

Abstract
Alcohol intoxication results in marked reductions in brain glucose metabolism, which we hypothesized reflect not just its GABAergic enhancing effects but also metabolism of acetate as an alternative brain energy source. To test this hypothesis we separately assessed the effects of alcohol intoxication on brain glucose and acetate metabolism using Positron Emission Tomography (PET). We found that alcohol intoxication significantly decreased whole brain glucose metabolism (measured with FDG) with the largest decrements in cerebellum and occipital cortex and the smallest in thalamus. In contrast, alcohol intoxication caused a significant increase in [1-11C]acetate brain uptake (measured as standard uptake value, SUV), with the largest increases occurring in cerebellum and the smallest in thalamus. In heavy alcohol drinkers [1-11C]acetate brain uptake during alcohol challenge trended to be higher than in occasional drinkers (p <0.06) and the increases in [1-11C]acetate uptake in cerebellum with alcohol were positively associated with the reported amount of alcohol consumed (r=0.66, p<0.01). Our findings corroborate a reduction of brain glucose metabolism during intoxication and document an increase in brain acetate uptake. The opposite changes observed between regional brain metabolic decrements and regional increases in [1-11C]acetate uptake support the hypothesis that during alcohol intoxication the brain may rely on acetate as an alternative brain energy source and provides preliminary evidence that heavy alcohol exposures may facilitate the use of acetate as an energy substrate. These findings raise the question of the potential therapeutic benefits that increasing plasma acetate concentration (ie ketogenic diets) may have in alcoholics undergoing alcohol detoxification.
Keywords: Glia, PET, FDG, alcoholism, withdrawal, acetate
1. Introduction
Low to moderate doses of alcohol (0.25–0.75 g/kg) result in significant reductions in glucose metabolism in the human brain (range 10–30%) that are not associated with the behavioral effects seen with intoxication (Volkow et al., 1990). Moreover, in alcoholics we showed much greater reduction in regional brain glucose metabolism during intoxication than in healthy controls despite the fact that the alcoholics in contrast to the controls showed no evidence of behavioral intoxication (Volkow et al., 1990). This led us to postulate that brain glucose metabolic decrements could reflect utilization of acetate as an alternative source of energy for the brain during alcohol intoxication (Volkow et al., 2006b). Acetate is an accepted marker of glial metabolism (Wyss et al., 2011) and it is readily taken up by the brain where it is predominantly metabolized by glia (Cruz et al., 2005). However, plasma acetate concentration is constitutively low (about 0.2 to 0.3 mM), whereas that of glucose is typically high (about 5 mM); so under normal physiological conditions acetate brain metabolism is one order of magnitude lower than glucose metabolism (Dienel et al., 2001; Kammula and Fong, 1973). In contrast, during alcohol intoxication the concentration of acetate in blood increases significantly (around 0.5–1 mM) (Korri et al., 1985; Orrego et al., 1988) to levels that could support 10–20% of the total brain metabolic rate (Waniewski and Martin, 1998). In this study we test the hypothesis that during alcohol intoxication there is an increase in acetate metabolism in the human brain using [1-11C]acetate and Positron Emission Tomography (PET).
[1-11C]Acetate has been proposed as a PET ligand to assess glial metabolism in brain (Lanz et al., 2012; Wyss et al., 2009). Indeed stimulation of the rodent and the human brain increases the brain uptake of [1-11C]acetate (Wyss et al., 2009), consistent with autoradiographic studies showing increases in acetate metabolism in activated brain regions (Cruz et al., 2005; Dienel et al., 2007). The kinetics of [1-11C] acetate in brain are confounded by the loss of 11C as [11C]CO2 when [1-11C]acetate is oxidized through the tricarboxylic (TCA) cycle. Despite this confound, the initial uptake of [1-11C]acetate (peaks ~20 minutes) appears to reflect acetate, such that higher uptake is associated with higher acetate metabolism (Wyss et al., 2009).
Here we used PET and [1-11C]acetate to measure acetate metabolism after placebo and during alcohol intoxication both in occasional social drinkers (OSD) and in heavy drinkers (HD). As an indicator of acetate brain metabolism we quantified the standardized uptake values (SUV) of [1-11C]acetate. In parallel we conducted a second study to assess the effects of acute alcohol intoxication on brain glucose metabolism using PET and 2-deoxy-2-[18F]fluoro-D-glucose (18FDG) in occasional OSD to identify the brain regions that were most sensitive to alcohol’s metabolic effects. We used a 0.75 g/kg alcohol dose, which is roughly equivalent to three drinks for a 50 kg person and are within the doses consumed socially (Stinson et al., 1998). We hypothesized that acetate metabolism (as measured by SUV of [1-11C]acetate) in brain would be higher during acute alcohol intoxication and that the larger increases would occur in regions that showed the largest decreases in glucose metabolism. We also hypothesized that acetate metabolism would be associated with alcohol exposures and greater for HD than for OSD.
2 Material and Methods
2.1 Subjects
The study on the effects of alcohol on brain glucose metabolism was done in 15 healthy OSD (12 males, 32 ±7 years of age, BMI 25 ±3, Education 14 ±2 years) who were recruited to serve as comparison to the [1-11C]acetate studies. The study on the effects of alcohol intoxication on acetate metabolism was done in 16 healthy male OSD (37 ±7 years old, BMI 26 ±4, Education 13 ±2 years; 4 tobacco smokers) and 15 healthy male HD (37 ±9 years old, BMI 28 4, Education 12 ±1 years; 5 tobacco smokers). Inclusion criteria for all the subjects were: ability to understand and give informed consent, being 18–50 years of age. Inclusion criteria for OSD were having a prior experience with alcohol and regular use of no more than 1 drink per day. Inclusion criteria for HD were: history of 5+ drinks per day at least on 2 or more occasions per week. Exclusion criteria for subjects in both studies were: 1) urine positive for psychotropic drugs; 2) present or past history of dependence on alcohol or other drugs of abuse (except nicotine and allowed diagnosis of alcohol abuse though not dependence for HD); 3) present or past history of neurological or psychiatric disorder; 4) use of psychoactive medications in the past month (i.e., opiate analgesics, stimulants, sedatives); 5) use of prescription (non-psychiatric) medication(s), i.e., antihistamines; 6) medical conditions that may alter cerebral function; 7) cardiovascular and metabolic diseases and 8) history of head trauma with loss of consciousness of more than 30 minutes. We excluded subjects who had never been intoxicated since we did not want the experimental procedure to be their first exposure to alcohol intoxication. Subjects that met DSM IV diagnosis of alcohol dependence were excluded. Subjects were instructed to discontinue any over the counter medication two weeks prior to the PET scan, and were asked to abstain from alcohol for 48 hours prior to the placebo and alcohol PET scanning session. Self-reports were used to determine if they had consumed any alcohol during the two days that preceded the study. Signed informed consents were obtained from the subjects prior to participation as approved by the Committee on Research in Human Subjects at Stony Brook University
2.2 Alcohol & Placebo Administration
Subjects drank the alcohol (0.75 g/kg mixed in a caffeine-free diet soda) or the placebo (caffeine free diet soda) within a 20 minutes period under blind conditions. For this purpose we used a specialized drinking container with an alcohol-containing lid that provided the smell of alcohol and delivered the same volume of liquid for both conditions. Participants were injected with the radiotracer 40 minutes after initiating the alcohol consumption (20 minutes after completion of drinking). This timing was selected to correspond to the ascending limb of the blood alcohol curves and the time when peak concentration of alcohol are reached in the human brain after its oral administration (Hetherington et al., 1999).
