

Abstract
Diseases of the mitochondria generally affect cells with high-energy demand, and appear to most profoundly affect excitatory cells that have localized high energy requirements, such as neurons and cardiac and skeletal muscle cells. Complex I of the mammalian mitochondrial respiratory chain is a very large, 45 subunits enzyme, and functional deficiency of complex I is the most frequently observed cause of oxidative phosphorylation (OXPHOS) disorders. Impairment of complex I results in decreased cellular energy production and is responsible for a variety of human encephalopathies, myopathies and cardiomyopathies. Complex I deficiency may be caused by mutations in any of the seven mitochondrial or 38 nuclear genes that encode complex I subunits or by mutations in various other nuclear genes that affect complex I assembly or function. Mouse models that faithfully mimic human complex I disorders are needed to better understand the role of complex I in health and disease and for evaluation of potential therapies for mitochondrial diseases. In this review we discuss existing mouse models of mitochondrial complex I dysfunction, focusing on those with similarities to human mitochondrial disorders. We also discuss some of the noteworthy murine genetic models in which complex I genes are not disrupted, but complex I dysfunction is observed, along with some of the more popular chemical compounds that inhibit complex I function and are useful for modeling complex I deficiency in mice.
Keywords: Mitochondria, Mouse model, Oxidative phosphorylation, Complex I, Gene targeting
1. Introduction
Eukaryotic cells typically contain hundreds of maternally inherited mitochondria that function via intact mitochondrial and nuclear genomes to fuel metabolism (Shoffner, 1996). Mitochondria provide energy for eukaryotic cells via oxidative phosphorylation (OXPHOS), producing the majority of cellular ATP in non-proliferating cells. OXPHOS is accomplished by 5 enzymes of the mitochondrial respiratory chain and involves coupling of electron transfer through a chain of oxidoreductase reactions to the pumping of hydrogen ions across the inner mitochondrial membrane to create an electrochemical gradient that is used to produce cellular energy in the form of ATP. This is achieved via four properly assembled and folded protein complexes of the electron transport chain (ETC), designated complexes I–IV. The fifth respiratory chain protein, complex V, uses this proton gradient to convert ADP to ATP. Human mtDNA is a 16.6kb closed circular DNA containing 37 genes: 22 tRNA genes and 2 rRNA genes that function in the translation of 13 mitochondrial-encoded polypeptides. These 13 mitochondrial polypeptide genes along with 75 nuclear genes, synthesized in the cytosol and imported into mitochondria, encode the 88 known structural subunits of the five respiratory chain complexes (van den Heuvel and Smeitink, 2001; Ugalde et al., 2004). Thus, mutations in mitochondrial proteins encoded by nDNA or mtDNA can lead to impairment of mitochondrial functions. Mitochondria contain multiple mitochondrial genomes, divide independently of the cell cycle, and still segregate normally during mitosis (Schon and Manfredi, 2003). In contrast to nDNA mutations, mtDNA mutations are maternally inherited, often heteroplasmic (co-existence of both normal and mutant mtDNA genomes within a given cell or tissue) and the disease phenotype is observed only when a threshold level of heteroplasmy is exceeded. Mutations of mitochondrial proteins occur in humans with an estimated incidence as high as 1:5,000 live births (Wallace, 1992; Bourgeron et al., 1995; Haas et al., 2007). Mitochondrial defects are associated with metabolic/bioenergetics related diseases ranging from childhood disorders such as progressive encephalomyopathy, Leber's hereditary optic neuropathy (LHON), and Leigh syndrome to geriatric diseases such as Alzheimer's and Parkinson's diseases. Tissues with high metabolic rates (e.g., brain, heart, muscle, liver and kidney) typically display the most severe dysfunction (De Vivo and Dimauro, 1990). Excitatory cells which require localized high concentrations of ATP for membrane depolarization (e.g., neurons, skeletal and cardiac muscle cells), are particularly susceptible to impaired mitochondrial energy production (Schon, 2000).
At ~980kDa, complex I (CI, NADH:ubiquinone oxidoreductase) is by far the largest of the ETC complexes, containing 45 subunits. Seven of these subunits are encoded by mtDNA and 38 are nDNA-encoded (Loeffen et al., 2000; Sharma et al., 2009). Electrons from oxidation of NADH are passed from CI to coenzyme Q10 (coQ, or ubiquinone), and shuttled to complex III (CIII, ubiquinol-cytochrome c oxidoreductase). This electron transport is accompanied by CI extrusion of protons from the matrix into the intermembrane space. Electrons from oxidation of FADH2 by complex II (CII, succinate-ubiquinone oxidoreductase) are also transferred by coQ to complex III. CIII catalyzes the reduction of cytochrome c through oxidation of coQ while pumping protons to the intermembrane space. Cytochrome c delivers electrons to the terminal electron acceptor/ proton pump, complex IV (CIV, cytochrome c oxidase). Finally, complex V (CV, ATP synthase) utilizes this electrochemical gradient, translocating protons back to the matrix, to capture energy via phosphorylation of ADP.
Mouse models that faithfully recapitulate human mitochondrial diseases are needed for a variety of studies aimed at dissecting disease progression and testing of potential therapies. To date, there are few mouse models of human mitochondrial dysfunction; however, some of these are targeted mutations of nDNA that affect CI function (Torraco et al., 2009; Koene et al., 2011; Vempati et al., 2008). There is currently no proven technology for targeted mutation of mtDNA (Dunn et al., 2011). This short review will discuss mouse models of CI dysfunction, encompassing both genetic models and chemical inhibition models, including some of the notable mouse models in which genes other than those of CI subunit genes were modified, but CI deficiency was observed.
