

SUMMARY
Systemic dissemination of microbial pathogens permits microbes to spread from the initial site of infection to secondary target tissues and is responsible for most mortality due to bacterial infections. Dissemination is a critical stage of disease progression by the Lyme spirochete, Borrelia burgdorferi. However, many mechanistic features of the process are not yet understood. A key step is adhesion of circulating microbes to vascular surfaces in the face of the shear forces present in flowing blood. Using real-time microscopic imaging of the Lyme spirochete in living mice we previously identified the first bacterial protein (B. burgdorferi BBK32) shown to mediate vascular adhesion in vivo. Vascular adhesion is also dependent on host fibronectin (Fn) and glycosaminoglycans (GAGs). In the present study, we investigated the mechanisms of BBK32-dependent vascular adhesion in vivo. We determined that BBK32-Fn interactions (tethering) function as a molecular braking mechanism that permits the formation of more stable BBK32-GAG interactions (dragging) between circulating bacteria and vascular surfaces. Since BBK32-like proteins are expressed in a variety of pathogens we believe that the vascular adhesion mechanisms we have deciphered here may be critical for understanding the dissemination mechanisms of other bacterial pathogens.
Keywords: Lyme disease, Borrelia, spirochete, hematogenous dissemination, intravital microscopy, vascular adhesion, fibronectin, glycosaminoglycans, confocal microscopy, shear force, BBK32
INTRODUCTION
Lyme borreliosis is a multi-system, tick-transmitted infection caused by the spirochete Borrelia burgdorferi and several other closely related species (Stanek et al., 2012, Steere et al., 2004). Lyme borreliosis is the most prevalent vector-borne disease in the northern hemisphere and its reported incidence worldwide is steadily increasing (Wilske, 2005). B. burgdorferi is transmitted to the skin of vertebrate hosts during the blood meal of Ixodes ticks. Inoculation is typically followed by appearance of a skin lesion, transient spirochetemia, and vascular dissemination to multiple host tissues, including joints and the cardiovascular and nervous systems (Schmid, 1989).
Despite the health impacts of bacterial dissemination, the mechanisms involved in this process remain largely unknown. The recent development of dynamic fluorescence- and bioluminescence-based imaging methods for visualizing spirochete behaviour and dissemination in mouse models of Lyme disease have provided new approaches to study this process (Hyde et al., 2011, Moriarty et al., 2008, Norman et al., 2008, Lee et al., 2010, Harman et al., 2012). In situ imaging has also yielded critical insight into B. burgdorferi dissemination in ticks (Dunham-Ems et al., 2012, Dunham-Ems et al., 2009). Bioluminescence-based imaging of B. burgdorferi infection has provided important insight into the kinetics of bacterial dissemination and revealed a role for the adhesin BBK32 in early colonization of target tissues, including skin and joints (Hyde et al., 2011). Intravital microscopy-based fluorescent imaging approaches (see Videos S1 and S2) have identified host mechanisms mediating intravascular immune responses to disseminating B. burgdorferi (Lee et al., 2010) and have provided considerable insight into host and spirochete molecules and mechanisms underlying vascular adhesion of blood-borne B. burgdorferi (Moriarty et al., 2008, Norman et al., 2008). Intravital microscopy approaches have also identified BBK32 as a B. burgdorferi adhesin under the shear force conditions encountered in the mouse vasculature (Norman et al., 2008).
A critical step in dissemination is adhesion of circulating microbes to vascular surfaces prior to their transmigration into surrounding tissues where sites of secondary infection are established. Intravital microscopy studies of B. burgdorferi vascular adhesion and extravasation in vivo indicate that the early stages of this process bear an interesting resemblance to leukocyte recruitment, likely because the initiation steps of both forms of vascular interaction face a similar physical problem: the requirement for overcoming fluid shear forces during deceleration and arrest on vascular surfaces (Moriarty et al., 2008, Norman et al., 2008, Ley et al., 2007). Like leukocyte recruitment, B. burgdorferi vascular adhesion and extravasation is a progressive multi-stage process initiated by tethering then dragging/crawling interactions on extrajunctional surfaces of the endothelial glycocalyx, followed by stationary adhesion and/or extravasation at intercellular junctions (Fig. 1) (Moriarty et al., 2008, Norman et al., 2008).
Figure 1. Summary of current knowledge of the stages of infectious B. burgdorferi interaction with and escape from the microvasculature.

Based upon our previous work (Moriarty et al., 2008, Norman et al., 2008) we have proposed that tethering (previously referred to as transient) and dragging interactions together constitute the essential first step (initiation) of microvascular interactions. We also proposed (Moriarty et al., 2008, Norman et al., 2008) that initiation interactions are mechanistically distinct from downstream interaction events for two reasons: 1) stationary adhesions and transmigrating spirochetes localize to different sites on the endothelium than transient and dragging interactions, and 2) stationary adhesion appears to require host and/or spirochete molecules in addition to or other than BBK32, GAGs and Fn.
All shear force-regulated cell-cell adhesion mechanisms identified to date are mediated by interactions between glycosylated lectins (Thomas, 2009). Our intravital microscopy studies reveal that B. burgdorferi interactions with the vasculature in vivo are also regulated by carbohydrate-containing molecules, in this case glycosaminoglycans (GAGs) (Norman et al., 2008), and that the initiating tethering and dragging steps of B. burgdorferi vascular adhesion occur on the surface of the endothelial glycocalyx, which is rich in heparan sulphate (Moriarty et al., 2008, Wang et al., 2005) B. burgdorferi vascular interactions are potently inhibited by intravascular competition with antibodies to plasma fibronectin (pFn) (Norman et al., 2008), a molecule which can bind the GAG heparin, suggesting a fascinating biological role for pathogen pFn-recruitment in vascular adhesion. Fn is a complex, multidomain molecule (see Fig. 2A) that interacts with a wide range of cell types, molecules and many bacteria (Henderson et al., 2011, Pankov & Yamada, 2002). Intravascular competition with pFn antibodies or Fn-derived peptides impairs the tethering interactions that initiate B. burgdorferi vascular adhesion (Norman et al., 2008). Tethering interactions are not inhibited by a Fn-derived peptide carrying the sequence Arg-Gly-Asp (RGD), which mediates interaction of many cell types with Fn via integrins (see Fig. 2A). However, tethering interactions are inhibited by peptide sequences from the Fn HepII domain (FN C/H-II; see Fig. 2A), which binds to GAGs. The Fn HepII domain may block both Fn-dependent and –independent interactions by occupying glycosylated bacterial adhesion receptors in the vascular glycocalyx (Norman et al., 2008). Thus, initiation of B. burgdorferi vascular adhesion could be mediated by direct interaction of B. burgdorferi adhesins with GAGs, or indirectly by association of adhesin-recruited bridging pFn molecules with glycosylated endothelial receptors.
Figure 2. Effect of bbk32 disruption and Fn heparin-binding peptide on microvascular interactions of B. burgdorferi in vivo.
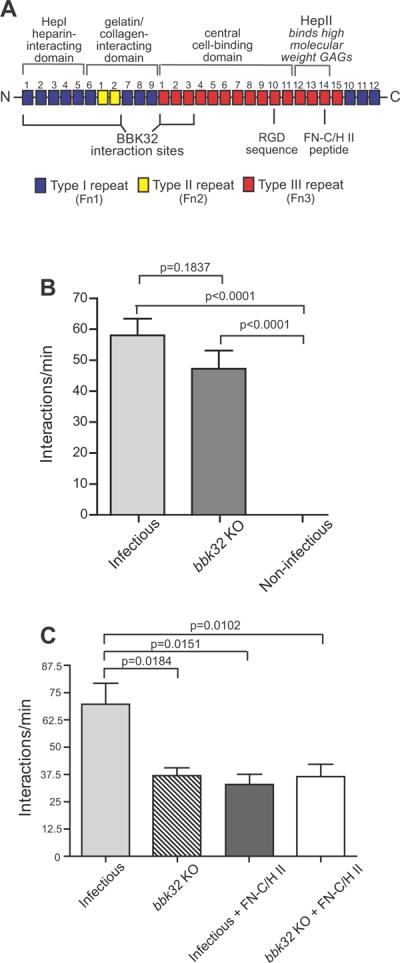
A) Schematic depicting the organization of plasma Fn (not to scale). Major domains are indicated by labels above the schematic. BBK32 interaction sites, and the locations of RGD and FN-C/H II heparin-binding peptides are indicated below the schematic. Numbers above each Fn repeat indicate repeat number, and Fn Type 1, 2 and 3 repeats are color-coded in blue, yellow and red, respectively. Information on Fn structure and the Fn regions where BBK32 interacts are from (Kim et al., 2004, Pankov & Yamada, 2002, Prabhakaran et al., 2009, Raibaud et al., 2005). B) Microvascular interaction rates in skin (transient and dragging interactions combined) are shown for three GFP-expressing B. burgdorferi strains, as analyzed by high acquisition rate spinning disk confocal intravital microscopy. Strains (see also Table S1): 1) GCB966: B31 derivative ML23 (infectious following transformation with a shuttle vector containing bbe22), 2) GCB971: parental with genetic disruption of bbk32 (bbk32 KO), 3) GCB705: non-infectious high passage control (B31-A derivative). A total of 10,387 interactions in 169 venules from 16 mice (n=6 infectious; n=5 bbk32 KO; n=5 non-infectious) were analyzed. Means and standard error bars are indicated for each experimental group. Statistical testing for significant differences among all experimental groups was performed using a non-parametric Kruskal-Wallis ANOVA (overall P value of <0.0001), followed by Dunn's multiple comparison test. *P<0.05, **P<0.01, ***P<0.001. All statistical data for pairwise comparison between groups may be found in Table S4. Microvascular interactions were measured between 5 and 45 minutes after spirochete injection. The average time after injection for recordings made with infectious, bbk32 KO, and non-infectious strains was 22.63−/+1.31, 23.32−/+2.02, and 23.19−/+2.07 min, respectively. C) Microvascular interaction rates in the knee joint are shown for two GFP-expressing B. burgdorferi strains in mice treated or untreated with the fibronectin heparin-binding peptide (FN-C/H II), as analyzed by high acquisition rate spinning disk confocal intravital microscopy. Strains (see also Table S1): 1) GCB966: parental B31 derivative ML23 (infectious following transformation with shuttle vector containing bbe22 and PflaB-gfp), 2) GCB971: GFP-expressing parental with genetic disruption of bbk32. The interactions in 84 venules from 12 mice (n=3 mice/experimental group) were analyzed. Microvascular interactions were measured between 10 and 50 minutes after spirochete injection. Means and standard error bars are indicated for each experimental group. Statistical testing for significant differences among all experimental groups was performed using a non-parametric Kruskal-Wallis ANOVA (overall P value of 0.0114). P-values obtained by two-tailed t-test with correction for unequal variance are indicated for selected pair-wise comparisons. All statistical data may be found in Table S4.