2.3 Behavioral Evaluation and measures of Alcohol in Plasma
Before placebo or alcohol, and at 10, 15, 30 and 85 minutes after placebo or alcohol administration, subjects were asked to evaluate on an analog scale (rated 1–10) their subjective sense of intoxication, high, sleepiness, and dizzy. At the end of the study subjects were asked to rate the effects of the drug for “liking” and “pleasant” (rated 1–10). Blood alcohol concentrations were measured before and 20, 40, 60, 80, 90, and 120 min after the initiation of alcohol administration using the enzymatic assay described by Lloyd (Lloyd et al., 1978).
2.4 PET scans
PET scans were conducted with a whole-body, high-resolution positron emission tomograph (Siemens/CTI ECAT HR+, with 4.6 × 4.6 × 4.2 mm NEMA (National Electrical Manufacturers Association) collected on a 3D mode. Procedures for positioning of the subjects in the scanner, for transmission and emission scans for 18FDG have been published (Wang et al., 1993) and for [1-11C]acetate we used the same procedures that we have used for [11C]raclopride scans (Volkow et al., 1993). For arterial sampling we used an automated device (Ole Dick, Denmark) that sampled every 2.5 seconds for the first 2 minutes and then every minute from 2–5 minutes and then at 10, 15, 20, 30, 45, and 60 minutes
For the 18FDG scans (study to measure brain glucose metabolism), subjects were injected with 4–6 mCi of 18FDG and one twenty minutes emission scan was started 35 minutes after injections. Subjects were scanned over a two day period tested with placebo on one day and with alcohol on the other with the order of the scans randomly assigned. Images were reconstructed using a filtered back projection (Hann filter with a 4.9 mm Kernel FWHM). The 18FDG scans were transformed into metabolic images as previously described (Volkow et al., 1990) and metabolic rates were computed using an extension of Sokoloff's model (Phelps et al., 1979). In addition we also obtained regional SUV for FDG since that was the model we used to quantify the acetate measures.
For the [1-11C]acetate scans, subjects were injected with 8–10 mCi of [1-11C]acetate and dynamic emission scans were obtained immediately after radiotracer injection for a total of 60 minutes using the following time frames: 1 × 10 sec; 12 × 5 sec; 1 × 20 sec; 1 × 30 sec; 8 × 1 min; 4 × 5 min; 4 × 7.5 min. Each subject underwent two [1-11C]acetate scans two hours apart from each other; the first scan after placebo and the second after alcohol administration. To estimate brain acetate metabolism we used the SUV on the averaged images obtained between 3 and 20 minutes after injection. This initial uptake of acetate in brain has been shown to correspond with the initial metabolism of acetate through the TCA cycle (Wyss et al., 2009).
For all scans subjects were tested while lying in the supine position in a dimly lit room with noise kept to a minimum and the only intervention was the periodic assessment for self-report of drug effects. To ensure that subjects did not fall asleep they were monitored throughout the procedure and were asked to keep their eyes open.
2.5 Analysis
Regions of interest (ROI) were selected using a neuroanatomically template of 423 non-overlapping regions based on the Talairach and Tournoux's atlas (Talairach and Tournoux, 1988). Values for the cortical, subcortical and cerebellar regions were computed using the weighted average from the different slices where the regions were obtained and grouped into 7 composite regions, which included frontal, parietal, temporal, occipital cortices, striatum, and cerebellum. An estimate of whole-brain metabolism was obtained by averaging the values from all of the regions of interest. For the 18FDG scans these ROIs were delineated directly in the metabolic images and for the [1-11C]acetate scans these were delineated in the summed images obtained from 3 to 60 minutes.
For statistical comparisons on the 18FDG study we used a repeated measures analysis of variance (ANOVA) with two conditions (placebo versus alcohol). Effects sizes for the effects of alcohol on regional brain glucose metabolism were quantified with Cohen’s d (Cohen, 1988). To assess if alcohol effects differed between regions we used a repeated ANOVA (percent change in metabolism in each of the seven ROI). In addition findings were corroborated with Statistical Parametric Mapping (SPM2)(Friston et al., 1994). For this purpose metabolic images (normalized to the individual’s whole brain metabolism) were spatially adjusted to the Montreal Neurological Institute (MNI) stereotactic space using the PET template provided in SPM and subsequently smoothed with an 8 mm isotropic Gaussian kernel. We used a within-subjects ANOVA model with two conditions (placebo vs alcohol). Statistical significance was set using simultaneously a voxel-level threshold p <0.005 (uncorrected) and a minimum cluster volume of 200 voxels; only regions that survive corrections for multiple comparisons using a family-wise error correction (pFWE<0.05) are reported as significant.
For statistical comparisons on the [1-11C]acetate study we used a mixed model ANOVA with group as the between factor (OSD versus HD) and condition (placebo versus alcohol) to assess regional effects of alcohol on brain acetate metabolism and to compare these changes between OSD and HD. Effects sizes for the effects of alcohol on acetate SUV were quantified with Cohen’s d (Cohen, 1988). Note that we were unable to accurately co-register the [1-11C]acetate images to the SPM template (activity outside the brain interfered with the correct delineation of the cortical boundaries by the computer), which is why we were unable to corroborate findings with SPM.
Pearson product moment correlation analyses were performed to assess the association between alcohol-induced regional changes in [1-11C]acetate (% change from placebo) and the history of alcohol use in HD and between regional changes in [1-11C]acetate (% change from placebo) and changes in behavior (all subjects).
We set level of significance to p <0.05 for a priori hypothesis (1) acetate metabolism would be increased throughout the brain during alcohol intoxication and the largest increases would occur in the areas were alcohol reduced glucose metabolism the most; (2) increases in acetate metabolism with alcohol would be greater in HD than in OSD and (3) acetate metabolism would be associated with drinking behavior. To avoid type one errors from multiple comparisons (7 brain regions) we used a Bonferroni correction (p <0.007) for exploratory analysis.