2. Complex I structure
CI is an L-shaped structure (Walker, 1995), located in the inner mitochondrial membrane and protruding into the matrix, and is composed of 3 functional domains or modules: 1) the N module, involved in binding and oxidation of NADH; 2) the Q module, responsible for electron transfer to coQ; and 3) the P module, involved in proton translocation (Mimaki et al., 2011). A hypothetical structure for CI protein subcomplexes was proposed (Antonicka et al., 2003), and more recent work with E. coli (Efremov et al., 2010), yeast (Hunte et al., 2010) and mammalian CI (Lazarou et al., 2009; Vogel et al., 2007) helped to resolve the formation of assembly subcomplexes which combine in a specific order to give rise to the ultimate structure of CI (reviewed by Mimaki et al., 2011). Proper assembly of CI is a complex multi-step process and requires at least nine nDNA-encoded assembly factors/chaperones that are not present in the final assembled complex (reviewed by McKenzie and Ryan, 2010; Nouws et al., 2011; Mimaki et al., 2011). CI contains three detergent-separable fractions: the flavin mononucleotide protein fraction (FP), the iron-sulfur (Fe-S) protein fraction (IP) and the hydrophobic protein fraction (HP) (Belogrudov & Hatefi, 1994). The catalytic domain is located within the IP fraction and mutations in this region are often associated with mitochondrial myopathies and encephalopathies (see reviews by Wallace, 1992; Loeffen et al., 2000; van den Heuvel and Smeitink, 2001; Ugalde et al., 2004). Large, hydrophobic protein subunits of CI, including seven mitochondrial-encoded proteins, are located in the HP which resides in the inner mitochondrial membrane and contains two Fe-S clusters (Belogrudov and Hatefi, 1994). The nuclear-encoded IP proteins are referred to as NADH dehydrogenase:ubiquinone Fe-S proteins (NDUFS1–8); the nuclear-encoded FP proteins are referred to as NADH dehydrogenase:ubiquinone flavoproteins (NDUFV1–3). All of the human nuclear-encoded CI subunits have the prefix “NDU” (NADH dehydrogenase:ubiquinone); the seven mtDNA-encoded CI subunits have the prefix “ND” (NADH dehydrogenase).
3. Complex I and disease
The most frequently observed OXPHOS disorders are associated with CI dysfunction (Kirby et al., 1999). This is logical in that CI is a very large, highly conserved, multi-subunit protein that must undergo a complex, multi-step assembly process. Furthermore, although CI plays a crucial role as an entry point of electrons into the ETC, CII is also an entry point for electrons (i.e., CI and CII act in parallel to a degree). The remaining complexes, CIII-CV, are functionally arranged in series; therefore, deleterious mutations in CIII-CV might reasonably be expected to have a higher probability of embryonic lethality. In addition, since seven of the 13 peptides encoded by mtDNA are CI subunits, CI is statistically much more likely than the other respiratory chain complexes to be affected by mtDNA mutations, including the 4977bp “common deletion” and mutations in tRNA or rRNA genes that globally affect mitochondrial protein synthesis (Cortopassi and Wang, 1995). It has been hypothesized that many disease causing mtDNA mutations may be considered only mild to moderately deleterious, and that truly severe mtDNA mutations are filtered out of the female germline during oogenesis (Fan et al., 2008). Additional support for this derives from the observation that the more severe mtDNA disease-associated mutations such as NARP (Neuropathy, ataxia, and retinitis pigmentosa) remain heteroplasmic, whereas milder mtDNA mutations (such as LHON) can exist in the homoplasmic state without imminent fatality (Fan et al., 2008). It is also possible that severe homoplasmic mtDNA mutations that escape this female germline filtering mechanism may be embryonic lethal.
Interestingly, CI is a major contributor to the formation of mitochondrial reactive oxygen species (ROS), an important source of cellular stress (Kushnareva et al., 2002), that was shown to be elevated in animal models that affect or inhibit CI function (e.g., rotenone treatment; Li et al., 2003).
CI deficiency was shown to play a central role in Parkinson's disease (PD) and in aging-related neurodegeneration (Wallace, 1992). Predisposition to PD and Alzheimer's disease (AD) is linked to certain mtDNA haplogroups (reviewed by Coskun et al., 2011), and in PD a CI deficiency in brain and other tissues such as skeletal muscle and platelets is well established (Bindoff et al., 1991; reviewed by Mounsey and Teismann, 2010). However, in AD the most consistent finding has been an inhibition of CIV (Canevari et al., 1999; Crouch et al., 2005).
An important class of CI deficiencies results from defects in assembly, through mutations in CI subunits critical for proper assembly or mutations in associated assembly factors that are not present in the holoenzyme (Lazarou et al., 2009; McKenzie and Ryan, 2010).There are a number of known mutations in nuclear-encoded CI subunit genes that are associated with mitochondrial dysfunction in humans, including assembly factors (Koene et al., 2011; Mimaki et al., 2011). The nuclear-encoded NDUFS4 gene is a hotspot for mutations which are associated with Leigh disease or Leigh-like symptoms, and is the only nuclear-encoded CI subunit thus far that has been successfully targeted for creation of knock-out and knock-in mutant mice (Kruse et al., 2008; Ingraham et al., 2009). The NDUFS4 gene is conserved across two distinct domains in a variety of species, a structural domain and a serine phosphorylation consensus site domain (Papa et al., 1996), and resides in the N module of CI, involved in binding and oxidizing NADH (Lazarou et al., 2007). The structural domain may also play a role in CI assembly, as mutations in the NDUFS4 gene affect CI assembly and function (Scacco et al., 2003). The second NDUFS4 conserved domain encodes a serine phosphorylation site that is responsive to protein kinase A (PKA) and cAMP-mediated phosphorylation. Specific phosphorylation of an 18kDa protein (NDUFS4) was found to occur in response to cAMP-dependent kinase phosphorylation and was also shown to activate CI (Pearson and Kemp, 1991; Papa et al., 1999; Scacco et al., 2000). PKA localization and subsequent cAMP-dependent phosphorylation were shown to occur in mitochondria (Sardanelli et al., 1996) in conjunction with phosphorylation of the conserved serine in NDUFS4 (Sardanelli et al., 1996; Scott and McCartney, 1994). Therefore, NDUFS4 likely regulates some aspect of electron transport from NADH + H+ to coQ via cAMP phosphorylation.
4. Mouse models of complex I dysfunction
4.1 Mitochondrial DNA-encoded models
No stable genetically engineered mouse lineage currently exists that harbors a specific mutation in any of the seven mtDNA-encoded CI subunit genes. The so-called “mutator mouse”, which expresses a mutant catalytic subunit of Pol gamma (Pol γ), the only known DNA polymerase present in mitochondria, accumulates point mutations and deletions in mtDNA with age (Trifunovic et al., 2004). Considering that seven of the 13 mtDNA-encoded peptides are CI subunits, one would expect to see disruption of OXPHOS in these mice, and that premise is borne out (Trifunovic et al., 2005). In addition, disruptions in any of the two mitochondrial rRNA or 22 tRNA genes involved in mitochondrial protein translation would be expected to impact CI more so than the other respiratory chain complexes in mutator mice. Although, CI dysfunction is observed in mutator mice, the presence of myriad mutations randomly spread throughout the mtDNA genome also leads to dysfunction of all of the respiratory chain complexes. (Fan et al., 2008). As such, they were able to generate transmitochondrial mice harboring an insertion in the ND6 gene of subunit of CI that completely abolished its function. However, the proportion of the heteroplasmic ND6 mutation declined with each generation and was undetectable after four generations. This supported the contention that an active mechanism removes severe mutations during oogenesis (Fan et al., 2008).