B. burgdorferi expresses several known and putative adhesins that interact with Fn and/or GAGs and might contribute to vascular adhesion, including the outer membrane lipoprotein BBK32, RevA, RevB, CRASP-1, Bgp, DbpA/B and BB0347 (Antonara et al., 2011, Fischer et al., 2006, Probert et al., 2001, Brissette et al., 2009, Hallstrom et al., 2010, Parveen & Leong, 2000, Parveen et al., 2003, Fraser et al., 1997). Using comparative genetic and gain-of-function approaches coupled with intravital microscopy, we determined that BBK32, which binds both Fn and GAGs, is sufficient to restore vascular interactions to a high passage, non-adhesive, non-infectious B. burgdorferi strain in vivo, and is thus a vascular adhesin (Norman et al., 2008). Fn-binding BBK32 orthologues are found in other pathogens including Streptococcus pyogenes and Staphylococcus aureus (Raibaud et al., 2005, Kim et al., 2004, Henderson et al., 2011, Norris et al., 2011) but the potential contribution of these BBK32-like adhesins to vascular adhesion has not been determined. Many pathogens encode multiple Fn-binding proteins which are partially or fully functionally redundant with one another, and pathogen Fn-binding proteins often confer additional functions, such as complement evasion (Henderson et al., 2011, Zipfel et al., 2007).
BBK32 is the first bacterial protein which has been shown to mediate vascular adhesion in vivo (Norman et al., 2008). BBK32 interacts with multiple substrates, including Fn, heparan and dermatan sulfate GAGs, and fibrinogen (Probert et al., 2001, Kim et al., 2004, Raibaud et al., 2005, Prabhakaran et al., 2009, Fischer et al., 2006). BBK32 binds Fn via the Fn N-terminal HepI, gelatin-binding domains and Fn3 modules, but is not known to interact with the C-terminal HepII GAG-binding sequences of Fn implicated in vascular adhesion of B. burgdorferi (Fig. 2A). BBK32 peptides induce conformational changes in soluble Fn in vitro that aggregate Fn into adhesive superfibronectin arrays (Prabhakaran et al., 2009). Genetic disruption of bbk32 does not reduce infectivity in mice inoculated with a high infectious dose of B. burgdorferi (Li et al., 2006). However, infection with more naturally relevant doses indicates that bbk32 disruption significantly attenuates infectivity and dissemination to target tissues, including skin and joints (Seshu et al., 2006, Hyde et al., 2011). Although we have identified BBK32 as a vascular adhesin, it is not yet known whether B. burgdorferi encodes additional vascular adhesins. Furthermore, the mechanisms by which BBK32 confers vascular adhesion are not yet well-defined. Specifically, it is not yet known whether BBK32 GAG-binding, Fn-binding, or both, are responsible for its ability to confer vascular adhesion under fluid shear force.
In this study, we explore the ability of several known and putative B. burgdorferi Fn-binding proteins to functionally substitute for BBK32 in vascular adhesion in vivo. We also define the contributions of BBK32 Fn- and GAG-binding to the initiation steps of B. burgdorferi vascular adhesion
RESULTS
Disruption of bbk32 has a modest effect upon B. burgdorferi microvascular interactions and results in interactions that are resistant to Fn HepII peptide
We showed previously using intravital microscopy that BBK32 is sufficient to restore the initiation steps of microvascular adhesion (tethering and dragging interactions) to the non-adhesive, non-infectious B. burgdorferi strain B31-A (Norman et al., 2008). To determine whether BBK32 is required for vascular interaction of B. burgdorferi, we examined the properties of a bbk32-disrupted strain, GCB971 (JS315/pJW201) and its infectious parent, GCB966 (ML23/pJW201) (Seshu et al., 2006, Wu et al., 2011, Hyde et al., 2011, Labandeira-Rey & Skare, 2001) in microvascular interactions using intravital microscopy (Norman et al., 2008, Moriarty et al., 2008) (see Videos S1 and S2; see Table S1 for strain information). We found that genetic disruption of bbk32 reduced transient (tethering) and dragging microvascular interactions in the skin by about 20%, a reduction that did not reach statistical significance (Fig. 2B). Thus, in spite of the complete disruption of BBK32 expression in this strain (Seshu et al., 2006), about 80% of the microvascular interactions remained, implying that BBK32 was not the only adhesin involved in interactions with the skin vascular endothelium.
Recent evidence indicates that genetic disruption of bbk32 in an otherwise infectious strain significantly impairs colonization of dissemination target tissues, including skin and joints, in early stages of infection (Hyde et al., 2011). Using GCB966 and GCB971, we found that genetic disruption of bbk32 reduced initiating interactions in the joint of mice, where BBK32 is especially important for dissemination of B. burgdorferi (Hyde et al., 2011), by about 50% (P=0.0184; Fig 2C). In spite of the complete disruption of BBK32 expression in this strain (Seshu et al., 2006), at least 50% of the microvascular interactions remained in both joint and skin, implying that BBK32 was not the only adhesin involved in interactions with the vascular endothelium. These data also suggest that there are tissue-specific differences in the BBK32 dependence of vascular interactions.
To determine if infectious B. burgdorferi encodes additional Fn adhesins that can functionally substitute for BBK32, we analyzed the effect of the Fn C-terminal HepII heparin-binding peptide (FN-C/H II: KNNQKSEPLIGRKKT; Fig 2A) on B. burgdorferi vascular interactions for infectious parental and the bbk32-deficient strain (Fig. 2C). This peptide, which inhibits Fn-mediated cell adhesion and heparan sulphate binding (Drake et al., 1993), specifically reduces microvascular interactions for infectious B. burgdorferi in skin by ~50% (Norman et al., 2008). The peptide was injected immediately before spirochete inoculation as previously described (Norman et al., 2008). As we noted in flank skin (Norman et al., 2008), the infectious parental strain showed a significant, approximately 50% reduction in interactions in the joint in the presence of the Fn HepII peptide (Fig. 2C). In contrast, no additive decrease in interactions was observed for the bbk32 mutant in the presence of this peptide. No significant differences were found in the interaction rates for the peptide-treated parental strain, and the treated and untreated bbk32 knockout strain. We conclude that BBK32-mediated initiation of vascular adhesion is dependent on interactions with glycosylated endothelial receptors which are blocked by the HepII peptide, and that BBK32 and interactions mediated by the Fn HepII peptide are part of the same vascular adhesion pathway. These data also indicate that as yet unidentified vascular adhesins encoded by infectious B. burgdorferi mediate BBK32-independent vascular adhesion by an interaction mechanism that is distinct from the binding pathway targeted by the Fn HepII peptide. Other GAG-binding proteins such as DbpA/B (Benoit et al., 2011) and Bgp (Parveen et al., 2006) could potentially play a role in Fn-independent microvascular interactions.
RevA, RevB and BB0347 do not restore vascular interactions to non-adhesive spirochetes in vivo, and bind Fn more weakly than BBK32
The results of the above bbk32 disruption experiments (Fig. 2) implied that additional, functionally redundant B. burgdorferi adhesins also likely mediate microvascular interactions in vivo. Therefore we examined the ability of other B. burgdorferi proteins reported or predicted to recognize Fn to restore microvascular interactions to a non-infectious, non-adhesive strain in vivo, using a gain-of-function system. We previously used a gain-of-function approach to identify BBK32 as a vascular adhesin in skin (Norman et al., 2008). Candidate vascular adhesins RevA, RevB and BB0347 (Brissette et al., 2009, Fraser et al., 1997) were constitutively expressed from independent plasmids in a fluorescent non-adherent B. burgdorferi strain (Fig 3A). Antibodies to RevA and RevB are detected in the sera of infected mice, and RevA is known to be expressed on the surface of B. burgdorferi, although recent evidence indicates that RevA is not involved in endothelial interactions under static conditions in vitro (Carroll et al., 2001, Brissette et al., 2009, Schmit et al., 2011). BB0347 is a predicted Fn-binding protein whose expression and localization have not previously been characterized (Fraser et al., 1997). An intact bb0347 gene is present in the non-infectious strain we used for our studies (Norman et al., 2008); however, whether the protein is expressed in this high passage strain (GCB706) is unknown. One copy of the revA gene is present but the other is absent from the non-infectious strain; expression is unknown. The revB gene is absent. RevA, RevB and BB0347 were expressed in our gain-of-function strains, as determined by Western blotting of lysates derived from strains expressing C-terminally Flag-tagged versions of each protein (Fig. S1). Treatment with proteinase K (Fig. S1A) or pronase (Fig. S1B) indicated that at least 50% of the expressed RevA and almost all of the RevB were surface localized, as expected. In contrast, BB0347 was resistant to both proteinase K and pronase, suggesting that this protein is either not surface localized or is resistant to these proteases for other reasons.
Figure 3. Role of BBK32 and other B. burgdorferi Fn-binding proteins in microvascular interactions in vivo and in evasion of intravascular immune clearance.