3. Results
3.1 Effects of Alcohol on Brain Glucose Metabolism
Acute alcohol administration decreased whole brain glucose metabolism from 40.4 ± 4 micromol/100g/min with placebo to 35.6 ± 5 during intoxication (11.5% ± 11; p <0.005; Effect size=0.82) (Figure 1). Repeated ANOVA with regions as within-subject measure showed a significant region effect (F=5.5, df 6, 84, p <0.001) indicating that the differences varied between regions. Alcohol induced decreases were largest in cerebellum (−16% ± 11; Effect size=1.33) and occipital cortex (−16% ± 10; Effect size=1.81), and smallest in thalamus (−7% ± 17; Effect size=0.61) and striatum (−8% ± 13; Effect size=0.80) (Figure 2A). The individual values for the absolute metabolic measures for these regions are included in Supplemental Figure 1.
Figure 1.

Axial planes for regional brain metabolic images obtained with FDG in a subject during placebo and during alcohol intoxication (20 minutes after alcohol consumption).
Figure 2.

A. Regional brain glucose metabolic values in the preselected large regions of interest during placebo and during alcohol intoxication (20 minutes after alcohol consumption). Metabolic values in all regions were significantly lower for alcohol than for placebo (p <0.001); B. Results from the SPM analysis on the normalized glucose metabolic image Alcohol > Placebo (p<0.005, uncorrected) superimposed on the surface of the right cerebrum (lateral and medial) and the cerebellum (posterior and anterior) of the human Colin template.
The regional quantification of FDG using SUV yielded similar regional changes with alcohol with the largets decreases occuring in cerebellum (−17% ± 15), occipital cortex (− 18% ± 13) and the smallest in thalamus (−9% ± 21) and striatum (−10% ± 18). SPM analysis on the absolute metabolic images showed significant decreases througout the brain with alcohol intoxication (data not shown). The SPM on the normalized metabolic images (normalized to whole brain metabolism), which were done to hihgligth regional effects confirmed that after controlling for global decreases there were significant decreases in occipital cortex and cerebellum (p <0.001) but not in subcortical regions (Figure 2B and Supplemental Figure 2).
3.2 Effects of Alcohol on Brain Acetate Metabolism
Plasma alcohol concentration and behavioral effects
Plasma alcohol concentrations peaked around 60 minutes after its administration and did not differ between OSD and HD (Figure 3B).
Figure 3.

A. Axial planes for brain images obtained with [1-11C]acetate (averaged between 3–60 minutes) during placebo and during alcohol intoxication (20 minutes after alcohol consumption) in an occasional social drinker (OSD) and in a heavy drinker (HD). B. Plasma alcohol concentration for the OSD and for the HD at different times after its administration.
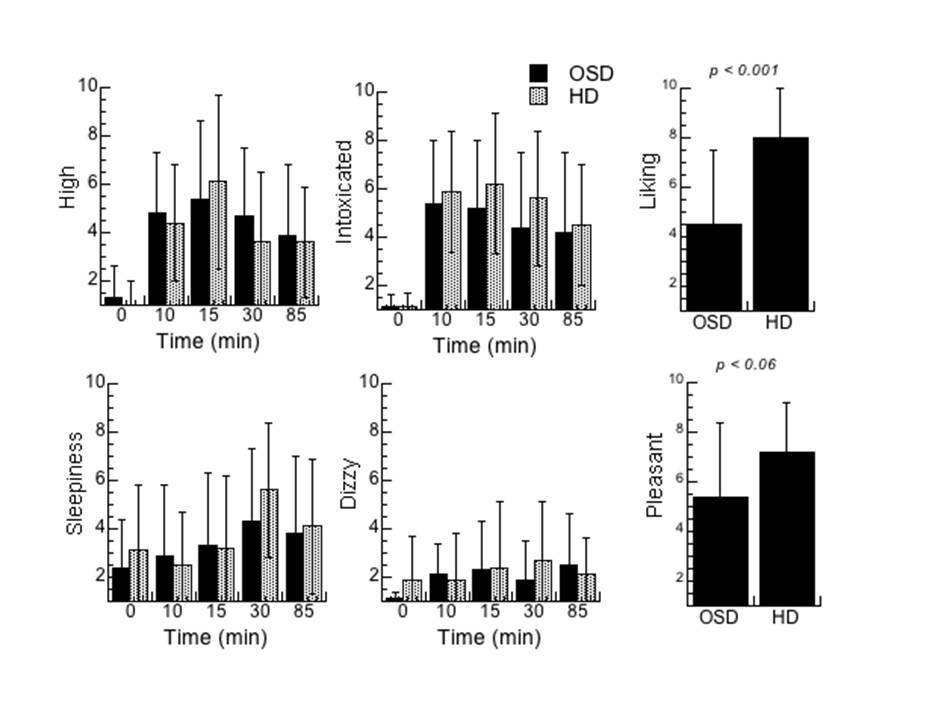
Acute alcohol increased self-reports of “intoxication”, “high”, “dizzy” and “sleepy” to the same extent in OSD and HD as evidence by a significant Drug Effect but a lack of Drug by Group Interaction effect (Supplemental Figure 3). However the self-reports taken at the end of the study for “liking” were significantly higher for HD than OSD (p < 0.001) and also showed a trend for higher scores on “pleasant” for HD than for OSD ( P < 0.06) (Supplemental Figure 3).
Effects of Alcohol on Brain [1-11C]Acetate Uptake
[1-11C]Acetate distributed throughout cortical and subcortical brain regions (Figure 3) with peak uptake occurring around 20 minutes after administration and with higher peak values for the alcohol than the placebo scans (Figures 3A and 4). Individual regional values are included in Supplemental Figure 4. The SUV values for [1-11C]acetate obtained for the placebo and alcohol scans showed overall higher values for HD than OSD but these differences only showed a trend of significance for the alcohol scans (F=3.9; p=0.06) (Figure 4).
Figure 4.

A Average time activity curves of [1-11C]acetate in cerebellum for the placebo and the alcohol scans in Occasional Social Drinkers (OSD) and in Heavy Drinkers (HD). B. Regional values for [1-11C]acetate SUV in OSD and in HD. All regions showed significantly higher [1-11C]acetate SUV with alcohol than with placebo (p <0.001). There was a trend for the values on the alcohol scans to be higher in the HD than in OSD (p<0.06).
Alcohol increased [1-11C]acetate brain uptake significantly to a similar extent in OSD (15% ± 11; p <0.001; Effects Size=0.82) and HD (16% ± 14; p<0.001; Effect size=0.74) (Figure 3 and 4). Since there were no differences between the groups we averaged the data for the two groups. Repeated ANOVA with regions as the within subject measure showed a significant region effect (F=4.7, df 6, 174, p <0.001) indicating that the differences varied between regions. The largest increases where in cerebellum (17% ± 14; p<0.0001; Effect size=0.77) and the smallest in thalamus (11% ± 12; p < 0.0001; Effect size=0.56) (Figure 4).
Measurements of C-11 in plasma from the sampled arterial blood also showed a higher concentration for the alcohol than the placebo scans during the first 20 minutes of the scan (12%) (Supplemental Figure 5).