Developing mouse models of mtDNA mutations is hampered by the inability to manipulate the mtDNA genome in a directed fashion in organello, relying instead on screening of naturally occurring or induced random mutations followed by efforts to propagate and transfer mutation-bearing mitochondria into embryonic stem cells or single-cell zygotes for mouse production (Dunn et al., 2011). Transmitochondrial mice produced in this way also include “xenomitochondrial” mice harboring homoplasmic mtDNA of a foreign species of mouse (e.g., M. terricolor) on the nuclear background of the common laboratory strain, M.m. domesticus (McKenzie et al., 2004).
4.2 Mouse models of nuclear-encoded complex I subunits
Currently, the only CI subunit gene that has been successfully targeted in mice is the nuclear-encoded NDUFS4 gene. In humans, NDUFS4 mutations are associated Leigh Disease, an extremely debilitating encephalomeylopathy usually diagnosed in infants, that progresses rapidly and almost always is fatal within 2–3 years after diagnosis.
4.2.1 Ndufs4 knockout mouse
The first genetic knock-out model of CI deficiency was produced by the Palmiter laboratory (Kruse et al., 2008). The gene construct was a conditional allele of the NDUFS4 gene with exon 2 flanked by loxP sites, such that deletion of exon 2 created a frameshift with no mature NDUFS4 protein produced (Kruse et al., 2008). Deletion of exon 2 gave rise to a severe Leigh-like phenotype in all mice. Heterozygous NDUFS4 knock-out mice were phenotypically normal; however, the homozygous mice displayed symptoms that were very comparable to Leigh disease including encephalopathy, blindness, ataxia, elevated lactate levels in serum, and the mice died at about 7 weeks of age. Reduced levels of intact CI were observed, suggesting a role for NDUFS4 in CI assembly or stability (Kruse et al., 2008). A tissue-specific knock-out of NDUFS4 in neurons and glia (NesKO mice) resulted in a similar phenotype to whole body knock-out with growth retardation, ataxia, breathing abnormalities, and progressive neuronal deterioration and gliosis in specific brain areas, with death at 7 weeks (Quintana et al., 2010). Recently, it was shown that in some tissues the formation of respiratory supercomplexes stabilized by CIII still allowed formation of active CI in NDUFS4 knock-out mice, although CI function was only about 50% of normal (Calvaruso et al., 2012).
4.2.2 Ndufs4 truncated knock-in
An NDUFS4 gene construct lacking the terminal 10–15 amino acids of the complete protein was used to make a knock-in mutation in mice. Fetuses homozygous for the mutant gene (NDUFS4−/−) did in utero, but heterozygous (NDUFS4+/−) mice displayed symptoms of CI deficiency including lactic acidosis in brain and neuromotor decline. Oxygen consumption and CI activity were reduced in NDUFS4+/− mitochondria, although the mice appeared normal and showed none of the severe Leigh-like symptoms seen in the Palmiter NDUFS4 knock-out mice (Ingraham et al., 2009).
4.3 Complex I deficiency associated with other mouse models
CI deficiency is noted or is a primary feature of several genetic models in which CI subunits are not targeted. Perhaps the most useful of these models are the apoptosis inducing factor deficient and tissue-specific knock-out models, due to the relationship between AIF and CI function.
4.3.1 Harlequin mouse (AIF deficient)
Apoptosis inducing factor 1 (AIF), a 57 kDa mitochondrial flavoprotein located in the intermembrane space, is a caspase-independent cell death effector that, after release from mitochondria, translocates to the nucleus where it participates in chromatin condensation and DNA fragmentation. AIF-deficient cells exhibit reduced OXPHOS capacity and reduced content of respiratory complexes, particularly CI (Vahsen et al., 2004), suggesting a role for AIF in CI assembly or maintenance. Attempts to create knockout mouse models of AIF failed due to embryonic lethality. The Harlequin mouse, originally identified as a model for late-onset neurodegeneration, was shown to express 10–20% of the normal concentration of AIF due to a retroviral insertion into the AIF gene (Klein et al., 2002). The Harlequin mouse shows a marked reduction in OXPHOS and CI function in retina and brain with a phenotype resembling CI deficiency (Benit et al., 2008). It is noteworthy that no human patient with CI deficiency has ever been shown to harbor a mutation in the AIF gene.
4.3.2 Tissue-specific AIF deficient mice
Embryonic lethality of whole-body knockout of AIF expression prompted the generation of tissue-specific mouse models. Using a targeting construct that flanked exon 7 of the (x-linked) AIF gene with loxP sites, conditional mutants (AIFlox/lox) were created. By mating females from this line with male mice expressing cre-recombinase under control of tissue specific promoters, both muscle- and liver-specifc knock-out mice were produced. Creatine kinase promoter-driven cre expression (Mck-cre) was used to knock-out AIF in skeletal and cardiac muscle (Joza et al., 2005). These mice exhibited deficient CI expression and activity giving rise to skeletal muscle atrophy and severe dilated cardiomyopathy with eventual heart failure. Albumin promoter-driven cre expression (Alb-cre) was used for liver-specific AIF knockout. Although these mice appeared healthy, a liver-specific decrease in CI, CIV and CV was noted (Pospisilik et al., 2007). Interestingly, both muscle- and liver-specific AIF knock-out mice showed reduced adiposity, improved glucose tolerance and increased insulin sensitivity, contradictory to the hypothesis that OXPHOS defects might contribute to type 2 diabetes and obesity (Pospisilik et al., 2007).
4.3.3 Neurodegeneration models
CI deficiency is a hallmark of PD (Swerdlow et al., 1996; Schapira et al., 1998; Mounsey and Teismann, 2010). Using cytoplast transfer with mitochondria derived from PD patients, (Swerdlow et al., 1996) showed that the complex I defect was transferred to cybrids via mtDNA. The neurodegeneration seen in PD is caused by selective degeneration of dopaminergic neurons in the substantia nigra pars compacta (Forno, 1996; Dawson and Dawson, 2003) and, in addition to multiple nuclear gene interactions, exposure to environmental toxins (insecticides, herbicides and fungicides) that inhibit mitochondrial complex I activity has been implicated (Chung et al., 2003). A common experimental mouse model of PD that was in use for many years is the neurotoxin 6-hydroxydopamine (6-OHDA) (Langston et al., 1983), which, like PD, also causes neurodegeneration selectively in dopaminergic neurons.