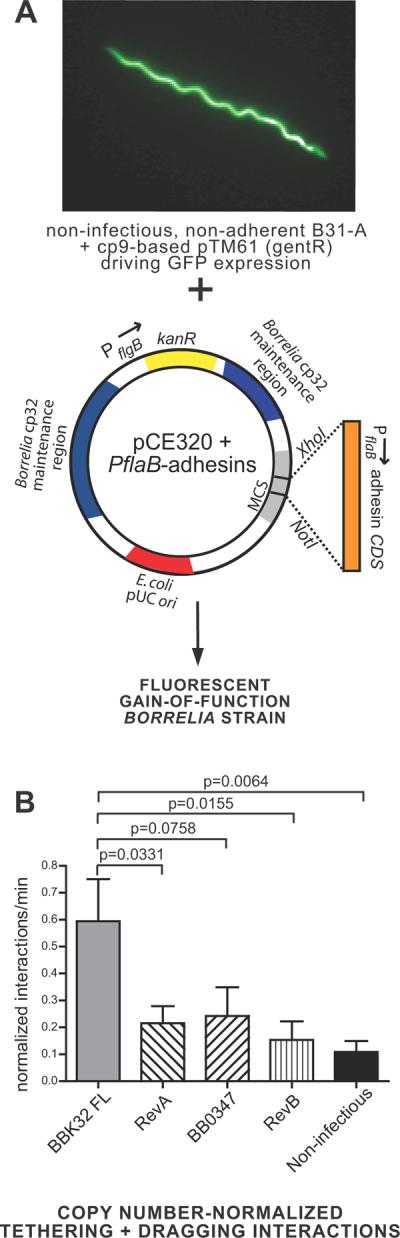
A) Schematic depicting the gain-of-function model system used to study the role of individual B. burgdorferi adhesins in vascular interactions in vivo. A GFP-expressing high passage non-adherent strain was transformed with a cp32-based pCE320 shuttle vector (Eggers 2002) bearing coding sequences for the adhesin of interest under the control of the constitutive flaB promoter. B) Microvascular interaction rates (tethering and dragging interactions combined) normalized to flaB copy number in blood collected from each animal following intravital microscopy. Normalization was performed by dividing the tethering and dragging interaction rate values measured in each vessel by the flaB copy number measured in the blood from the same mouse where intravital microscopy was performed. For unadjusted interaction rates and copy number data, see Fig. S2. Interactions in 101 venules from 27 mice (n=6 BBK32 FL; n=6 RevA; n=5 BB0347; n=6 RevB; n=4 non-infectious) were analyzed. Means and standard error bars are indicated for each experimental group. Microvascular interactions were measured between 5 and 25 minutes after spirochete injection. The average time after injection for recordings made with BBK32 FL, RevA, BB0347, RevB and non-infectious strains was 11.96−/+0.95, 11.88−/+1.19, 12.11−/+1.10, 13.25−/+0.93, and 13.50−/+0.77 min, respectively. Statistical testing for significant differences among all experimental groups was performed using a non-parametric Kruskal-Wallis ANOVA (overall P value of 0.0076). P-values are indicated for selected pair-wise comparisons. All statistical data may be found in Table S4. Strains used were GCB1585 (BBK32 FL), GCB1570 (RevA), GCB1574 (BB0347), GCB1586 (RevB) and GCB706 (non-adherent parent).
We examined the abilities of plasmid-expressed RevA, RevB and BB0347 to confer vascular interactions to the non-adherent B. burgdorferi strain in the skin in vivo. These gain of function strains displayed altered the levels of blood-borne spirochetes in mice (Fig. S2A). Therefore, following analysis of microvascular interaction rates recorded by intravital microscopy (Fig. S2B, C), we normalized the interaction rates measured in each vessel by dividing by the flaB copy number measured in the blood of the same mouse where interactions were recorded. The resulting data (Fig. 3B) indicated that none of these adhesins significantly restored microvascular interactions. Gain of function experiments were performed in skin, where we first identified a role for BBK32, Fn and GAGs in the B. burgdorferi vascular adhesion mechanism (Norman et al., 2008). In contrast, as we have previously reported (Norman et al., 2008), gain-of-function expression of BBK32 restored tethering and dragging interaction rates to levels observed for an infectious B. burgdorferi strain (Fig. 3B and S2). Therefore, we conclude that RevA, RevB and BB0347 do not act as vascular adhesins in the context of the fluid shear forces associated with blood flow and under the conditions of this experiment. These results support BBK32 as the major Fn-binding protein in B. burgdorferi that is involved in vascular interactions.
To investigate the mechanistic basis for the ability of BBK32, but not RevA, RevB or BB0347, to act as a vascular adhesin, we quantitatively studied the Fn-binding properties of these adhesins using surface plasmon resonance (SPR). The interaction of various concentrations of each of the adhesins with immobilized Fn is shown in Fig. S3 and the data are summarized in Table 1. The equilibrium binding constants (KD) for RevA, RevB and BB0347 were 46, 1,447 and 34 times higher, respectively than the KD (1.9 × 10−8) observed for full length BBK32, indicating that these adhesins bind Fn with substantially reduced affinity compared to BBK32. RevB binding to Fn was very weak (KD of 27.5 μM), suggesting that soluble Fn-binding may not be a major function of this protein. BB0347, which has been previously reported only as a putative Fn-binding protein based upon primary sequence (Fraser et al., 1997), bound immobilized Fn with an affinity similar to that of RevA. The dissociation rate constants for all four proteins differed by less than a factor of two, whereas major differences occurred in the forward rate constants. Thus, differences in affinity of these proteins for Fn was due primarily to differences in the rate of association, and not to differences in interaction stability (half-life). We postulate that the lower affinity interactions of RevA, RevB and BB0347 with Fn compared to the BBK32-Fn interaction are likely important factors in their inability to restore vascular interactions to non-adherent spirochetes. In addition, our data suggesting that BB0347 may not be surface localized would also be a decisive factor in precluding its participation in endothelial interactions.
Table 1.
Dissociation constants and kinetic data for various Fn- and dermatan sulfate-binding proteins interacting with Fn or dermatan sulfate, as determined by surface plasmon resonance.
| Fn binding protein | Binding partners | Kon (105s−1M−1) | Koff (s−1) | KD (μM) |
|---|---|---|---|---|
| RevA | Fn | 1.71 ± 0.220 | 0.15 ± 0.023 | 0.88 ± 0.03 |
| RevB | Fn | 0.04 ± 0.015a | 0.11 ± 0.010a | 27.5 ± 6.14a |
| BB0347 | Fn | 2.65 ± 0.492 | 0.16 ± 0.028 | 0.64 ± 0.02 |
| BBK32 | Fn | 52.1 ± 2.1 | 0.10 ± 0.054 | 0.019 ± 0.009 |
| BBK32 | Dermatan sulfate | 1.12 ± 0.115 | 0.02 ± 0.006 | 0.20 ± 0.03 |
Because the binding of RevB to Fn is weak, the determined values should be considered estimates.
Since BBK32 binds both Fn and GAGs, and vascular adhesion is dependent on Fn and GAGs (Fischer et al., 2006, Norman et al., 2008), we also measured the affinity of BBK32 for GAGs (Fig. S3, Table 1). We found that BBK32 GAG-binding was at least one order of magnitude weaker than BBK32 Fn-binding. BBK32 bound Fn with rapid association and dissociation kinetics (Table 1). However, BBK32-GAG interactions had a five-fold longer half-life than BBK32-Fn interactions (t½ = 34.65s versus t½ = 6.93s, respectively, as calculated from Koff measurements provided in Table 1). It should be noted that interaction half-lives measured by SPR are likely longer than the interaction half-lives that would be expected under the elevated fluid shear forces found in the vasculature. These data suggest that although the highest affinity target of BBK32 is Fn, BBK32-GAG interactions are more stable once formed. This is an important point bearing on the mechanistically distinct roles of Fn and GAG binding in transient and dragging interactions, to be discussed below.
BBK32 Fn-binding sequences mediate the initial tethering step of microvascular adhesion whereas GAG-binding sequences are critical for subsequent dragging interactions
We have shown previously that the initiation steps of microvascular interactions in vivo are regulated by both Fn and GAGs (Norman et al., 2008), and that BBK32 independently binds both Fn and GAGs under static conditions in vitro (Fischer et al., 2006). Therefore, it was critical to characterize the contributions of BBK32 Fn and GAG binding to B. burgdorferi vascular adhesion.
We have shown recently that deletion of BBK32 amino acids 45–68 reduces BBK32-dependent binding of B. burgdorferi to immobilized dermatan sulfate GAGs in vitro by ~60%, without disrupting Fn interactions, and that deletion of residues 158–182 reduces Fn binding by ~85% without disrupting GAG interactions (Fig. 4A) (Lin et al., 2012). These BBK32 variants are stably expressed and localized to the surface of the spirochete when expressed in the non-infectious B. burgdorferi strain B314 (Lin et al., 2012). T o investigate the relative contributions of BBK32 GAG- and Fn-binding sequences to microvascular interactions in vivo, we expressed full-length BBK32 (FL BBK32) and deletion mutants Δ45–68 and Δ158–182 in the GFP-expressing B31A-derived non-infectious B. burgdorferi strain GCB706.
Figure 4. Schematic of BBK32 deletion mutants and models of BBK32 vascular adhesion.

A) Deletions disrupting GAG- and Fn-binding in BBK32 (Lin et al., 2012). B) Models of the BBK32 vascular adhesion mechanism (not to scale). Model 1: Adhesion to glycosylated endothelial receptors mediated by direct BBK32-GAG binding. Model 2: Adhesion to glycosylated endothelial receptors mediated indirectly by Fn-bridged BBK32-GAG binding.