Correlation between Brain [1-11C] Acetate and the Reported Alcohol Use in HD
For HD, the correlation analysis between the doses of alcohol consumed (beers per day) and SUV was positively correlated both for the placebo and the alcohol scans (Table 1); the larger the amount consumed the higher the SUV. In addition alcohol-induced SUV changes (% change from placebo) also showed significant positive correlations for most brain regions (r >0.56, p < 0.05) and strongest for cerebellum (Table 1). The larger the amount of alcohol consumed the greater the increases in [1-11C]acetate brain uptake with alcohol intoxication as compared to placebo.
Table 1.
Correlations between: SUV during the alcohol scan and self-reports of “Intoxication” (all subjects); SUV for placebo and SUV for alcohol scans and the alcohol doses used (number of beers consumed per day) in the Heavy Drinkers (HD); and percent change in SUV (with respect to placebo) and alcohol doses used in HD.
| SUV Alcohol vs “Intoxication” All Subjects |
SUV Placebo vs Doses Used HD |
SUV Alcohol vs Doses Used HD |
% Change SUV vs Doses Used HD |
|
|---|---|---|---|---|
| Frontal | R=−0.46, p=0.009 | R=0.68, p=0.005 | R=0.54, p=0.04 | R=0.65, p<0.02 |
| Parietal | R=−0.41, p=0.02 | R=0.53, p=0.05 | R=0.49, NS | R=0.51, p<0.08 |
| Temporal | R=−0.49, p=0.006* | R=0.66, p=0.007 | R=0.53, p=0.007 | R=065, p<0.02 |
| Occipital | R=−0.46, p=0.009 | R=0.60, p-0.02 | R=0.60, p-0.04 | R=0.62, p<0.03 |
| Striatum | R=−0.49, p=0.005* | R=0.63, p=0.02 | R=0.51, NS | R=0.64, p<0.02 |
| Thalamus | R=−0.33, p=0.07 | R=0.65, p=0.01 | R=0.64, p=0.01 | R=0.55, p<0.06 |
| Cerebellum | R=−0.40, p=0.03 | R=0.54, p=0.04 | R=0.39, NS | R=0.66, p<0.01 |
Significant after Bonferroni correction P <0.006 for exploratory analysis
Correlation between Brain [1-11C]Acetate and Behavioral Effects of Alcohol (exploratory analysis)
The regional changes in [1-11C]Acetate with alcohol intoxication (% change from placebo) were not correlated with changes in behavioral effects of alcohol. However the SUV values obtained during the alcohol scan showed a negative correlation with self-reports of “intoxication” (peak effects) and this effect was significant after Bonferroni correction in striatum and temporal cortex (Table 1). The higher the SUV measures the lower the self-reports of “intoxication”. The correlations with self-report of “sleepiness”, “high” or “dizzy” were not significant.
4. Discussion
Here we show a significant decrease in brain glucose metabolism and a significant increase in brain uptake of [1-11C]acetate with acute alcohol administration that is consistent with our hypothesis that during alcohol intoxication the brain may use acetate as a substrate for energy production. Though we did not corroborate our hypothesis that the effects of alcohol on brain acetate metabolism would be significantly higher in HD than in OSD we showed a trend towards greater increases in cerebellum of HD than OSD (p <0.06). Also in the HD the amount of alcohol consumed correlated with brain acetate uptake (placebo and alcohol scans) that is, the greater the reported history of alcohol consumption the greater the brain [1-11C]acetate uptake.
The increases in brain [1-11C]acetate uptake with acute alcohol administration at doses that reduced brain glucose metabolism and in a regional pattern opposite to the glucose metabolic changes (largest changes in cerebellum and smallest in thalamus) support the hypothesis that during alcohol intoxication the brain uses acetate as a substrate for energy production. Glucose is the major substrate for brain metabolism and is used by neurons and glia (Nehlig et al., 2004). The mechanism(s) by which acute alcohol decreases brain glucose metabolism are unclear (Handa et al., 2000; Volkow et al., 2006b) but have been linked with inhibition of glucose transport into the brain (Abdul Muneer et al., 2010), inhibition of glucose uptake by neurons and glia (Singh et al., 1990), and/or inhibition of glucose phosphorylation by hexokinases (Pawlosky et al., 2010).
Increases in plasma acetate concentration during alcohol intoxication could trigger the reductions in brain glucose uptake and phosphorylation (Korri et al., 1985). Indeed in rodents infusion of alcohol or of acetate were associated with an inhibition of brain glucose uptake and elevated blood acetate concentrations were associated with a decrease in glucose phosphorylation (Pawlosky et al., 2010). Acetate is a good substrate for the glial monocarboxylate transporter MCT1 (KM 1.6 ± 0.4 mM) (Rae et al., 2012) and for acetyl-CoA synthetase, which is the first enzymatic step for acetate metabolism (2.9 nmol/min per milligram of protein) (Waniewski and Martin, 1998). Thus, when acetate concentration in plasma increases, such as during alcohol intoxication, it is possible that its increased uptake by glial cells may inhibit their use of glucose and favor acetate metabolism.
However, alcohol also facilitates GABAergic neurotransmission (Krystal et al., 2006) and therefore would be expected to decrease neuronal activity as is the case for other GABEergic enhancing drugs (Volkow et al., 1995). Thus it is likely that alcohol-induced reductions in brain glucose metabolism reflect a combination of alcohol’s GABAergic actions and its effects on glucose transport and phosphorylation.
Findings of decreases in brain glucose metabolism with acute alcohol are very consistent in the preclinical and clinical literature (Volkow et al., 1990; Volkow et al., 2006b). However, the magnitude of the reported brain metabolic decreases varies among investigators. For example, whole-brain metabolic reductions of 26% were reported with 0.75 g/kg of alcohol (Wang et al., 2000), 25% with 40 g iv (approximately 0.6 g/kg iv) (Schreckenberger et al., 2004), 18% with 1 g/kg (Volkow et al., 1990), whereas De Wit reported 3% decreases with 0.5 g/kg (de Wit et al., 1990). In preclinical studies the magnitude of the effect is influenced by the time at which the measurements are made after alcohol consumption (Lyons et al., 1998), by the past alcohol history of the animals (Porrino et al., 1998) and by the doses given (Williams-Hemby and Porrino, 1997). In humans, imaging studies have also shown that alcohol-induced changes in brain glucose metabolism are sensitive to histories of alcohol use (Volkow et al., 1990), doses given (Volkow et al., 2006a) and gender (Wang et al., 2003). Indeed we had reported that in alcoholics the reduction in glucose metabolism with alcohol administration were significantly larger than in non-alcoholic controls despite the fact that, different from the controls, they showed no evidence of behavioral intoxication (Volkow et al., 1990). This finding led us to hypothesize that the elevated levels of acetate in plasma observed in chronic heavy alcohol users (Roine et al., 1988) may favor the use of acetate as an energy substrate by brain. While not significant, there was a trend toward higher brain [1-11C]acetate uptake in HD than in OSD for the alcohol scans. Similarly brain [1-11C]acetate uptake (placebo and alcohol scans), as well as the increases with alcohol (% change from placebo) were associated with the amount of alcohol consumed, being greater in those that reported the highest doses of alcohol use. Thus this suggests that with chronic alcohol exposures the brain may rely increasingly on acetate as an alternative energy source. This is clinically relevant for a lack of energy substrates upon alcohol discontinuation has been reported to contribute to the clinical symptoms during acute alcohol withdrawal (Derr, 1984). Indeed acetate infusion was reported to abate ethanol withdrawal in rodents (Derr et al., 1981), which lead us to speculate that perhaps increasing acetate levels in plasma could help minimize the adverse effects of alcohol withdrawal in alcoholics. Ketogenic diets increase acetate in plasma and thus, studies that evaluate them during alcohol withdrawal would allow to test their potential value for alcohol detoxification.