There are a number of other mouse disease models in which a CI deficiency has been shown. A mouse model of human motor neuron disease, the “wobbler” mouse, shows a profound decrease in oxygen consumption in motor neurons of the cervical spinal cord, and inhibition of CI in these neurons, but not in motor neurons of the lumbar spinal cord (Santoro et al., 2004) which are not affected in this model. The wobbler mouse carries a missense mutation in Vps54, a protein involved in vesicular trafficking in eukaryotic cells (Schmitt-John et al., 2005). The CI deficiency was confined to a small set of neurons and was demonstrated using a histochemical method, but inhibition of other respiratory chain enzymes was not measured by this technique. Interestingly, the CI deficiency in the cervical motor neurons correlates with the onset of the wobbler phenotype (Jung et al., 2002). Mice heterozygous for the homeobox gene Engrailed-1 (En1) show reduced expression of CI subunits NDUFS1 and NDUFS3 in brain (Alvarez-Fischer et al., 2011). Testicular nuclear receptor 4 (TR4) modulates complex I activity via transcriptional regulation of NDUFAF1, a CI assembly factor. Mice lacking TR4 (TR4−/−) presented a mitochondrial myopathy that was rescued by restoring NDUFAF1 levels, with a concomitant increase in CI activity (Liu et al., 2011). Lindfors et al. (2011) showed that the anorectic mouse (anx/anx), which exhibits poor feeding behavior and dies of starvation early in life (Maltais et al., 1984), displays increased oxidative stress and impaired CI efficiency related to downregulation of the Ndufaf1 gene, possibly affecting CI assembly in the appetite-regulating peptidergic neurons in the arcuate nucleus of the hypothalamus, which undergoes neurodegeneration in these mice (Lindfors et al., 2011).
4.4 Other genetic approaches
A mouse model of LHON was made using adeno-associated virus (AAV)-mediated delivery of an allotopic expression construct for mutated version of human ND4, a mtDNA encoded CI subunit (Qi et al., 2007). The coding sequence of the ND4 gene in this construct was changed to nuclear codon usage for nuclear expression and contained an arginine-to-histidine substitution at amino acid residue 340, and an N-terminal mitochondrial targeting signal peptide for transport into mitochondria. Intraocular injection of the AAV-ND4/R340H construct yielded mice with a phenotype similar to LHON, whereas mice treated with the control vector (AAV-wild type ND4 construct) did not show the disease phenotype (Qi et al., 2007). In an earlier study, this group used intraocular injection of ribozymes targeted against the mRNA for NDUFA1 to induce an LHON-like phenotype and showed rescue from the disease phenotype after AAV-mediated delivery of a human SOD2 (superoxide dismutase 2) gene construct (Qi et al., 2004).
5. Chemical inhibition of complex I
Rotenone, a botanical compound found naturally in the roots and stems of certain plants, is commonly used as an insecticide/pesticide, and inhibits CI via specific binding to subunit ND1 (Singer and Ramsay, 1994). Rotenone has neurotoxic effects in humans and other mammals and has been linked to the development of Parkinson's disease (Spivey, 2011). Rotenone exposure in mice and rats is widely viewed as a model of a neurotoxin-induced neuropathology that closely mimics features of Parkinson's disease (Betarbet et al., 2000). The major effect of rotenone exposure is the inhibition of CI and generation of ROS which has been demonstrated in several different cell types (Barrientos and Moraes, 1999). However, as with other chemical inhibitors of CI, rotenone has various other detrimental effects on cellular processes that are unrelated to mitochondrial dysfunction. For example, in addition to CI inhibition, rotenone inhibits cellular proliferation by inhibiting microtubule assembly (Brinkley et al., 1974) and also inhibits proteasome formation (Choi et al., 2010). Another common inhibitor of CI, MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), has been used in animal studies to induce mitochondrial CI dysfunction in brain. Unlike rotenone, which is lipophilic and readily crosses biological membranes, MPTP shows specificity to dopaminergic neurons, utilizing dopamine transporters for gaining access to cells (Heikkila et al., 1985). Both rotenone and MPTP are strong CI inhibitors, but only MPTP has been linked to a form of human parkinsonism and, due to its specificity to dopaminergic neurons, has been widely accepted in modeling PD in rodents (Dauer and Przedborski, 2003). Although rotenone, MPTP and (more recently) paraquat have been investigated for modeling PD, paraquat and rotenone showed no specificity to the dopamine transporter and paraquat was a weak CI inhibitor compared to rotenone or MPTP (Richardson et al., 2005).
6. Conclusions
The earliest versions of mouse models of CI deficiency were produced by treatment with chemical inhibitors of CI, such as rotenone. These types of models are still in use and have certain advantages, for example, in studies of CI deficiency induced by environmental toxins. Genetic models in which genes other than CI subunit genes are mutated, but with CI deficiency as a central feature (such as the Harlequin mouse), represent another class of mouse models. Although a clear CI deficiency may be shown in these models, one cannot discount the possibility that other subtle effects on metabolic or signaling pathways may affect the phenotype in ways that are not easily discerned.
The most faithful mouse models of human mitochondrial disease caused by a CI deficiency would be expected to arise from direct targeting of CI subunit genes. Indeed, the conditional NDUFS4 knockout mouse of Palmiter (Kruse et al., 2008) is perhaps the best characterized with a phenotype very similar to an expected Leigh-like syndrome. The Pinkert NDUFS4 heterozygous mutant mouse (Ingraham et al., 2009) shows little resemblance to a Leigh-like phenotype and has a near normal life span. The differences noted between these two NDUFS4 lines may relate to the different strains of mice that were used in gene targeting efforts. Interestingly, although the Palmiter mouse showed a Leigh-like phenotype in homozygous knockouts, the Pinkert NDUFS4 homozygous mutant mice showed embryonic lethality. There are few mouse models of CI disorders caused by mtDNA mutations, due to the technological limitations of manipulating mtDNA in vivo. Although technologies for targeting of nuclear genes are well established and somewhat routine, techniques for manipulation of mtDNA in a directed fashion are lacking. Such technology would need to overcome a number of hurdles: mitochondria are too small for efficient microinjection of DNA targeting constructs; electroporation voltages that allow for passage of large DNA constructs (> 7 kb) into isolated mitochondria would likely destroy mitochondrial membrane integrity (Collombet et al., 1997); recombination of mammalian mtDNA is likely a rare event (Eyre-Walker & Awadalla, 2001); and most cells contain hundreds to thousands of mtDNA genomes, a significant proportion of which would need to be genetically modified to impart a disease phenotype (Pinkert & Trounce, 2002, 2007). Developing the techniques for mitochondrial gene targeting is a focus of a handful of laboratories; however, this technology is desperately needed to accurately recapitulate in mice – the mitochondrial diseases of humans that are caused by specific mtDNA mutations.