BBK32 interacts independently with both Fn and GAGs (Fig. 4A, Fig. S3, Table 1) (Fischer et al., 2006), providing two potential pathways for microvascular interactions (Fig. 4B). In the direct GAG binding pathway BBK32 would interact directly with glycosylated receptors through its GAG-binding sequences. In the indirect pathway BBK32 would bind to glycosylated receptors by interacting directly with Fn via its Fn binding sequences; Fn would interact with glycosylated receptors through heparin-binding sequences. The end result is a ternary complex with Fn bridging BBK32 and glycosylated endothelial receptors. The differences in BBK32 equilibrium and kinetic parameters for binding to Fn versus GAGs (Table 1) suggested the possibility that BBK32-Fn and BBK32-GAG interactions might function at mechanically distinct steps of vascular adhesion under fluid shear force. We therefore conducted experiments to determine the relative contributions of BBK32 GAG- and Fn binding to different steps of vascular adhesion in vivo (Fig. 5).
Figure 5. Specific contributions of BBK32 Fn- and GAG-binding sequences to different steps of B. burgdorferi vascular interactions in vivo.

Strains used in these experiments: 1) GCB1585:non-infectious expressing full-length BBK32 in trans (BBK32 FL), 2) GCB1580: non-infectious expressing BBK32 Δ45–68 in trans (BBK32 Δ45–68), 3) GCB1583: non-infectious expressing BBK32 Δ158–182 in trans (BBK32 Δ158–182), and 4) GCB706: non infectious parent, B31-A (non-infectious). A-B) Microvascular interaction rates (A: tethering, and B: dragging interaction) for B. burgdorferi strains expressing BBK32 mutants described in Fig. 4A, as analyzed by high acquisition rate spinning disk confocal intravital microscopy performed in flank skin,normalized to flaB copy number in blood collected from each animal following intravital microscopy. Normalization was performed by dividing the tethering and dragging interaction rate values measured in each vessel by the flaB copy number measured in the blood from the same mouse where intravital microscopy was performed. For unadjusted interaction and copy number data, see Fig. S4. C) Ratios of dragging to tethering interaction rates, calculated in each vessel where interacting B. burgdorferi were observed. Note: the dragging to tethering ratio could not be calculated in vessels where no interactions were detected, or where no tethering interactions were observed. Therefore, for Panel C, dragging to tethering ratios were calculated from the data for a subset of venules shown in A and B. Ratios were calculated using crude (non-normalized) interaction rate data (see Fig. S4 for unadjusted data). For A-B, interactions in 106 venules from 24 mice (n=6 BBK32 FL; n=6 BBK32 Δ45–68; n=4 BBK32 Δ158–182; n=4 non-infectious) were analyzed. Microvascular interactions were measured between 5 and 25 minutes after spirochete injection. The average time after injection for recordings made with BBK32 FL, BBK32 Δ45–68, BBK32 Δ158–182 and non-infectious strains was 11.96−/+0.95, 12.74−/+0.84, 12.25−/+1.01, and 13.5−/+0.77 min, respectively. Means and standard error bars are indicated for each experimental group. Statistical testing for significant differences among all experimental groups was performed using a non-parametric Kruskal-Wallis ANOVA (P values of 0.0033, 0.0021 and 0.0103 for A, B and C, respectively). P-values are indicated for selected pair-wise comparisons. All statistical data may be found in Table S4.
Disruption of Fn (Δ158–182) or GAG binding sequences (Δ45–68) in BBK32 resulted in similar levels of expression and surface localization as the wild-type protein in gain of function strains, suggesting no major defect in folding of these mutant proteins (Lin et al., 2012). The structural integrity of the mutant proteins is also supported by the ability of each protein to bind either Fn or GAGs at wild-type levels. Nonetheless, disruption of BBK32 sequences altered the levels of blood-borne gain-of-function B. burgdorferi strains in mice (Fig. S4A). Therefore, following analysis of microvascular interaction rates recorded by intravital microscopy (Fig. S4A, B), we normalized the interaction rates measured in each vessel by dividing by the flaB copy number measured in the blood of the same mouse where interactions were recorded (Fig. 5A, B). The resulting data suggested that BBK32 GAG- and Fn-binding sequences mediate two different steps of vascular adhesion (Fig. 5A, B).
First, deletion of BBK32 Fn-binding sequences 158–182, but not GAG-binding sequences 45–68, reduced the tethering interaction stage of B. burgdorferi vascular interactions by 58% (Fig. 5A) The comparison between the two deletion mutants gave a P value of 0.0529, very close to significance at the 95% confidence level. Second, while deletion of BBK32 Fn-binding sequences (Δ158–182) did not reduce dragging interaction rates substantially further compared to tethering interaction rates (compare Fig. 5A and 5B), deletion of GAG-binding sequences 45–68 reduced dragging, but not tethering interactions by 66% compared to full-length BBK32. These observations suggested that BBK32 Fn-binding plays a critical role in the tethering step of vascular adhesion, whereas BBK32 GAG-binding is important for dragging.
We also analyzed the data by a more direct approach that did not require any data normalization based upon flaB copy number in the blood. We calculated the ratio of dragging to tethering interactions in every blood vessel where vascular associations could be observed (Fig. 5C). These calculations provided measurements of interaction properties that were independent of differences in copy number among BBK32 variants. Using this approach, we found that disruption of BBK32 GAG-binding sequences (45–68) reduced the mean dragging to tethering ratio 3-fold, to levels found in non-infectious B. burgdorferi (Fig. 5C), supporting our conclusion that BBK32 GAG-binding mediates dragging. In contrast, disruption of BBK32 Fn-binding sequences (158–183) did not reduce the ratio of dragging to tethering spirochetes, confirming that deficiency in BBK32 Fn-binding specifically impairs tethering. The difference in the dragging to tethering ratio of the GAG-binding (45–68) versus the Fn-binding (158–183) mutant was 5-fold with a P value of 0.0048. Collectively, these observations indicate that the initial tethering step of B. burgdorferi vascular adhesion is mediating by BBK32 Fn binding, and that subsequent dragging interactions are mediated by BBK32 association with GAGs.
DISCUSSION
The initiating steps enabling vascular adhesion and transmigration of bacterial pathogens under fluid shear forces are poorly understood, despite their importance for our understanding of blood-borne pathogen dissemination mechanisms. The predominant reason for this has been the lack of methodologies to study pathogen-vascular interactions in real time in the natural setting of a living host. This obstacle has been recently overcome through the use of fluorescent spirochetes and intravital imaging (Lee et al., 2010, Moriarty et al., 2008, Norman et al., 2008). Using this approach we previously identified the first bona fide bacterial vascular adhesin, the B. burgdorferi lipoprotein BBK32 (Norman et al., 2008). Our current work (summarized in Fig. 6) demonstrates that the first two steps of B. burgdorferi vascular adhesion, tethering and dragging, are mediated respectively by Fn- and GAG-binding sites in BBK32, which exhibit distinct affinities and kinetics of ligand binding. The high affinity of BBK32-Fn interactions may be critical for overcoming fluid shear forces in the bloodstream during the first, tethering step of vascular interactions. Our data also suggest that B. burgdorferi encodes additional, unidentified vascular adhesin(s) that may mediate adhesion by a pathway independent of Fn-bridged GAG binding (Fig. 2).
Figure 6. Proposed BBK32 vascular adhesion mechanism.

Schematic summarizing the proposed two-step model of BBK32-mediated initiation of vascular adhesion. Insets depict the interaction pathways responsible for tethering (left) and dragging (right). Depicted in each inset are BBK32 (top, yellow and blue: BBK32 GAG- and Fn-binding sequences, respectively), plasma Fn (middle, green) and glycosylated endothelial receptors (bottom, red). The schematics depicting each member of the interaction pathway are not drawn to scale. Text boxes beside the schematics for tethering and dragging list known and predicted properties of each stage of the interaction mechanism, which are derived from the studies reported here and previously (Norman et al., 2008).
BBK32 is the predominant Fn-binding vascular adhesin but B. burgdorferi expresses other adhesin(s) that promote microvascular interactions
Our data indicate that although BBK32 remains the only B. burgdorferi vascular adhesin identified to date, it is not the only molecule capable of mediating vascular adhesion. Disruption of BBK32 expression in an infectious strain reduced interactions by ~20 and 50% in skin and joints, respectively (Fig. 2). Interestingly, differences in the BBK32 dependence of vascular interactions in joints and skin mirror previously described differences in the BBK32 dependence of dissemination kinetics in these tissues (Hyde et al., 2011). Hyde et al. reported that bbk32 disruption has a stronger and earlier effect on dissemination to joints during infection than is observed in skin. Specifically, at days 7–10 of infection with 103 Borrelia, dissemination to joints by bbk32-disrupted bacteria is reduced by an average of ~70%, while dissemination to skin is reduced by an average of ~35%. Therefore, both vascular interaction and dissemination appear to be approximately two-fold more dependent on BBK32 in joints compared to skin. The smaller overall effect of bbk32 disruption on vascular interactions (this study) compared to dissemination (Hyde et al.) may be because our intravital microscopy method is not as sensitive as low inoculum studies of infection kinetics in detecting functional differences between bbk32-disrupted and WT strains; alternatively, BBK32 may play roles in addition to vascular adhesion which are important for infectivity in skin and joints.
The observation that bbk32 disruption does not abolish vascular interactions in either skin or joints indicates that additional, as yet unidentified B. burgdorferi molecules can mediate vascular interactions in vivo. One candidate vascular adhesin is CRASP-1 (cspA/bba68), which has recently been shown to bind Fn in vitro (Hallstrom et al., 2010); it is unknown whether CRASP-1, like RevA, RevB and BB0347, binds Fn with reduced affinity, or if this protein's affinity for Fn approaches the nanomolar-range affinity of BBK32 for Fn (Table 1). However, our data indicate that Fn or the glycosylated receptors targeted by the Fn HepII peptide may not play a role in vascular adhesion in the absence of BBK32 (Fig. 2). Fn-independent vascular adhesin(s) could target alternative macromolecules on the surface of endothelial cells or recruit other soluble bridging factors in the bloodstream, thereby providing a variety of interaction targets to more efficiently mediate vascular interaction and escape. Analysis of additional putative B. burgdorferi adhesins (see (Antonara et al., 2011, Norris et al., 2010)) using our intravital imaging system will help to address this issue.