The cerebellum had the largest increase in acetate metabolism and the largest decreases in glucose metabolism with alcohol. This suggests that the cerebellum may favor acetate over glucose to a greater extent during alcohol intoxication than other brain regions. The mechanism(s) responsible for this cerebellar response is unclear but could reflect regional differences in the concentration of enzymes involved in alcohol and/or acetate metabolism. Interestingly the Allen Brain Atlas browser indicates higher concentrations of the genes that encode for dehydrogenase (ADH) and acetaldehyde dehydrogenase (ALDH), which are the enzymes that convert alcohol to acetaldehyde and acetaldehyde to acetate respectively, in cerebellum than in most brain regions (http://human.brain-map.org/). Also in rodents differences in the concentration of ALDH in cerebellum have been reported between strains that differed in their sensitivity to ethanol (Zimatkin and Lindros, 1989; Zimatkin and Deitrich, 1995). A differential sensitivity of the cerebellum to alcohol’s effects on energy metabolism could contribute to its susceptibility to ethanol toxicity from compromised energy production (Fattoretti et al., 2003; Jaatinen and Rintala, 2008) and glial damage (Aschner and Allen, 2000).
The exploratory analysis showed a negative correlation between [1-11C]acetate’s SUV in striatum and temporal cortex and self-reports of intoxication during the alcohol scans; such that the higher the scores of intoxication the lower the [1-11C]acetate uptake. In preclinical studies a similar negative association was reported between the rate of brain acetate metabolism and the sensitivity to alcohol’s hypnotic effects (Zimatkin et al., 2011), which indicates that genetic differences may also contribute to inter individual differences in brain acetate metabolism and that this in turn may modulate the sensitivity to alcohol’s intoxicating effects.
In our studies we did not see differences in self-reports of intoxication or high between OSD and HD, which is distinct from the lower behavioral measures of intoxication we had reported when we compared alcoholics with non-alcoholic subjects (Volkow et al., 1990). However this may reflect the fact that we previously tested alcoholics whereas in this study we excluded HD who were alcohol dependent as per DSM. Thus, our HD may have not developed the behavioral tolerance to the effects of alcohol seen in alcoholics.
5. Limitations
Though we interpret brain [1-11C]acetate uptake to reflect acetate metabolism, acetate is also metabolized through the glutamate glutamine cycle (Chowdhury et al., 2007). However, the transfer of labeled glutamine to neurons is slow relative to the 20-min uptake of [1-11C]acetate in brain (Wyss et al., 2009). We use [1-11C]acetate’s SUV over the first 20 minutes as estimate of acetate metabolism but the exact relationship is unclear and we cannot rule out that increases in cerebral blood flow (CBF) may have contributed to the increased uptake of [1-11C]acetate during alcohol intoxication. However, it is unlikely that CBF increases account for our findings since CBF increases with alcohol intoxication are predominantly observed in cortical regions (frontal, temporal) but not in cerebellum (Khalili-Mahani et al., 2011) (Volkow et al., 1988); whereas the cerebellum showed the largest increases in [1-11C]acetate uptake with intoxication.
[1-11C]Acetate is rapidly metabolized in the TCA cycle with the concomitant release of 11CO2, which readily crosses the blood brain barrier. Since the concentration of C-11 in plasma during the alcohol scans was higher than for placebo we cannot rule out that the increases in SUV reflect higher 11CO2 or other C-11 labeled metabolites. However, this is unlikely to account for the regional effects since increases in radiotracer delivery would have resulted in homogeneous effects throughout the brain whereas we show significant differences in [1-11C]acetate uptake between regions that mirrored the regional effects observed for glucose metabolism (but in the opposite direction). Nonetheless the use of 13C/1H magnetic resonance spectroscopy in conjunction with [2-13C]acetate would be a better technique to measure brain acetate metabolism (Bluml et al., 2002; Deelchand et al., 2009).
Because of total radiation exposures we were unable to assess the effects of alcohol on brain glucose metabolism and on acetate metabolism in the same subjects. Also we did not measure the effects of alcohol intoxication on brain glucose metabolism in HD. However, we had previously performed such studies and reported that alcoholics showed significantly greater reductions in whole brain glucose metabolism than controls (double the magnitude) and this effect was largest in cerebellum (Volkow et al., 1990), which is also the region that showed the largest increases in [1-11C]acetate uptake with alcohol.
Finally we have no test-retest measures for brain [1-11C]acetate uptake and thus we cannot determine its reproducibility. However the consistency of the increases seen with alcohol both in HD and OSD indicates that this is a pharmacological effect rather than test-retest variability. We did not collect measures of plasma acetate concentration and thus we could not determine what the levels were or whether they differed between HD and OSD. However, from the literature we estimate that the acetate plasma levels may have been in the 0.5 to 1mM range (Nuutinen et al., 1985).
In summary, here we show that acute alcohol administration reduced brain glucose metabolism and increased brain uptake of [1-11C]acetate consistent with our hypothesis that during alcohol intoxication the brain relies on acetate as an alternative energy source for metabolism. HD tended to have higher brain [1-11C]acetate uptake than OSD (alcohol scans; p <0.06), which provides preliminary evidence that heavy alcohol exposures may favor the use of acetate as an energy substrate by the brain.
Supplementary Material
Supplemental Figure 1. Individual values for the regional brain metabolic measures after placebo and after alcohol in whole brain, cerebellum and occipital cortex (regions with the largest alcohol-induced decreases in metabolism); and in striatum and thalamus (regions where alcohol had the smallest effect).
{kind=link}
Supplemental Figure 2. SPM results for the comparisons on the normalized metabolic images showing areas where metabolism was significantly decreased after alcohol consumption when compared with placebo. Parameters set at pu <0.005 and cluster >200 voxels.
{kind=link}
Supplemental Figure 3. Self-reports on alcohol effects prior to (0 minutes) and at 10, 15, 30 and 85 minutes after its consumption for the occasional social drinkers (OSD) and the heavy drinkers (HD) and for self-reports of “liking” and “pleasant” scored at the end of the study. The one Factor for Group (OSD vs HD) repeated Drug (0, 10, 15, 30 and 85 minutes) ANOVA showed significant Drug effects for high (F=29, p <0.0001), intoxication (F=28, p <0.0001), sleepiness (F=4, 4,112, p=0.004) and dizzy (f=3.8, p=0.007) but the Interaction Effect was not significant for any of these measures, which indicates that they did not differ between HD and OSD. However, the ratings taken at the end of the study were significantly higher for “liking” for HD than OSD and showed a trend for “pleasant”. Values represent means and SD.