Figure 1. Schematic representation of oxidative phosphorylation and the mitochondrial respiratory chain enzyme complexes.
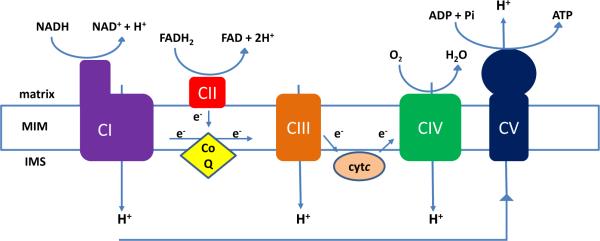
The respiratory chain enzymes, embedded in the mitochondrial inner membrane (MIM), couple the transport of electrons through the electron transport chain complexes (CI-CIV) to ATP production by CV. Protons extruded into the intermembrane space (IMS) by CI, CIII and CIV create an electrochemical gradient that is utilized by CV, capturing this energy by phosphorylation of ADP while translocating protons back to the mitochondrial matrix.
Other abbreviations: CoQ (coenzyme Q10, ubiquinone), cytC (cytochrome C).
Table 1.
Gene Mutations and Complex I Dysfunction.
| ND1 | LHON; ADPD-associated (?)* |
| ND4 | LHON |
| ND6 | LHON |
| Ndufs1 | LD; Leigh syndrome and leucoencephalopathy |
| Ndufs2 | LD; encephalomyopathy and hypertrophic cardiomyopathy |
| Ndufs3 | LD |
| Ndufs4 | LD |
| Ndufs6 | Mitochondrial encephalopathy |
| Ndufs7 | LD |
| Ndufs8 | LD |
| Ndufa1 | LD; Myoclonic epilepsy |
| Ndufa2 | LD |
| Ndufa11 | Encephalocardiomyopathy; Fatal infantile lactic acidosis |
| Ndufaf1 | Encephalocardiomyopathy |
| Ndufaf2 | Encephalomyopathy |
| Ndufaf3 | Lethal neonatal disease |
| Ndufaf4 | Fatal infantile lactic acidosis; Infantile mitochondrial encephalomyopathy |
| Ndufaf5 | Lethal neonatal disease |
| Ndufaf6 | LD |
ND1, 4 and 6 are mtDNA-encoded genes. LHON, Leber's hereditary Optic Neuropathy; LD, Leigh Disease; ADPD, Alzheimer's Disease/Parkinson's Disease.
polymorphism in haplogroup associated with risk factor for ADPD (Wallace, 2010).
Table 2.
Examples of mouse models of complex I dysfunction.
| mtDNA-encoded CI mutations | |
| Transmitochondrial CI mutant mice | (Fan et al., 2008) |
| Nuclear-encoded CI subunit gene targeting models | |
| NDUFS4 Knock out | (Kruse et al., 2008) |
| NDUFS4 knock-in | (Ingraham et al., 2009) |
| Other CI dysfunction genetic models | |
| Apoptosis Inducing Factor (AIF) models | |
| Harlequin mouse | (Klein et al., 2002) |
| Tissue-specific AIF knock outs | |
| Muscle-specific (MAIFKO) | (Joza et al., 2005) |
| Liver-specific (LAIFKO) | (Popisilik et al., 2007) |
| Neurodegeneration models | |
| Wobbler mouse | (Santori et al., 2004) |
| Parkinson's (MPTP) | (Langston et al., 1983) |
| Other genetic approaches | |
| Ribozymes against NDUFA1 mRNA | (Qi et al., 2004) |
| AAV allotopically expressed mutant ND4 | (Qi et al., 2007) |
| Chemical inhibition | |
| Rotenone | (Betarbet et al., 2000) |
| MPTP | (Langston et al., 1983) |
Acknowledgements
The authors express sincere appreciation to Ian A. Trounce, Kumiko Takeda and Kosta Steliou. Work in our laboratory was supported by NIH, NSF, the MitoCure Foundation, the Alabama Agricultural Experiment Station, and Auburn University.
Abbreviations
- AD
Alzheimer's disease
- AIF
apoptosis-inducing factor
- ATP
adenosine triphosphate
- CI
complex I, NADH:ubiquinone oxidoreductase
- coQ
coenzyme Q10, ubiquinone
- kDa
kilo Dalton
- LHON
Leber's hereditary optic neuropathy
- MIM
mitochondrial inner membrane
- mtDNA
mitochondrial DNA
- mTFA
mitochondrial transcription factor alpha
- NADH
nicotinamide adenine dinucleotide
- nDNA
nuclear DNA
- NDUFS4
NADH dehydrogenase:ubiquinone iron-sulfur protein4
- OXPHOS
oxidative phosphorylation
- PD
Parkinson's disease
- ROS
reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alvarez-Fischer D, Fuchs J, Castagner F, Stettler O, Massiani-Beaudoin O, Moya KL, Bouillot C, Oertel WH, Lombes A, Faigle W, Joshi RL, Hartmann A, Prochiantz A. Engrailed protects mouse midbrain dopaminergic neurons against mitochondrial complex I insults. Nat Neurosci. 2011;14:1260–1266. doi: 10.1038/nn.2916. [DOI] [PubMed] [Google Scholar]
- Antonicka H, Ogilvie I, Taivassalo T, Anitori RP, Haller RG, Vissing J, Kennaway NG, Shoubridge EA. Identification and characterization of a common set of complex I assembly intermediates in mitochondria from patients with complex I deficiency. J Biol Chem. 2003;278:43081–43088. doi: 10.1074/jbc.M304998200. [DOI] [PubMed] [Google Scholar]
- Barrientos A, Moraes CT. Titrating the effects of mitochondrial complex I impairment in the cell physiology. J Biol Chem. 1999;274:16188–16197. doi: 10.1074/jbc.274.23.16188. [DOI] [PubMed] [Google Scholar]
- Belogrudov G, Hatefi Y. Catalytic sector of complex I (NADH:ubiquinone oxidoreductase): subunit stoichiometry and substrate-induced conformation changes. Biochemistry. 1994;33:4571–4576. doi: 10.1021/bi00181a018. [DOI] [PubMed] [Google Scholar]
- Benit P, Goncalves S, Dassa EP, Briere JJ, Rustin P. The variability of the harlequin mouse phenotype resembles that of human mitochondrial-complex I-deficiency syndromes. PLoS One. 2008;3:e3208. doi: 10.1371/journal.pone.0003208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Bindoff LA, Birch-Machin MA, Cartlidge NEF, Parker WD, Jr., Turnbull DM. Respiratory chain abnormalities in skeletal muscle from patients with Parkinson's disease. J Neurol Sci. 1991;104:203–208. doi: 10.1016/0022-510x(91)90311-t. [DOI] [PubMed] [Google Scholar]
- Bourgeron T, Rustin P, Chretien D, Birch-Machin M, Bourgeois M, Viegas-Pequignot E, Munnich A, Rotig A. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet. 1995;11:144–149. doi: 10.1038/ng1095-144. [DOI] [PubMed] [Google Scholar]
- Brinkley BR, Barham SS, Barranco SC, Fuller GM. Rotenone inhibition of spindle microtubule assembly in mammalian cells. Exp Cell Res. 1974;85:41–46. doi: 10.1016/0014-4827(74)90210-9. [DOI] [PubMed] [Google Scholar]