A two-step BBK32 vascular adhesion mechanism
Our data indicate that initiation of B. burgdorferi vascular adhesion occurs in two distinct steps, tethering and dragging. Both steps target glycosylated receptors, but are mediated by distinct BBK32 ligand-binding sites and mechanisms. The details of the two-part vascular adhesion mechanism are schematically summarized in Figure 6.
BBK32-mediated tethering
The first step of vascular interaction, tethering, is a transient interaction characterized by rapid rounds of association and dissociation; it can occur anywhere along the length of the spirochete but typically does not involve the entire length of the bacterium (Moriarty et al., 2008). Evidence from this study and our previous work indicate that BBK32-regulated tethering is mediated by Fn-bridged interactions with glycosylated receptors. Tethering is inhibited by antibodies to plasma Fn, and by a GAG-binding peptide derived from the Fn HepII domain, but not by canonical integrin-interacting Fn RGD sequences (Fig. 2C) (Norman et al., 2008). Tethering is also inhibited by competition with a high molecular weight GAG with an average chain length longer than the minimal length required for high affinity HepII-GAG interactions. It is not inhibited by competitive inhibition with a low molecular weight GAG with an average chain length shorter than the minimum required for high affinity HepII-GAG interactions (Norman et al., 2008). These data suggest that tethering is mediated by glycosylated receptors targeted by the Fn HepII domain (Norman et al., 2008, Sharma et al., 1999, Ingham et al., 1990). Finally, competitive inhibition with the Fn HepII peptide impairs interaction rates for BBK32-expressing spirochetes, but not bbk32-null bacteria, indicating that this peptide and BBK32 act within the same interaction pathway (Fig. 2).
We found that specific disruption of BBK32 Fn-binding sequences disrupts the tethering step of vascular adhesion (Fig. 5) and that the affinity of BBK32 for Fn is an order of magnitude greater than BBK32 affinity for GAGs (Table 1). These observations imply that the high affinity of BBK32 for Fn may be essential for the ability of this protein to act as a vascular adhesin. High affinity Fn-binding-mediated tethering may be required to withstand the fluid shear forces encountered during endothelial tethering in the bloodstream and for deceleration of circulating spirochetes. Interestingly, BBK32-Fn complexes dissociate five times faster than BBK32-GAG complexes. The rapid kinetics of BBK32-Fn association and dissociation may provide an explanation for why tethering spirochetes undergo rapid rounds of association and dissociation with vessel walls, whereas dragging spirochetes linked to the endothelium through BBK32-GAG interactions do not (see below). It is noteworthy that the affinities of BBK32 for Fn (KD~2×10−8) and of Fn for heparin (KD~1×10−8) (Bentley et al., 1985) are similar to the binding affinities of other ligand-receptor pairs known to mediate vascular adhesion of leukocytes and platelets in vivo (Norman et al., 2008, Lyon et al., 2000, Klopocki et al., 2008); hence, these interactions are strong enough to withstand the fluid shear forces in postcapillary venules and capillaries, the major sites of B. burgdorferi vascular adhesion and extravasation (Moriarty et al., 2008). Intriguingly, the BBK32 sequences deleted in the tethering-deficient variant partially overlap with sequences previously demonstrated to induce formation in vitro of a highly adhesive form of Fn termed superfibronectin (Prabhakaran et al., 2009). We do not yet know if superfibronectin formation contributes to vascular adhesion in vivo. We conclude that BBK32-dependent tethering interactions with glycosylated receptors are mediated by a transient, high affinity, Fn-bridged binding mechanism.
BBK32-mediated dragging
The second step of B. burgdorferi vascular interactions is dragging, a slower, more stable form of association in which most of the length of the bacterium contacts the vascular surface (Moriarty et al., 2008). An important factor in dragging is BBK32 GAG-binding, since disruption of GAG binding sequences specifically impairs the dragging, but not the tethering step of vascular adhesion (Fig. 5). BBK32 likely mediates dragging by direct interactions with glycosylated sites on endothelial receptors, since dragging does not require Fn-binding sequences (Fig. 5). Dragging is inhibited by competition with both high and low molecular weight GAGs (Norman et al., 2008), suggesting that the adhesin-GAG interactions underlying dragging may be less specific than those required for tethering, and/or that tethering and dragging are mediated by interactions with distinct endothelial receptors. BBK32 confers Fn-independent binding to a broad range of mammalian cell types and GAGs (Fischer et al., 2006), supporting the hypothesis that dragging interactions may be less selective than tethering interactions. Interestingly, although BBK32 binds GAGs with substantially reduced affinity compared to BBK32-Fn binding, the half-life of BBK32-GAG interactions is five times longer than the half-life for BBK32-Fn binding (Table 1). This observation indicates that once formed, BBK32-GAG interactions are significantly more stable than BBK32-Fn interactions. The longer half-life and more relaxed specificity of BBK32-GAG binding may provide a mechanistic basis for the stability of dragging interactions.
Based on data presented here and previously, we believe that dragging generally follows and is dependent on tethering interactions. First, visual observation of numerous B. burgdorferi vascular interactions indicates that tethering precedes dragging chronologically (Moriarty et al., 2008, Norman et al., 2008). Second, a tethering-defective BBK32 mutant exhibits similar levels of reduction in both tethering and dragging (Fig. 5A,B), and competitive inhibition with the Fn HepII peptide reduces both tethering and dragging to the same extent (Norman et al., 2008). Finally, tethering appears to require an adhesin-ligand binding affinity an order of magnitude greater than the affinity of BBK32 for GAGs (Fig. S3, Table 1). Based upon these points we propose that establishing BBK32/GAG-mediated dragging interactions is energetically and or kinetically unfavourable in the absence of tethering interactions that would reduce both kinetic and energetic barriers to BBK32-GAG interactions under shear force.
Implications for vascular adhesion mechanisms of other bacterial pathogens
To date, BBK32 is the only bacterial adhesin shown to play a direct role in vascular adhesion under shear force in vivo. The studies reported here provide substantial insight into the BBK32-dependent vascular adhesion mechanism and identify a key role for Fn recruitment at the first step of vascular adhesion. BBK32 orthologues from staphylococcal and streptococcal species, and a number of adhesins from other invasive bacterial pathogens, bind Fn with an affinity similar to BBK32 (Schwarz-Linek et al., 2004, Heilmann et al., 2005, Tamura et al., 2006, Meenan et al., 2007, Fink et al., 2002, Christner et al., 2010, Henderson et al., 2011, Raibaud et al., 2005, Norris et al., 2011). Our findings raise the possibility that these bacterial Fn-binding proteins, like BBK32, confer vascular adhesion during blood-borne dissemination.
EXPERIMENTAL PROCEDURES
Ethics statement
This study was carried out in accordance with the principles outlined in the most recent policies and Guide to the Care and Use of Experimental Animals by The Canadian Council on Animal Care. Our animal protocol was approved by The Animal Care Committee of the University of Calgary.
Cloning of constructs for PflaB-driven expression of Fn adhesins in B. burgdorferi (Table S2)
All PflaB-adhesin expression cassettes were built by overlap extension PCR as previously described (Moriarty & Chaconas, 2009), ligated into cloning vector pJET1.2 (Fermentas, Burlington ON), as per manufacturers' instructions, and subsequently excised and ligated into the pCE320 shuttle vector (Eggers et al., 2002) via XhoI/NotI restriction digest. Cloning intermediates and final ligation products were used to transform Escherichia coli strain DH5α, followed by growth in Luria-Bertani broth or agar supplemented with kanamycin (50 μg/ml). The resulting constructs were purified from 100 ml cultures using a Qiagen midiprep kit (Qiagen, Toronto, ON), and sequenced before transformation into Borrelia. Sequencing was performed by the University of Calgary DNA Services, using primers B1703 and B1704 (derived from the pCE320 vector). All primers used for PCR amplification and sequencing are provided in Table S3.
The coding sequences for WT BBK32 and internal deletion mutants (see below) were fused to PflaB by overlap extension PCR using primers B1662, B1663, B1723, and B1725. Primers used to generate the PflaB-revA expression construct were B1723, B1736, B1737 and B1747. Primers used to generate the PflaB-revA+flag expression construct were B1723, B1736, B1737, B1738, B1739 and B1724. Primers used to generate the PflaB-bb0347 expression construct were B1723, B1728, B1729 and B1745. Primers used to generate the PflaB-bb0347+flag expression construct were B1723, B1728, B1729, B1730, B1731 and B1724. Primers used to generate the PflaB-revB expression construct were B1723, B1732, B1733 and B1799. Primers used to generate the PflaB-revB+flag expression construct were B1723, B1733, B1732, B1735, B1734 and B1724. The DNA templates used for PCR were as follows: PflaB (template pTM61), revA and bb0347 coding sequences (template B31 5A4 NP1 genomic DNA), revB coding sequences (template B31 5A4 genomic DNA), flag coding sequences (template pJL148SPA (Zeghouf et al., 2004).
PCR reaction conditions were as follows: 50 μl PCR reaction mixtures containing 3% DMSO, 1 mM dNTPs, 1 unit Phusion DNA polymerase (NEB, Pickering, ON), 1× HF buffer (NEB, Pickering, ON), 5 pmoles of each primer and 100 ng DNA template. PCR cycling conditions were: 98°C/3 min followed by 6–25 cycles of 98°C/15 sec, 55–60°C/30 sec, 68°C/1 min, followed by a 7 min final extension at 68°C. PflaB and adhesin coding sequences were amplified separately in the first round of PCR (25 cycles). In PCR round two, 2.5 μl of each round one product were mixed (5 μl final volume), and underwent an additional 6 PCR cycles to generate full-length product. In PCR round three, full-length expression cassettes were amplified in 25 PCR cycles using primers derived from the 5' end of PflaB and the 3' end of each adhesin's coding sequence, and 2.5 μl of PCR products from round two as a template.