{kind=link}
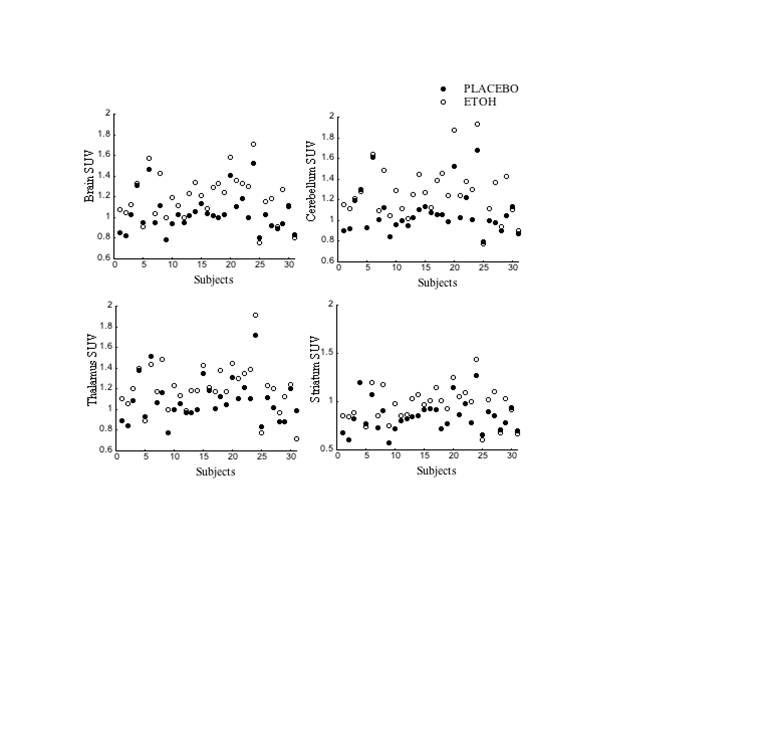
Supplemental Figure 4. Individual values for the SUV measures of [1-11C]acetate after placebo and after alcohol in whole brain, cerebellum (region with the largest alcoholinduced increases); and in striatum and thalamus (regions with the least SUV increases with alcohol).
{kind=link}
Supplemental Figure 5. Average time activity curves for the arterial concentration of C-11 for the placebo and the alcohol scans in OSD and in HD subjects.
{kind=link}
Highlights.
Acute alcohol intoxication decreases glucose metabolism, increases acetate uptake in the human brain.
PET imaging reveals acetate as an alternative energy source during alcohol intoxication.
By increasing plasma acetate, a ketonic diet may help prevent some of the adverse effects associated with alcohol withdrawal
Acknowledgements
We thank David Schlyer, Paul Vaska, Youwen Xu, Pauline Carter, Millard Jayne and Karen Apelskog for their contributions and Ruben Baler for editorial assistance. Research supported by NIH’s Intramural Research Program (NIAAA) and by DOE (DE-AC01-76CH00016).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdul Muneer PM, Alikunju S, Szlachetka AM, Haorah J. Inhibitory effects of alcohol on glucose transport across the blood-brain barrier leads to neurodegeneration: preventive role of acetyl-L: -carnitine. Psychopharmacology (Berl) 2010;214:707–718. doi: 10.1007/s00213-010-2076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M, Allen JW. Astrocytes in methylmercury, ammonia, methionine sulfoximine and alcohol-induced neurotoxicity. Neurotoxicology. 2000;21:573–579. [PubMed] [Google Scholar]
- Bluml S, Moreno-Torres A, Shic F, Nguy CH, Ross BD. Tricarboxylic acid cycle of glia in the in vivo human brain. NMR Biomed. 2002;15:1–5. doi: 10.1002/nbm.725. [DOI] [PubMed] [Google Scholar]
- Chowdhury GM, Gupta M, Gibson KM, Patel AB, Behar KL. Altered cerebral glucose and acetate metabolism in succinic semialdehyde dehydrogenase-deficient mice: evidence for glial dysfunction and reduced glutamate/glutamine cycling. J Neurochem. 2007;103:2077–2091. doi: 10.1111/j.1471-4159.2007.04887.x. [DOI] [PubMed] [Google Scholar]
- Cohen J. Statistical power analysis for the behavioral sciences. 2nd Ed. New Jersey: 1988. [Google Scholar]
- Cruz NF, Lasater A, Zielke HR, Dienel GA. Activation of astrocytes in brain of conscious rats during acoustic stimulation: acetate utilization in working brain. J Neurochem. 2005;92:934–947. doi: 10.1111/j.1471-4159.2004.02935.x. [DOI] [PubMed] [Google Scholar]
- de Wit H, Metz J, Wagner N, Cooper M. Behavioral and subjective effects of ethanol: relationship to cerebral metabolism using PET. Alcohol Clin Exp Res. 1990;14:482–489. doi: 10.1111/j.1530-0277.1990.tb00508.x. [DOI] [PubMed] [Google Scholar]
- Deelchand DK, Shestov AA, Koski DM, Ugurbil K, Henry PG. Acetate transport and utilization in the rat brain. J Neurochem. 2009;109(Suppl 1):46–54. doi: 10.1111/j.1471-4159.2009.05895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derr RF. The ethanol withdrawal syndrome: a consequence of lack of substrate for a cerebral Krebs-cycle. J Theor Biol. 1984;106:375–381. doi: 10.1016/0022-5193(84)90036-5. [DOI] [PubMed] [Google Scholar]
- Derr RF, Draves K, Derr M. Abatement by acetate of an ethanol withdrawal syndrome. Life Sci. 1981;29:1787–1790. doi: 10.1016/0024-3205(81)90189-2. [DOI] [PubMed] [Google Scholar]
- Dienel GA, Popp D, Drew PD, Ball K, Krisht A, Cruz NF. Preferential labeling of glial and meningial brain tumors with [2-(14)C]acetate. J Nucl Med. 2001;42:1243–1250. [PubMed] [Google Scholar]
- Dienel GA, Schmidt KC, Cruz NF. Astrocyte activation in vivo during graded photic stimulation. J Neurochem. 2007;103:1506–1522. doi: 10.1111/j.1471-4159.2007.04859.x. [DOI] [PubMed] [Google Scholar]
- Fattoretti P, Bertoni-Freddari C, Casoli T, Di Stefano G, Giorgetti G, Solazzi M. Ethanol-induced decrease of the expression of glucose transport protein (Glut3) in the central nervous system as a predisposing condition to apoptosis: the effect of age. Ann N Y Acad Sci. 2003;1010:500–503. doi: 10.1196/annals.1299.092. [DOI] [PubMed] [Google Scholar]
- Friston K, Holmes A, Worsley K, Poline J-P, Frith C, Frackowiak R. Statistical Parametric Maps in functional imaging: A general linear approach. Hum. Brain Mapp. 1994;2:189–210. [Google Scholar]
- Handa RK, DeJoseph MR, Singh LD, Hawkins RA, Singh SP. Glucose transporters and glucose utilization in rat brain after acute ethanol administration. Metab Brain Dis. 2000;15:211–222. doi: 10.1007/BF02674530. [DOI] [PubMed] [Google Scholar]
- Hetherington HP, Telang F, Pan JW, Sammi M, Schuhlein D, Molina P, Volkow ND. Spectroscopic imaging of the uptake kinetics of human brain ethanol. Magn Reson Med. 1999;42:1019–1026. doi: 10.1002/(sici)1522-2594(199912)42:6<1019::aid-mrm5>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Jaatinen P, Rintala J. Mechanisms of ethanol-induced degeneration in the developing, mature, and aging cerebellum. Cerebellum. 2008;7:332–347. doi: 10.1007/s12311-008-0034-z. [DOI] [PubMed] [Google Scholar]
- Kammula RG, Fong BC. Metabolism of glucose and acetate by the ovine brain in vivo. Am J Physiol. 1973;225:110–113. doi: 10.1152/ajplegacy.1973.225.1.110. [DOI] [PubMed] [Google Scholar]