- Calvaruso MA, Willems P, van den Brand M, Valsecchi F, Kruse S, Palmiter R, Smeitink J, Nijtmans L. Mitochondrial complex III stabilizes complex I in the absence of NDUFS4 to provide partial activity. Hum Mol Genet. 2012;21:115–120. doi: 10.1093/hmg/ddr446. [DOI] [PubMed] [Google Scholar]
- Canevari L, Clark JB, Bates TE. beta-amyloid fragment 25–35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett. 1999;457:131–134. doi: 10.1016/s0014-5793(99)01028-5. [DOI] [PubMed] [Google Scholar]
- Choi WS, Abel G, Klintworth H, Flavell RA, Xia Z. JNK3 mediates paraquatand rotenone-induced dopaminergic neuron death. J Neuropathol Exp Neurol. 2010;69:511–520. doi: 10.1097/NEN.0b013e3181db8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KK, Dawson VL, Dawson TM. New insights into Parkinson's disease. J Neurol. 2003;250(Suppl 3):III15–24. doi: 10.1007/s00415-003-1304-9. [DOI] [PubMed] [Google Scholar]
- Collombet J-M, Wheeler VC, Vogel F, Coutelle C. Introduction of plasmid DNA into isolated mitochondria by electroporation. A novel approach toward gene correction for mitochondrial disorders. J Biol Chem. 1997;272:5342–5347. doi: 10.1074/jbc.272.8.5342. [DOI] [PubMed] [Google Scholar]
- Cortopassi G, Wang E. Modelling the effects of age-related mtDNA mutation accumulation; complex I deficiency, superoxide and cell death. Biochim Biophys Acta. 1995;1271:171–176. doi: 10.1016/0925-4439(95)00025-y. [DOI] [PubMed] [Google Scholar]
- Coskun P, Wyrembak J, Schriner S, Chen HW, Marciniack C, Laferla F, Wallace DC. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophys Acta. 2012;1820:553–564. doi: 10.1016/j.bbagen.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch PJ, Blake R, Duce JA, Ciccotosto GD, L,i QX, Barnham KJ, Curtain CC, Cherny RA, Cappai R, Dyrks T, Masters CL, Trounce IA. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloidbeta1-42. J Neurosci. 2005;25:672–679. doi: 10.1523/JNEUROSCI.4276-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson's disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- De Vivo DC, Dimauro S. Mitochondrial defects of brain and muscle. Biol Neonate. 1990;58:54–69. doi: 10.1159/000243300. [DOI] [PubMed] [Google Scholar]
- Dunn DA, Cannon MV, Irwin MH, Pinkert CA. Animal models of human mitochondrial DNA mutations. Biochim Biophys Acta. 2011;1820:601–607. doi: 10.1016/j.bbagen.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremov RG, Baradaran R, Sazanov LA. The architecture of respiratory complex I. Nature. 2010;465:441–445. doi: 10.1038/nature09066. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A, Awadalla P. Does human mtDNA recombine? J Mol Evol. 2001;53:430–435. doi: 10.1007/s002390010232. [DOI] [PubMed] [Google Scholar]
- Fan W, Waymire KG, Narula N, Li P, Rocher C, Coskun PE, Vannan MA, Narula J, Macgregor GR, Wallace DC. A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science. 2008;319:958–962. doi: 10.1126/science.1147786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forno LS. Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol. 1996;55:259–272. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, Cohen BH. Mitochondrial disease: a practical approach for primary care physicians. Pediatrics. 2007;120:1326–1333. doi: 10.1542/peds.2007-0391. [DOI] [PubMed] [Google Scholar]
- Heikkila RE, Nicklas WJ, Vyas I, Duvoisin RC. Dopaminergic toxicity of rotenone and the 1-methyl-4-phenylpyridinium ion after their stereotaxic administration to rats: implication for the mechanism of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity. Neurosci Lett. 1985;62:389–394. doi: 10.1016/0304-3940(85)90580-4. [DOI] [PubMed] [Google Scholar]
- Hunte C, Zickermann V, Brandt U. Functional modules and structural basis of conformational coupling in mitochondrial complex I. Science. 2010;329:448–451. doi: 10.1126/science.1191046. [DOI] [PubMed] [Google Scholar]
- Ingraham CA, Burwell LS, Skalska J, Brookes PS, Howell RL, Sheu SS, Pinkert CA. NDUFS4: creation of a mouse model mimicking a Complex I disorder. Mitochondrion. 2009;9:204–210. doi: 10.1016/j.mito.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joza N, Oudit GY, Brown D, Benit P, Kassiri Z, Vahsen N, Benoit L, Patel MM, Nowikovsky K, Vassault A, Backx PH, Wada T, Kroemer G, Rustin P, Penninger JM. Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy. Mol Cell Biol. 2005;25:10261–10272. doi: 10.1128/MCB.25.23.10261-10272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C, Higgins CM, Xu Z. A quantitative histochemical assay for activities of mitochondrial electron transport chain complexes in mouse spinal cord sections. J Neurosci Methods. 2002;114:165–172. doi: 10.1016/s0165-0270(01)00524-6. [DOI] [PubMed] [Google Scholar]
- Kirby DM, Crawford M, Cleary MA, Dahl HH, Dennett X, Thorburn DR. Respiratory chain complex I deficiency: an underdiagnosed energy generation disorder. Neurology. 1999;52:1255–1264. doi: 10.1212/wnl.52.6.1255. [DOI] [PubMed] [Google Scholar]
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- Koene S, Willems PH, Roestenberg P, Koopman WJ, Smeitink JA. Mouse models for nuclear DNA-encoded mitochondrial complex I deficiency. J Inherit Metab Dis. 2011;34:293–307. doi: 10.1007/s10545-009-9005-x. [DOI] [PubMed] [Google Scholar]