Cloning of BBK32 internal deletion mutants (Table S2)
The cloning of these mutants is described elsewhere (Lin et al., 2012), and is summarized briefly here. pBBK32QC44 (Δ45–68): generated by inverse PCR, removing residues 45–68; residue 45 was mutated to make a new restriction site (Thr to Ala). pBBK32QC48 (Δ158–182): generated by inverse PCR, removing residues 159–181; residues 158 and 182 were mutated to make new restriction sites; residue 158 mutated from Asp to Ala; residue 182 mutated from Ile to Ser.
Generation and cultivation of B. burgdorferi strains
B. burgdorferi strains used in this study are described in Table S1. All strains were grown in BSK-II medium prepared in house (Barbour, 1984). Electrocompetent B. burgdorferi strains were prepared as described (Moriarty et al., 2008, Samuels, 1995). Liquid plating transformations were performed with 20–50 μg plasmid (pTM61, pTM242, pTM244, pTM246, pTM248, pTM255, pTM257 or pJW201) in the presence of antibiotic selection (100 μg/ml gentamycin, 200 μg/ml kanamycin, 50 μg/ml streptomycin, as required), as described previously (Bankhead & Chaconas, 2007, Yang et al., 2004). Genomic DNA from antibiotic-resistant B. burgdorferi clones identified after 10–12 days of selection was isolated using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI). DNA preparations were PCR-screened for the presence of pCE320 vector containing appropriately sized inserts as previously described (Tourand et al., 2006), using pCE320-derived primers B1703 and B1704, and for the presence of the kanamycin resistance gene, using primers B70 and B71.
Adhesin expression and localization in gain of function strains
Expression and probing of surface localization of RevA, RevB and BB0347, all carrying a C-terminal FLAG tag, was performed by Western blotting of B. burgdorferi strains (Table S1) following incubation in the presence or absence of proteinase K or pronase (Fig. S1). Protease treatment of spirochetes was performed as previously described (Probert & Johnson, 1998, Fischer et al., 2006, Norman et al., 2008). B. burgdorferi lysed in loading dye was analyzed on 12% SDS-polyacrylamide gels with a 5% stack. Following transfer to nitrocellulose membranes (Amersham Hydrobond-ECL), the blots were developed using mouse primary monoclonal antibody (Sigma F1804 - 200UG, Monoclonal anti-FLAG M2, affinity isolated antibody) at a 1:1000 dilution. The secondary antibody was peroxidase-conjugated, affinity purified donkey anti-Mouse IgG (Jackson ImmunoResearch Laboratories Code 715-035-151) diluted 1:5,000. The colorimetric substrate TMB (Sigma T0565) was used according to the manufacturer's instructions.
Preparation of Borrelia for direct bloodstream injection and intravital surgical preparations
Cultivation and preparation of Borrelia for intravenous inoculation was performed as described in detail previously (Moriarty et al., 2008). Washed GFP-expressing host-adapted B. burgdorferi strains grown in 1% mouse blood were injected intravenously via tail or jugular veins (2.5–5 × 108 bacteria per mouse), together with vessel counterstain (50 μl of 0.02% 70 kDa Texas Red dextran or 50 μg of AlexaFluor 555-conjugated CD31/PECAM-1 antibody) (Invitrogen, Burlington, ON; BD Biosciences, Mississauga, ON). For all experiments except those described in Fig. 2, intravital microscopy was performed in flank skin following inoculation with bacteria, as described in detail previously (Moriarty et al., 2008). For experiments shown in Fig. 2, intravital microscopy experiments were performed in surgically-prepared joints, as previously described (Andruski et al., 2008). Briefly, the anteriomedial aspect of the right knee of mice anesthetized with 10 mg/kg xylazine (MTC Pharmaceuticals, Cambridge, Ontario) and 200 mg/kg ketamine hydrochloride (Rogar/STB, Montreal, Quebec), was surgically exposed by removal of skin and connective tissue. The exposed knee joint of dorsally recumbent mice was perfused with warm buffer (135 mmol/l NaCl, 20 mmol/l NaHCO3, 5 mmol/l KCl, 1 mmol/l MgSO4·7H2O, pH 7.4), and the medial aspect was covered with a glass coverslip secured to the stage with vacuum grease. Intravital microscopy was performed in 5–6 week old C57BL/6 male mice from Charles River Laboratories (Montreal, QC). This study was carried out in accordance with the principles outlined in the most recent Guide to the Care and Use of Experimental Animals by the Canadian Council on Animal Care. Our animal protocol (M08131) was approved by The Animal Care Committee of the University of Calgary.
Intravital microscopy conditions and analysis
Vascular interactions of intravenously injected B. burgdorferi were recorded in 4–6 straight, unbranched, postcapillary venules (diameter 20–50 μm) of the knee joint capsule or skin from 5–25 minutes following spirochete injection, and spirochete interactions were quantified offline, as described in detail previously (Norman et al., 2008, Moriarty et al., 2008). Where vascular interaction rates were normalized to copy number, the interaction rates measured in each vessel were divided by the flaB copy number value measured in the blood of the same mouse where intravital microscopy was performed. Intravital microscopy was performed using a 20× 0.95 NA water immersion objective and previously described custom-built spinning disk microscope system (Quorum Technologies Inc., Guelph, ON) (Moriarty et al., 2008). Image acquisition at 14–15 fps was achieved by 2× binning, at maximum sensitivity range (255), using auto-contrast, LMM5 transmission of 100 and LMM5 laser power of 1.84 mV. Exposure time for capturing the fluorescent signal of GFP-expressing Borrelia was 50 ms. Exposure time for capturing signal from vessel counterstains Texas Red dextran or AlexaFluor 555-conjugated PECAM-1 antibody was 30 ms.
Quantitative PCR (qPCR) measurement of B. burgdorferi flaB copy number in blood and tissues of mice following intravital microscopy
One hour following intravital microscopy, 0.5–1 ml of heparinized blood was obtained by cardiac puncture, and flaB copy number in blood samples obtained from euthanized animals was measured by qPCR, as described previously (Lee et al., 2010). DNA was extracted from 100 μl blood samples using a Qiagen DNeasy kit (Qiagen, Toronto, ON) according to manufacturer's instruction. qPCR was performed in triplicate for each DNA sample and for a logarithmic dilution series of flaB-containing DNA standard plasmid pTM222, using 400 nM each of primers to flaB (B1672 and B1673). qPCR was performed on 2 μl of sample DNA in a 25 μl reaction volume in a Bio-Rad CFX96 real-time PCR detection system. Reactions were performed in 1× iQ SsoFast EvaGreen Supermix (Bio-Rad Laboratories, Mississauga, Ontario), prepared according to manufacturer's instructions. PCR conditions: Step 1: 98°C 2 min; Step 2: 40 cycles of 98°C 5 sec, 59.2°C 5 sec; Step 3: 65°C 5 sec; Step 4: melt curve analysis over melting range 65°C to 95°C. The number of copies of flaB measured in each experimental sample was normalized to the number of μg of total extracted DNA in each sample, rather than the volume of blood, to avoid errors resulting from variations in DNA extraction efficiency from small volumes. Since most of the DNA in any sample is host-derived and not spirochetal, normalization of copy number by the number of μg of DNA analyzed by qPCR provides copy number values which take into account differences in sample processing efficiency.
Fn peptide intravital microscopy experiments
Borrelia strains were prepared and injected as described above, together with 100 μg of FN-C/H II peptide (Sigma Canada, Oakville, ON; catalogue number F7049), as previously described (Norman et al., 2008). Intravital microscopy was performed in knee joints, as described above.
Expression and purification of recombinant proteins
The constructs for the expression of His6-tagged RevA, RevB, or BB0347 were generated using the vector pQE30 (Qiagen, Alencia, CA). The genes were amplified by PCR using the primers shown in Table S3, based upon the published genome sequence (Casjens et al., 2000, Fraser et al., 1997). Primers were engineered to introduce a BamHI site at the start of each gene fragment and a stop codon followed by a SalI site at the end of each fragment. PCR products were sequentially digested with BamHI and SalI and then ligated into pQE30 cut with BamHI and SalI. E. coli strains DH5α, M15 and their transformants were grown in Luria-Bertani broth or agar supplemented with ampicillin (100 μg/ml) when necessary. The soluble forms of all of recombinant proteins were purified from E. coli as recently described (Benoit et al., 2011).
Surface Plasmon Resonance (SPR)
The interaction of Fn or dermatan sulfate (DS) with RevA, RevB, BB0347 or BBK32, was analyzed using surface plasmon resonance and a Biacore 3000 instrument (GE Healthcare, Piscataway, NJ). Briefly, 1 μg of Fn was conjugated on a CM5 chip (GE Healthcare, Piscataway, NJ). A control flow cell was injected with phosphate-buffered saline without Fn. Then, 10 μL of serial dilutions of RevA or BB0347 (0, 0.0625, 0.125, 0.25, 0.5, 1 μM), RevB (0, 1.875, 3.75, 7.5, 15, 30μM), or 5μL of BBK32 (0, 6.25, 12.5, 25, 50, 100 nM) were injected into the control cell and flow cell coated with immobilized Fn, at a flow rate and temperature of 10μL/min and 25°C. To measure the affinity of BBK32 binding to DS, DS was biotinylated using EZ-Link Sulfo-NHS-Biotinylation Kits (Pierce, Rockford, IL). Then, 1 μg of biotinylated DS was conjugated on an SA chip (GE Healthcare, Piscataway, NJ). A control flow cell was injected with phosphate-buffered saline without DS. Then, 10 μL of serial dilutions of BBK32 (0, 0.0625, 0.125, 0.25, 0.5, 1μM, Fig. S3, Table 1) was injected into the control cell and flow cell coated with immobilized DS, at 10 μL/min and 25°C. Negative control flow cell data were subtracted from the sensogram data. To obtain the kinetic parameters of the interaction, the sensogram data were fitted by BIAevaluation software version 3.0 using the one step biomolecular association reaction model (1:1 Langmuir model), which resulted in optimal mathematical fits with the lowest Chi values.