- Khalili-Mahani N, van Osch MJ, Baerends E, Soeter RP, de Kam M, Zoethout RW, Dahan A, van Buchem MA, van Gerven JM, Rombouts SA. Pseudocontinuous arterial spin labeling reveals dissociable effects of morphine and alcohol on regional cerebral blood flow. J Cereb Blood Flow Metab. 2011;31:1321–1333. doi: 10.1038/jcbfm.2010.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korri UM, Nuutinen H, Salaspuro M. Increased blood acetate: a new laboratory marker of alcoholism and heavy drinking. Alcohol Clin Exp Res. 1985;9:468–471. doi: 10.1111/j.1530-0277.1985.tb05585.x. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Staley J, Mason G, Petrakis IL, Kaufman J, Harris RA, Gelernter J, Lappalainen J. Gamma-aminobutyric acid type A receptors and alcoholism: intoxication, dependence, vulnerability, and treatment. Arch Gen Psychiatry. 2006;63:957–968. doi: 10.1001/archpsyc.63.9.957. [DOI] [PubMed] [Google Scholar]
- Lanz B, Uffmann K, M TW, Weber B, Buck A, Gruetter R. A two-compartment mathematical model of neuroglial metabolism using [1-(11)C] acetate. J Cereb Blood Flow Metab. 2012;32:548–559. doi: 10.1038/jcbfm.2011.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd B, Burrin J, Smythe P, Alberti KG. Enzymic fluorometric continuous-flow assays for blood glucose, lactate, pyruvate, alanine, glycerol, and 3-hydroxybutyrate. Clin Chem. 1978;24:1724–1729. [PubMed] [Google Scholar]
- Lyons D, Whitlow CT, Porrino LJ. Multiphasic consequences of the acute administration of ethanol on cerebral glucose metabolism in the rat. Pharmacol Biochem Behav. 1998;61:201–206. doi: 10.1016/s0091-3057(98)00089-6. [DOI] [PubMed] [Google Scholar]
- Nehlig A, Wittendorp-Rechenmann E, Lam CD. Selective uptake of [14C]2-deoxyglucose by neurons and astrocytes: high-resolution microautoradiographic imaging by cellular 14C-trajectography combined with immunohistochemistry. J Cereb Blood Flow Metab. 2004;24:1004–1014. doi: 10.1097/01.WCB.0000128533.84196.D8. [DOI] [PubMed] [Google Scholar]
- Nuutinen H, Lindros K, Hekali P, Salaspuro M. Elevated blood acetate as indicator of fast ethanol elimination in chronic alcoholics. Alcohol. 1985;2:623–626. doi: 10.1016/0741-8329(85)90090-4. [DOI] [PubMed] [Google Scholar]
- Orrego H, Carmichael FJ, Israel Y. New insights on the mechanism of the alcohol-induced increase in portal blood flow. Can J Physiol Pharmacol. 1988;66:1–9. doi: 10.1139/y88-001. [DOI] [PubMed] [Google Scholar]
- Pawlosky RJ, Kashiwaya Y, Srivastava S, King MT, Crutchfield C, Volkow N, Kunos G, Li TK, Veech RL. Alterations in brain glucose utilization accompanying elevations in blood ethanol and acetate concentrations in the rat. Alcohol Clin Exp Res. 2010;34:375–381. doi: 10.1111/j.1530-0277.2009.01099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps ME, Huang SC, Hoffman EJ, Selin C, Sokoloff L, Kuhl DE. Tomographic measurement of local cerebral glucose metabolic rate in humans with (F-18)2-fluoro-2-deoxy-D-glucose: validation of method. Ann Neurol. 1979;6:371–388. doi: 10.1002/ana.410060502. [DOI] [PubMed] [Google Scholar]
- Porrino LJ, Whitlow CT, Samson HH. Effects of the self-administration of ethanol and ethanol/sucrose on rates of local cerebral glucose utilization in rats. Brain Res. 1998;791:18–26. doi: 10.1016/s0006-8993(97)01519-9. [DOI] [PubMed] [Google Scholar]
- Rae C, Fekete AD, Kashem MA, Nasrallah FA, Broer S. Metabolism, Compartmentation, Transport and Production of Acetate in the Cortical Brain Tissue Slice. Neurochem Res. 2012 doi: 10.1007/s11064-012-0847-5. [DOI] [PubMed] [Google Scholar]
- Roine RP, Korri UM, Ylikahri R, Penttila A, Pikkarainen J, Salaspuro M. Increased serum acetate as a marker of problem drinking among drunken drivers. Alcohol Alcohol. 1988;23:123–126. [PubMed] [Google Scholar]
- Schreckenberger M, Amberg R, Scheurich A, Lochmann M, Tichy W, Klega A, Siessmeier T, Grunder G, Buchholz HG, Landvogt C, Stauss J, Mann K, Bartenstein P, Urban R. Acute alcohol effects on neuronal and attentional processing: striatal reward system and inhibitory sensory interactions under acute ethanol challenge. Neuropsychopharmacology. 2004;29:1527–1537. doi: 10.1038/sj.npp.1300453. [DOI] [PubMed] [Google Scholar]
- Singh SP, Snyder AK, Eman S. Effects of ethanol on hexose uptake by cultured rat brain cells. Alcohol Clin Exp Res. 1990;14:741–745. doi: 10.1111/j.1530-0277.1990.tb01238.x. [DOI] [PubMed] [Google Scholar]
- Stinson F, Yi H, Grant B, Chou P, Dawson D, Pickering R. U.S. alcohol epidemiologic data reference manual. 1st ed. vol. 6. Bethesda, MD: National Institute on Alcohol Abuse and Alcoholism; 1998. Nov, Drinking in the United States: Main findings from the 1992 National Longitudinal Alcohol Epidemiologic Survey (NLAES) [Google Scholar]
- Talairach J, Tournoux P. Co-planar Stereotaxic Atlas of the Human Brain. New York, NY: Thieme Medical Publishers; 1988. [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ, Dewey SL, Schlyer D, MacGregor R, Logan J, Alexoff D, Shea C, Hitzemann R, et al. Reproducibility of repeated measures of carbon-11-raclopride binding in the human brain. J Nucl Med. 1993;34:609–613. [PubMed] [Google Scholar]