- Kruse SE, Watt WC, Marcinek DJ, Kapur RP, Schenkman KA, Palmiter RD. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 2008;7:312–320. doi: 10.1016/j.cmet.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnareva Y, Murphy AN, Andreyev A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem J. 2002;368:545–553. doi: 10.1042/BJ20021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- Lazarou M, McKenzie M, Ohtake A, Thorburn DR, Ryan MT. Analysis of the assembly profiles for mitochondrial- and nuclear-DNA-encoded subunits into complex I. Mol Cell Biol. 2007;27:4228–4237. doi: 10.1128/MCB.00074-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Thorburn DR, Ryan MT, McKenzie M. Assembly of mitochondrial complex I and defects in disease. Biochim Biophys Acta. 2009;1793:78–88. doi: 10.1016/j.bbamcr.2008.04.015. [DOI] [PubMed] [Google Scholar]
- Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278:8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- Lindfors C, Nilsson IA, Garcia-Roves PM, Zuberi AR, Karimi M, Donahue LR, Roopenian DC, Mulder J, Uhlen M, Ekstrom TJ, Davisson MT, Hokfelt TG, Schalling M, Johansen JE. Hypothalamic mitochondrial dysfunction associated with anorexia in the anx/anx mouse. Proc Natl Acad Sci U S A. 2011;108:18108–18113. doi: 10.1073/pnas.1114863108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Lee YF, Chou S, Uno H, Li G, Brookes P, Massett MP, Wu Q, Chen LM, Chang C. Mice lacking TR4 nuclear receptor develop mitochondrial myopathy with deficiency in complex I. Mol Endocrinol. 2011;25:1301–1310. doi: 10.1210/me.2010-0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffen JL, Smeitink JA, Trijbels JM, Janssen AJ, Triepels RH, Sengers RC, van den Heuvel LP. Isolated complex I deficiency in children: clinical, biochemical and genetic aspects. Hum Mutat. 2000;15:123–134. doi: 10.1002/(SICI)1098-1004(200002)15:2<123::AID-HUMU1>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Maltais LJ, Lane PW, Beamer WG. Anorexia, a recessive mutation causing starvation in preweanling mice. J Hered. 1984;5:468–472. doi: 10.1093/oxfordjournals.jhered.a109987. [DOI] [PubMed] [Google Scholar]
- McKenzie M, Ryan MT. Assembly factors of human mitochondrial complex I and their defects in disease. IUBMB Life. 2010;62:497–502. doi: 10.1002/iub.335. [DOI] [PubMed] [Google Scholar]
- McKenzie M, Trounce IA, Cassar CA, Pinkert CA. Production of homoplasmic xenomitochondrial mice. Proc Natl Acad Sci U S A. 2004;101:1685–1690. doi: 10.1073/pnas.0303184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimaki M, Wang X, McKenzie M, Thorburn DR, Ryan MT. Understanding mitochondrial complex I assembly in health and disease. Biochim Biophys Acta. 2011 doi: 10.1016/j.bbabio.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Mounsey RB, Teismann P. Mitochondrial dysfunction in Parkinson's disease: pathogenesis and neuroprotection. Parkinsons Dis. 2010;2011:617472. doi: 10.4061/2011/617472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouws J, Nijtmans LG, Smeitink JA, Vogel RO. Assembly factors as a new class of disease genes for mitochondrial complex I deficiency: cause, pathology and treatment options. Brain. 2011;135:12–22. doi: 10.1093/brain/awr261. [DOI] [PubMed] [Google Scholar]
- Papa S, Sardanelli AM, Cocco T, Speranza F, Scacco SC, Technikova-Dobrova Z. The nuclear-encoded 18 kDa (IP) AQDQ subunit of bovine heart complex I is phosphorylated by the mitochondrial cAMP-dependent protein kinase. FEBS Lett. 1996;379:299–301. doi: 10.1016/0014-5793(95)01532-9. [DOI] [PubMed] [Google Scholar]
- Papa S, Sardanelli AM, Scacco S, Technikova-Dobrova Z. cAMP-dependent protein kinase and phosphoproteins in mammalian mitochondria. An extension of the cAMP-mediated intracellular signal transduction. FEBS Lett. 1999;444:245–249. doi: 10.1016/s0014-5793(99)00070-8. [DOI] [PubMed] [Google Scholar]
- Pearson RB, Kemp BE. Protein kinase phosphorylation site sequences and consensus specificity motifs: tabulations. Methods Enzymol. 1991;200:62–81. doi: 10.1016/0076-6879(91)00127-i. [DOI] [PubMed] [Google Scholar]
- Pinkert CA, Trounce IA. Production of transmitochondrial mice. Methods. 2002;26:348–357. doi: 10.1016/S1046-2023(02)00041-5. [DOI] [PubMed] [Google Scholar]
- Pinkert CA, Trounce IA. Generation of transmitochondrial mice: development of xenomitochondrial mice to model neurodegenerative diseases. Methods Cell Biol. 2007;80:549–569. doi: 10.1016/S0091-679X(06)80027-0. [DOI] [PubMed] [Google Scholar]
- Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, Esterbauer H, Kozlov A, Kahn CR, Kroemer G, Rustin P, Burcelin R, Penninger JM. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell. 2007;131:476–491. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- Qi X, Lewin AS, Sun L, Hauswirth WW, Guy J. SOD2 gene transfer protects against optic neuropathy induced by deficiency of complex I. Ann Neurol. 2004;56:182–191. doi: 10.1002/ana.20175. [DOI] [PubMed] [Google Scholar]
- Qi X, Sun L, Lewin AS, Hauswirth WW, Guy J. The mutant human ND4 subunit of complex I induces optic neuropathy in the mouse. Invest Ophthalmol Vis Sci. 2007;48:1–10. doi: 10.1167/iovs.06-0789. [DOI] [PubMed] [Google Scholar]
- Quintana A, Kruse SE, Kapur RP, Sanz E, Palmiter RD. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc Natl Acad Sci U S A. 2010;107:10996–11001. doi: 10.1073/pnas.1006214107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JR, Quan Y, Sherer TB, Greenamyre JT, Miller GW. Paraquat neurotoxicity is distinct from that of MPTP and rotenone. Toxicol Sci. 2005;88:193–201. doi: 10.1093/toxsci/kfi304. [DOI] [PubMed] [Google Scholar]