Statistics
For quantitative analysis of intravital microscopy data, mean and standard error values for different variables were calculated and plotted graphically for all vessels from all mice using GraphPad Prism 4.03 (GraphPad Software, Inc., San Diego, CA). Statistical significance was calculated in GraphPad Prism using a non-parametric 1 way ANOVA Kruskal-Wallis test. Where numbers of data points were not sufficiently large for valid pairwise comparison of values using a non-parametric Dunn's multiple comparison post-test, pair-wise comparisons were performed using a two-tailed Student t-test, with correction for unequal variance. Confidence intervals were 95%. All statistical data are provided in Table S4.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Genevieve Chaconas for technical support, Lawrence Stern for kindly allowing us to use his surface plasmon resonance apparatus, Christian Eggers and Justin Radolf for providing vector pCE320, the Greenblatt laboratory for providing pJL148SPA, and Jenifer Coburn and Moriarty lab members for helpful comments on the manuscript. Intravital microscopy experiments were performed in the Live Cell Imaging Facility of the Calvin, Phoebe and Joan Snyder Institute of Infection, Immunity and Inflammation, and we are especially grateful for the support of Jenn Amon and the Facility's Scientific Director, Pina Colarusso. GC acknowledges salary support as the Canada Research Chair in the Molecular Biology of Lyme Borreliosis and in the form of a Scientist Award from the Alberta Heritage Foundation for Medical Research. TJM was partially supported by a postdoctoral fellowship from the Canadian Institutes of Health Research. This research was supported by the Canadian Institutes of Health Research (GC, MOP-53086), the U.S. National Institutes of Health (GC and JL, AI1093104; JTS, AI058086), the Canada Research Chairs Program (GC), Alberta Heritage Fund for Medical Research (GC), the Banting Research Foundation (TJM), Bertha Rosenstadt Endowment Fund (TJM) and Natural Sciences and Engineering Research Council of Canada (TJM).
REFERENCES
- Andruski B, McCafferty DM, Ignacy T, Millen B, McDougall JJ. Leukocyte trafficking and pain behavioral responses to a hydrogen sulfide donor in acute monoarthritis. American journal of physiology. 2008;295:R814–820. doi: 10.1152/ajpregu.90524.2008. [DOI] [PubMed] [Google Scholar]
- Antonara S, Ristow L, Coburn J. Adhesion mechanisms of Borrelia burgdorferi. Advances in experimental medicine and biology. 2011;715:35–49. doi: 10.1007/978-94-007-0940-9_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankhead T, Chaconas G. The role of VlsE antigenic variation in the Lyme disease spirochete: persistence through a mechanism that differs from other pathogens. Molecular microbiology. 2007;65:1547–1558. doi: 10.1111/j.1365-2958.2007.05895.x. [DOI] [PubMed] [Google Scholar]
- Barbour AG. Isolation and cultivation of Lyme disease spirochetes. Yale J Biol Med. 1984;57:521–525. [PMC free article] [PubMed] [Google Scholar]
- Benoit VM, Fischer JR, Lin YP, Parveen N, Leong JM. Allelic variation of the Lyme disease spirochete adhesin DbpA influences spirochetal binding to decorin, dermatan sulfate, and mammalian cells. Infection and immunity. 2011;79:3501–3509. doi: 10.1128/IAI.00163-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley KL, Klebe RJ, Hurst RE, Horowitz PM. Heparin binding is necessary, but not sufficient, for fibronectin aggregation. A fluorescence polarization study. The Journal of biological chemistry. 1985;260:7250–7256. [PubMed] [Google Scholar]
- Brissette CA, Bykowski T, Cooley AE, Bowman A, Stevenson B. Borrelia burgdorferi RevA antigen binds host fibronectin. Infection and immunity. 2009;77:2802–2812. doi: 10.1128/IAI.00227-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JA, El-Hage N, Miller JC, Babb K, Stevenson B. Borrelia burgdorferi RevA antigen is a surface-exposed outer membrane protein whose expression is regulated in response to environmental temperature and pH. Infection and immunity. 2001;69:5286–5293. doi: 10.1128/IAI.69.9.5286-5293.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casjens S, Palmer N, van Vugt R, Huang WH, Stevenson B, Rosa P, Lathigra R, Sutton G, Peterson J, Dodson RJ, Haft D, Hickey E, Gwinn M, White O, Fraser CM. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs in an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Molecular microbiology. 2000;35:490–516. doi: 10.1046/j.1365-2958.2000.01698.x. [DOI] [PubMed] [Google Scholar]
- Christner M, Franke GC, Schommer NN, Wendt U, Wegert K, Pehle P, Kroll G, Schulze C, Buck F, Mack D, Aepfelbacher M, Rohde H. The giant extracellular matrix-binding protein of Staphylococcus epidermidis mediates biofilm accumulation and attachment to fibronectin. Molecular microbiology. 2010;75:187–207. doi: 10.1111/j.1365-2958.2009.06981.x. [DOI] [PubMed] [Google Scholar]
- Drake SL, Varnum J, Mayo KH, Letourneau PC, Furcht LT, McCarthy JB. Structural features of fibronectin synthetic peptide FN-C/H II, responsible for cell adhesion, neurite extension, and heparan sulfate binding. The Journal of biological chemistry. 1993;268:15859–15867. [PubMed] [Google Scholar]
- Dunham-Ems SM, Caimano MJ, Eggers CH, Radolf JD. Borrelia burgdorferi requires the alternative sigma factor RpoS for dissemination within the vector during tick-to-mammal transmission. PLoS pathogens. 2012;8:e1002532. doi: 10.1371/journal.ppat.1002532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham-Ems SM, Caimano MJ, Pal U, Wolgemuth CW, Eggers CH, Balic A, Radolf JD. Live imaging reveals a biphasic mode of dissemination of Borrelia burgdorferi within ticks. J Clin Invest. 2009 doi: 10.1172/JCI39401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers CH, Caimano MJ, Clawson ML, Miller WG, Samuels DS, Radolf JD. Identification of loci critical for replication and compatibility of a Borrelia burgdorferi cp32-based shuttle vector for the expression of fluorescent reporters in the Lyme disease spirochete. Molecular microbiology. 2002;43:281–295. doi: 10.1046/j.1365-2958.2002.02758.x. [DOI] [PubMed] [Google Scholar]
- Fink DL, Green BA, St Geme JW., 3rd The Haemophilus influenzae Hap autotransporter binds to fibronectin, laminin, and collagen IV. Infection and immunity. 2002;70:4902–4907. doi: 10.1128/IAI.70.9.4902-4907.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer JR, Leblanc KT, Leong JM. Fibronectin binding protein BBK32 of the Lyme disease spirochete promotes bacterial attachment to glycosaminoglycans. Infection and immunity. 2006;74:435–441. doi: 10.1128/IAI.74.1.435-441.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, White O, Ketchum KA, Dodson R, Hickey EK, Gwinn M, Dougherty B, Tomb JF, Fleischmann RD, Richardson D, Peterson J, Kerlavage AR, Quackenbush J, Salzberg S, Hanson M, van Vugt R, Palmer N, Adams MD, Gocayne J, Weidman J, Utterback T, Watthey L, McDonald L, Artiach P, Bowman C, Garland S, Fujii C, Cotton MD, Horst K, Roberts K, Hatch B, Smith HO, Venter JC. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997;390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- Hallstrom T, Haupt K, Kraiczy P, Hortschansky P, Wallich R, Skerka C, Zipfel PF. Complement regulator-acquiring surface protein 1 of Borrelia burgdorferi binds to human bone morphogenic protein 2, several extracellular matrix proteins, and plasminogen. The Journal of infectious diseases. 2010;202:490–498. doi: 10.1086/653825. [DOI] [PubMed] [Google Scholar]
- Harman MW, Dunham-Ems SM, Caimano MJ, Belperron AA, Bockenstedt LK, Fu HC, Radolf JD, Wolgemuth CW. The heterogeneous motility of the Lyme disease spirochete in gelatin mimics dissemination through tissue. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:3059–3064. doi: 10.1073/pnas.1114362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilmann C, Hartleib J, Hussain MS, Peters G. The multifunctional Staphylococcus aureus autolysin aaa mediates adherence to immobilized fibrinogen and fibronectin. Infection and immunity. 2005;73:4793–4802. doi: 10.1128/IAI.73.8.4793-4802.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson B, Nair S, Pallas J, Williams MA. Fibronectin: a multidomain host adhesin targeted by bacterial fibronectin-binding proteins. FEMS microbiology reviews. 2011;35:147–200. doi: 10.1111/j.1574-6976.2010.00243.x. [DOI] [PubMed] [Google Scholar]
- Hyde JA, Weening EH, Chang M, Trzeciakowski JP, Hook M, Cirillo JD, Skare JT. Bioluminescent imaging of Borrelia burgdorferi in vivo demonstrates that the fibronectin-binding protein BBK32 is required for optimal infectivity. Molecular microbiology. 2011;82:99–113. doi: 10.1111/j.1365-2958.2011.07801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham KC, Brew SA, Atha DH. Interaction of heparin with fibronectin and isolated fibronectin domains. The Biochemical journal. 1990;272:605–611. doi: 10.1042/bj2720605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Singvall J, Schwarz-Linek U, Johnson BJ, Potts JR, Hook M. BBK32, a fibronectin binding MSCRAMM from Borrelia burgdorferi, contains a disordered region that undergoes a conformational change on ligand binding. The Journal of biological chemistry. 2004;279:41706–41714. doi: 10.1074/jbc.M401691200. [DOI] [PubMed] [Google Scholar]
