCN1) associated with dominant and recessive myotonia congenita. Neurology. 1996;47:993–998. doi: 10.1212/wnl.47.4.993. [DOI] [PubMed] [Google Scholar]
- Zhang XD, et al. ATP inhibition of CLC-1 is controlled by oxidation and reduction. J Gen Physiol. 2008;132:421–8. doi: 10.1085/jgp.200810023. [DOI] [PMC free article] [PubMed] [Google Scholar]