- Santoro B, Bigini P, Levandis G, Nobile V, Biggiogera M, Botti F, Mennini T, Curti D. Evidence for chronic mitochondrial impairment in the cervical spinal cord of a murine model of motor neuron disease. Neurobiol Dis. 2004;17:349–357. doi: 10.1016/j.nbd.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Sardanelli AM, Technikova-Dobrova Z, Speranza F, Mazzocca A, Scacco S, Papa S. Topology of the mitochondrial cAMP-dependent protein kinase and its substrates. FEBS Lett. 1996;396:276–278. doi: 10.1016/0014-5793(96)01112-x. [DOI] [PubMed] [Google Scholar]
- Scacco S, Petruzzella V, Budde S, Vergari R, Tamborra R, Panelli D, van den Heuvel LP, Smeitink JA, Papa S. Pathological mutations of the human NDUFS4 gene of the 18-kDa (AQDQ) subunit of complex I affect the expression of the protein and the assembly and function of the complex. J Biol Chem. 2003;278:44161–44167. doi: 10.1074/jbc.M307615200. [DOI] [PubMed] [Google Scholar]
- Scacco S, Vergari R, Scarpulla RC, Technikova-Dobrova Z, Sardanelli A, Lambo R, Lorusso V, Papa S. cAMP-dependent phosphorylation of the nuclear encoded 18-kDa (IP) subunit of respiratory complex I and activation of the complex in serum-starved mouse fibroblast cultures. J Biol Chem. 2000;275:17578–17582. doi: 10.1074/jbc.M001174200. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Gu M, Taanman JW, Tabrizi SJ, Seaton T, Cleeter M, Cooper JM. Mitochondria in the etiology and pathogenesis of Parkinson's disease. Ann Neurol. 1998;44:S89–98. doi: 10.1002/ana.410440714. [DOI] [PubMed] [Google Scholar]
- Schmitt-John T, Drepper C, Mussmann A, Hahn P, Kuhlmann M, Thiel C, Hafner M, Lengeling A, Heimann P, Jones JM, Meisler MH, Jockusch H. Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat Genet. 2005;37:1213–1215. doi: 10.1038/ng1661. [DOI] [PubMed] [Google Scholar]
- Schon EA. Mitochondrial genetics and disease. Trends Biochem Sci. 2000;25:555–560. doi: 10.1016/s0968-0004(00)01688-1. [DOI] [PubMed] [Google Scholar]
- Schon EA, Manfredi G. Neuronal degeneration and mitochondrial dysfunction. J Clin Invest. 2003;111:303–312. doi: 10.1172/JCI17741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JD, McCartney S. Localization of A-kinase through anchoring proteins. Mol Endocrinol. 1994;8:5–11. doi: 10.1210/mend.8.1.8152430. [DOI] [PubMed] [Google Scholar]
- Sharma LK, Lu J, Bai Y. Mitochondrial respiratory complex I: structure, function and implication in human diseases. Curr Med Chem. 2009;16:1266–1277. doi: 10.2174/092986709787846578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoffner JM. Maternal inheritance and the evaluation of oxidative phosphorylation diseases. Lancet. 1996;348:1283–1288. doi: 10.1016/S0140-6736(96)09138-6. [DOI] [PubMed] [Google Scholar]
- Singer TP, Ramsay RR. The reaction sites of rotenone and ubiquinone with mitochondrial NADH dehydrogenase. Biochim Biophys Acta. 1994;1187:198–202. doi: 10.1016/0005-2728(94)90110-4. [DOI] [PubMed] [Google Scholar]
- Spivey A. Rotenone and paraquat linked to Parkinson's disease: human exposure study supports years of animal studies. Environ Health Perspect. 2011;119:A259. doi: 10.1289/ehp.119-a259a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Brown MR. Mitochondrial aging and dysfunction in Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:407–410. doi: 10.1016/j.pnpbp.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH, Parks JK, Miller SW, Tuttle JB, Trimmer PA, Sheehan JP, Bennett JP, Jr., Davis RE, Parker WD., Jr. Origin and functional consequences of the complex I defect in Parkinson's disease. Ann Neurol. 1996;40:663–671. doi: 10.1002/ana.410400417. [DOI] [PubMed] [Google Scholar]
- Torraco A, Diaz F, Vempati UD, Moraes CT. Mouse models of oxidative phosphorylation defects: powerful tools to study the pathobiology of mitochondrial diseases. Biochim Biophys Acta. 2009;1793:171–180. doi: 10.1016/j.bbamcr.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT, Larsson NG. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci U S A. 2005;102:17993–17998. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Ugalde C, Janssen RJ, van den Heuvel LP, Smeitink JA, Nijtmans LG. Differences in assembly or stability of complex I and other mitochondrial OXPHOS complexes in inherited complex I deficiency. Hum Mol Genet. 2004;13:659–667. doi: 10.1093/hmg/ddh071. [DOI] [PubMed] [Google Scholar]
- Vahsen N, Cande C, Briere JJ, Benit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, Feraud O, Debili N, Wissing S, Engelhardt S, Madeo F, Piacentini M, Penninger JM, Schagger H, Rustin P, Kroemer G. AIF deficiency compromises oxidative phosphorylation. Embo J. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Heuvel L, Smeitink J. The oxidative phosphorylation (OXPHOS) system: nuclear genes and human genetic diseases. Bioessays. 2001;23:518–525. doi: 10.1002/bies.1071. [DOI] [PubMed] [Google Scholar]
- Vempati UD, Torraco A, Moraes CT. Mouse models of oxidative phosphorylation dysfunction and disease. Methods. 2008;46:241–247. doi: 10.1016/j.ymeth.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel RO, Smeitink JA, Nijtmans LG. Human mitochondrial complex I assembly: a dynamic and versatile process. Biochim Biophys Acta. 2007;1767:1215–1227. doi: 10.1016/j.bbabio.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Walker JE. Determination of the structures of respiratory enzyme complexes from mammalian mitochondria. Biochim Biophys Acta. 1995;1271:221–227. doi: 10.1016/0925-4439(95)00031-x. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen. 2010;51:440–450. doi: 10.1002/em.20586. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Diseases of the mitochondrial DNA. Annu Rev Biochem. 1992;61:1175–1212. doi: 10.1146/annurev.bi.61.070192.005523. [DOI] [PubMed] [Google Scholar]