- Klopocki AG, Yago T, Mehta P, Yang J, Wu T, Leppanen A, Bovin NV, Cummings RD, Zhu C, McEver RP. Replacing a lectin-domain residue in L-selectin enhances binding to P-selectin glycoprotein ligand-1 but not to 6-sulfo-sialyl Lewis x. The Journal of biological chemistry. 2008;283:11493–11500. doi: 10.1074/jbc.M709785200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labandeira-Rey M, Skare JT. Decreased infectivity in Borrelia burgdorferi strain B31 is associated with loss of linear plasmid 25 or 28-1. Infection and immunity. 2001;69:446–455. doi: 10.1128/IAI.69.1.446-455.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WY, Moriarty TJ, Wong CH, Zhou H, Strieter RM, van Rooijen N, Chaconas G, Kubes P. An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nature immunology. 2010;11:295–302. doi: 10.1038/ni.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- Li X, Liu X, Beck DS, Kantor FS, Fikrig E. Borrelia burgdorferi lacking BBK32, a fibronectin-binding protein, retains full pathogenicity. Infection and immunity. 2006;74:3305–3313. doi: 10.1128/IAI.02035-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y-P, Chen Q, Dufour NP, Fischer JR, Leong JM. Borrelia burgdorferi BBK32is a bifunctional adhesin with glycosaminoglycan- and fibronectin-binding activities that each confer binding to distinct mammalian cell types. Manuscript in preparation. 2012 [Google Scholar]
- Lyon M, Rushton G, Askari JA, Humphries MJ, Gallagher JT. Elucidation of the structural features of heparan sulfate important for interaction with the Hep-2 domain of fibronectin. The Journal of biological chemistry. 2000;275:4599–4606. doi: 10.1074/jbc.275.7.4599. [DOI] [PubMed] [Google Scholar]
- Meenan NA, Visai L, Valtulina V, Schwarz-Linek U, Norris NC, Gurusiddappa S, Hook M, Speziale P, Potts JR. The tandem beta-zipper model defines high affinity fibronectin-binding repeats within Staphylococcus aureus FnBPA. The Journal of biological chemistry. 2007;282:25893–25902. doi: 10.1074/jbc.M703063200. [DOI] [PubMed] [Google Scholar]
- Moriarty TJ, Chaconas G. Identification of the determinant conferring permissive substrate usage in the telomere resolvase, ResT. The Journal of biological chemistry. 2009;284:23293–23301. doi: 10.1074/jbc.M109.023549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty TJ, Norman MU, Colarusso P, Bankhead T, Kubes P, Chaconas G. Real-time high resolution 3D imaging of the lyme disease spirochete adhering to and escaping from the vasculature of a living host. PLoS pathogens. 2008;4:e1000090. doi: 10.1371/journal.ppat.1000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman MU, Moriarty TJ, Dresser AR, Millen B, Kubes P, Chaconas G. Molecular mechanisms involved in vascular interactions of the Lyme disease pathogen in a living host. PLoS pathogens. 2008;4:e1000169. doi: 10.1371/journal.ppat.1000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris NC, Bingham RJ, Harris G, Speakman A, Jones RP, Leech A, Turkenburg JP, Potts JR. Structural and functional analysis of the tandem beta-zipper interaction of a Streptococcal protein with human fibronectin. The Journal of biological chemistry. 2011;286:38311–38320. doi: 10.1074/jbc.M111.276592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris SJ, Coburn J, Leong JM, Hu LT, Hook M. Pathobiology of Lyme disease Borrelia. In: Samuels DS, Radolf JD, editors. Borrelia: Molecular Biology, Host Interaction and Pathogenesis. Horizon Scientific Press; Norwich, UK: 2010. pp. 293–325. [Google Scholar]
- Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115:3861–3863. doi: 10.1242/jcs.00059. [DOI] [PubMed] [Google Scholar]
- Parveen N, Caimano M, Radolf JD, Leong JM. Adaptation of the Lyme disease spirochaete to the mammalian host environment results in enhanced glycosaminoglycan and host cell binding. Molecular microbiology. 2003;47:1433–1444. doi: 10.1046/j.1365-2958.2003.03388.x. [DOI] [PubMed] [Google Scholar]
- Parveen N, Cornell KA, Bono JL, Chamberland C, Rosa P, Leong JM. Bgp, a secreted glycosaminoglycan-binding protein of Borrelia burgdorferi strain N40, displays nucleosidase activity and is not essential for infection of immunodeficient mice. Infection and immunity. 2006;74:3016–3020. doi: 10.1128/IAI.74.5.3016-3020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parveen N, Leong JM. Identification of a candidate glycosaminoglycan-binding adhesin of the Lyme disease spirochete Borrelia burgdorferi. Molecular microbiology. 2000;35:1220–1234. doi: 10.1046/j.1365-2958.2000.01792.x. [DOI] [PubMed] [Google Scholar]
- Prabhakaran S, Liang X, Skare JT, Potts JR, Hook M. A novel fibronectin binding motif in MSCRAMMs targets F3 modules. PLoS ONE. 2009;4:e5412. doi: 10.1371/journal.pone.0005412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probert WS, Johnson BJ. Identification of a 47 kDa fibronectin-binding protein expressed by Borrelia burgdorferi isolate B31. Molecular microbiology. 1998;30:1003–1015. doi: 10.1046/j.1365-2958.1998.01127.x. [DOI] [PubMed] [Google Scholar]
- Probert WS, Kim JH, Hook M, Johnson BJ. Mapping the ligand-binding region of Borrelia burgdorferi fibronectin-binding protein BBK32. Infection and immunity. 2001;69:4129–4133. doi: 10.1128/IAI.69.6.4129-4133.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raibaud S, Schwarz-Linek U, Kim JH, Jenkins HT, Baines ER, Gurusiddappa S, Hook M, Potts JR. Borrelia burgdorferi binds fibronectin through a tandem beta-zipper, a common mechanism of fibronectin binding in Staphylococci, Streptococci, and spirochetes. The Journal of biological chemistry. 2005;280:18803–18809. doi: 10.1074/jbc.M501731200. [DOI] [PubMed] [Google Scholar]
- Samuels DS. Electrotransformation of the spirochete Borrelia burgdorferi. Methods in molecular biology (Clifton, N.J. 1995;47:253–259. doi: 10.1385/0-89603-310-4:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid G. Epidemiology and clinical similarities of human spirochetal diseases. Rev Infect Dis. 1989;11:S1460–1469. doi: 10.1093/clinids/11.supplement_6.s1460. [DOI] [PubMed] [Google Scholar]
- Schmit VL, Patton TG, Gilmore RD., Jr. Analysis of Borrelia burgdorferi surface proteins as determinants in establishing host cell interactions. Frontiers in microbiology. 2011;2:141. doi: 10.3389/fmicb.2011.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz-Linek U, Hook M, Potts JR. The molecular basis of fibronectin-mediated bacterial adherence to host cells. Molecular microbiology. 2004;52:631–641. doi: 10.1111/j.1365-2958.2004.04027.x. [DOI] [PubMed] [Google Scholar]
- Seshu J, Esteve-Gassent MD, Labandeira-Rey M, Kim JH, Trzeciakowski JP, Hook M, Skare JT. Inactivation of the fibronectin-binding adhesin gene bbk32 significantly attenuates the infectivity potential of Borrelia burgdorferi. Molecular microbiology. 2006;59:1591–1601. doi: 10.1111/j.1365-2958.2005.05042.x. [DOI] [PubMed] [Google Scholar]
- Sharma A, Askari JA, Humphries MJ, Jones EY, Stuart DI. Crystal structure of a heparin- and integrin-binding segment of human fibronectin. EMBO J. 1999;18:1468–1479. doi: 10.1093/emboj/18.6.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanek G, Wormser GP, Gray J, Strle F. Lyme borreliosis. Lancet. 2012;379:461–473. doi: 10.1016/S0140-6736(11)60103-7. [DOI] [PubMed] [Google Scholar]
- Steere AC, Coburn J, Glickstein L. The emergence of Lyme disease. J Clin Invest. 2004;113:1093–1101. doi: 10.1172/JCI21681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura GS, Hull JR, Oberg MD, Castner DG. High-affinity interaction between fibronectin and the group B streptococcal C5a peptidase is unaffected by a naturally occurring four-amino-acid deletion that eliminates peptidase activity. Infection and immunity. 2006;74:5739–5746. doi: 10.1128/IAI.00241-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas WE. Mechanochemistry of receptor-ligand bonds. Current opinion in structural biology. 2009;19:50–55. doi: 10.1016/j.sbi.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Tourand Y, Bankhead T, Wilson SL, Putteet-Driver AD, Barbour AG, Byram R, Rosa PA, Chaconas G. Differential telomere processing by Borrelia telomere resolvases in vitro but not in vivo. Journal of bacteriology. 2006;188:7378–7386. doi: 10.1128/JB.00760-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nature immunology. 2005;6:902–910. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]
- Wilske B. Epidemiology and diagnosis of Lyme borreliosis. Annals of medicine. 2005;37:568–579. doi: 10.1080/07853890500431934. [DOI] [PubMed] [Google Scholar]
- Wu J, Weening EH, Faske JB, Hook M, Skare JT. Invasion of eukaryotic cells by Borrelia burgdorferi requires β1 integrins and Src kinase activity. Infection and immunity. 2011;79:1338–1348. doi: 10.1128/IAI.01188-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XF, Pal U, Alani SM, Fikrig E, Norgard MV. Essential role for OspA/B in the life cycle of the Lyme disease spirochete. J Exp Med. 2004;199:641–648. doi: 10.1084/jem.20031960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeghouf M, Li J, Butland G, Borkowska A, Canadien V, Richards D, Beattie B, Emili A, Greenblatt JF. Sequential Peptide Affinity (SPA) system for the identification of mammalian and bacterial protein complexes. J Proteome Res. 2004;3:463–468. doi: 10.1021/pr034084x. [DOI] [PubMed] [Google Scholar]
- Zipfel PF, Wurzner R, Skerka C. Complement evasion of pathogens: common strategies are shared by diverse organisms. Molecular immunology. 2007;44:3850–3857. doi: 10.1016/j.molimm.2007.06.149. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.