- Volkow ND, Hitzemann R, Wolf AP, Logan J, Fowler JS, Christman D, Dewey SL, Schlyer D, Burr G, Vitkun S, et al. Acute effects of ethanol on regional brain glucose metabolism and transport. Psychiatry Res. 1990;35:39–48. doi: 10.1016/0925-4927(90)90007-s. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Mullani N, Gould L, Adler SS, Guynn RW, Overall JE, Dewey S. Effects of acute alcohol intoxication on cerebral blood flow measured with PET. Psychiatry Res. 1988;24:201–209. doi: 10.1016/0165-1781(88)90063-7. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Begleiter H, Porjesz B, Fowler JS, Telang F, Wong C, Ma Y, Logan J, Goldstein R, Alexoff D, Thanos PK. High levels of dopamine D2 receptors in unaffected members of alcoholic families: possible protective factors. Arch Gen Psychiatry. 2006a;63:999–1008. doi: 10.1001/archpsyc.63.9.999. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Franceschi D, Fowler JS, Thanos PP, Maynard L, Gatley SJ, Wong C, Veech RL, Kunos G, Kai Li T. Low doses of alcohol substantially decrease glucose metabolism in the human brain. Neuroimage. 2006b;29:295–301. doi: 10.1016/j.neuroimage.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Hitzemann R, Fowler JS, Pappas N, Lowrimore P, Burr G, Pascani K, Overall J, Wolf AP. Depression of thalamic metabolism by lorazepam is associated with sleepiness. Neuropsychopharmacology. 1995;12:123–132. doi: 10.1016/0893-133X(94)00068-B. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Volkow ND, Fowler JS, Franceschi D, Wong CT, Pappas NR, Netusil N, Zhu W, Felder C, Ma Y. Alcohol intoxication induces greater reductions in brain metabolism in male than in female subjects. Alcohol Clin Exp Res. 2003;27:909–917. doi: 10.1097/01.ALC.0000071740.56375.BA. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Volkow ND, Franceschi D, Fowler JS, Thanos PK, Scherbaum N, Pappas N, Wong CT, Hitzemann RJ, Felder CA. Regional brain metabolism during alcohol intoxication. Alcohol Clin Exp Res. 2000;24:822–829. [PubMed] [Google Scholar]
- Wang GJ, Volkow ND, Roque CT, Cestaro VL, Hitzemann RJ, Cantos EL, Levy AV, Dhawan AP. Functional importance of ventricular enlargement and cortical atrophy in healthy subjects and alcoholics as assessed with PET, MR imaging, and neuropsychologic testing. Radiology. 1993;186:59–65. doi: 10.1148/radiology.186.1.8416587. [DOI] [PubMed] [Google Scholar]
- Waniewski RA, Martin DL. Preferential utilization of acetate by astrocytes is attributable to transport. J Neurosci. 1998;18:5225–5233. doi: 10.1523/JNEUROSCI.18-14-05225.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams-Hemby L, Porrino LJ. I. Functional consequences of intragastrically administered ethanol in rats as measured by the 2-[14C]deoxyglucose method. Alcohol Clin Exp Res. 1997;21:1573–1580. [PubMed] [Google Scholar]
- Wyss MT, Magistretti PJ, Buck A, Weber B. Labeled acetate as a marker of astrocytic metabolism. J Cereb Blood Flow Metab. 2011;31:1668–1674. doi: 10.1038/jcbfm.2011.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss MT, Weber B, Treyer V, Heer S, Pellerin L, Magistretti PJ, Buck A. Stimulation-induced increases of astrocytic oxidative metabolism in rats and humans investigated with 1-11C-acetate. J Cereb Blood Flow Metab. 2009;29:44–56. doi: 10.1038/jcbfm.2008.86. [DOI] [PubMed] [Google Scholar]
- Zimatkin S, Lindros KO. A histochemical study of the distribution of aldehyde dehydrogenase activity in brain structures of rats with genetically different alcohol-related behaviour. Alcohol. 1989;6:321–325. doi: 10.1016/0741-8329(89)90090-6. [DOI] [PubMed] [Google Scholar]
- Zimatkin SM, Deitrich RA. Aldehyde dehydrogenase activities in the brains of rats and mice genetically selected for different sensitivity to alcohol. Alcohol Clin Exp Res. 1995;19:1300–1306. doi: 10.1111/j.1530-0277.1995.tb01615.x. [DOI] [PubMed] [Google Scholar]
- Zimatkin SM, Oganesian NA, Kiselevski YV, Deitrich RA. Acetate-dependent mechanisms of inborn tolerance to ethanol. Alcohol Alcohol. 2011;46:233–238. doi: 10.1093/alcalc/agr014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Individual values for the regional brain metabolic measures after placebo and after alcohol in whole brain, cerebellum and occipital cortex (regions with the largest alcohol-induced decreases in metabolism); and in striatum and thalamus (regions where alcohol had the smallest effect).
Supplemental Figure 2. SPM results for the comparisons on the normalized metabolic images showing areas where metabolism was significantly decreased after alcohol consumption when compared with placebo. Parameters set at pu <0.005 and cluster >200 voxels.
Supplemental Figure 3. Self-reports on alcohol effects prior to (0 minutes) and at 10, 15, 30 and 85 minutes after its consumption for the occasional social drinkers (OSD) and the heavy drinkers (HD) and for self-reports of “liking” and “pleasant” scored at the end of the study. The one Factor for Group (OSD vs HD) repeated Drug (0, 10, 15, 30 and 85 minutes) ANOVA showed significant Drug effects for high (F=29, p <0.0001), intoxication (F=28, p <0.0001), sleepiness (F=4, 4,112, p=0.004) and dizzy (f=3.8, p=0.007) but the Interaction Effect was not significant for any of these measures, which indicates that they did not differ between HD and OSD. However, the ratings taken at the end of the study were significantly higher for “liking” for HD than OSD and showed a trend for “pleasant”. Values represent means and SD.
Supplemental Figure 4. Individual values for the SUV measures of [1-11C]acetate after placebo and after alcohol in whole brain, cerebellum (region with the largest alcoholinduced increases); and in striatum and thalamus (regions with the least SUV increases with alcohol).
Supplemental Figure 5. Average time activity curves for the arterial concentration of C-11 for the placebo and the alcohol scans in OSD and in HD subjects.