

Abstract
Alzheimer disease (AD) is the leading cause of age-related dementia, affecting over 5 million people in the United States alone. AD patients suffer from progressive neurodegeneration that gradually impairs their memory, ability to learn, and carry out daily activities. Unfortunately, current therapies for AD are largely palliative and several promising drug candidates have failed in recent clinical trials. There is therefore an urgent need to improve our understanding of AD pathogenesis, create innovative and predictive models, and develop new and effective therapies. In this review we will discuss the potential of stem cells to aid in these challenging endeavors.
Because of the widespread nature of AD pathology, cell replacement strategies have been viewed as an incredibly challenging and unlikely treatment approach. Yet, recent work shows that transplantation of neural stem cells (NSCs) can improve cognition, reduce neuronal loss, and enhance synaptic plasticity in animal models of AD. Interestingly, the mechanisms that mediate these effects appear to involve neuroprotection and trophic support rather than neuronal replacement.
Stem cells may also offer a powerful new approach to model and study AD. Patient-derived induced pluriptotent stem cells (iPSCs), for example, may help to advance our understanding of disease mechanisms. Likewise, studies of human embryonic and neural stem cells are helping to decipher the normal functions of AD-related genes; revealing intriguing roles in neural development.
Keywords: Neurodegeneration, Transplantation, disease modeling, iPSC, pluripotent, synapse, neural development, neurogenesis
Introduction
Alzheimer disease (AD), the most common form of age-related dementia, gradually destroys a person’s memory and ability to learn and reason. As our population ages, the incidence of this disease is expected to grow dramatically such that by 2050 as many as 115 million people worldwide will have developed dementia 1. Despite the prevalence of AD and extensive research, current therapies provide no long-term benefits. There is therefore a critical need to explore new approaches to treat and understand this disorder. In this review, we will first examine the potential use of stem cells to treat Alzheimer disease and discuss key hurdles in the development of a viable therapeutic approach. We will then review the growing use of pluripotent and reprogrammed stem cells to model and investigate this disorder.
The widespread loss of neurons and synaptic connectivity that occurs in AD appears to be driven by the accumulation of toxic species of the beta-amyloid (Aβ) peptide. Studies of both familial AD patients and animal models have provided a great deal of support for the notion that Aβ accumulation drives many of the downstream components of the disease including the development of neurofibrillary tangles, neuronal and synaptic loss, and cognitive dysfunction (Figure 1) 2–5. As a result, AD drug development has focused primarily on targeting Aβ production or enhancing its clearance. Unfortunately, such approaches have thus far failed in late stage clinical trials 6, 7.
Figure 1. Stem cell-based therapies could potentially treat AD by targeting several different stages of disease pathogenesis.

The amyloid cascade hypothesis 2 argues that overproduction and/or decreased clearance of beta-amyloid (Aβ) drives all other downstream components of AD including neurofibrillary tangles, neuronal and synaptic loss, and cognitive dysfunction (blue arrows). While many drugs in development only target the initial accumulation of Aβ, stem cell-based therapies could intervene at multiple stages of this cascade. Neural stem cells in particular (green), can modulate synaptic plasticity and provide robust neuroprotective and neurotrophic activity. Likewise, various stem cell populations can promote anti-inflammatory signals, slowing disease progression. Capitalizing on the unique migratory capacity of NSCs, genetic-modification of stem cells (purple) could also be used to concurrently target Aβ and tangle pathology or enhance neurotrophic and neuroprotective capacity. Given the complex nature of AD, these kind of combinatorial strategies will likely be needed.
Stem cell transplantation for AD?
Research on the potential use of stem cell transplantation for AD has lagged far behind that of many other neurodegenerative disorders, likely as a result of the widespread nature of AD pathology. For many disorders, stem cell-based therapies have aimed to replace missing or defective cells. Transplantation of mesencephalic fetal tissue and neural stem cells for Parkinson’s disease (PD) for example, has aimed to replace dopaminergic neurons of the substantia nigra that degenerate in this disorder 8, 9. Unfortunately, in AD multiple neuronal systems and neurotransmitter phenotypes are affected, making cell-replacement strategies an extremely challenging approach. For effective cell replacement strategies for AD, neural stem cells would first need to migrate to multiple areas of the brain and then differentiate and mature into multiple neuronal subtypes. These neurons would then also need to re-innervate appropriate targets and establish physiologically relevant afferent connectivity, in essence recapitulating much of the complex brain circuitry that develops in utero. Thus, cell-replacement approaches seem unlikely to succeed for a diffuse disorder such as AD. Despite this, recent studies have shown that neural stem cell transplantation can indeed provide meaningful functional benefits in mouse models of AD. In this review we will examine these studies and discuss several key mechanisms by which stem cell transplantation appear to influence AD.
Neurotrophic and neuroprotective activity
Despite the pathognomonic accumulation of plaques and tangles in AD, it is the loss of synapses that correlates most closely with dementia 10. Synapse loss also appears to precede neuronal loss in AD (Braak et al., 1997; Terry et al., 1991). One of the principle mechanisms by which synaptic strength and number is regulated is via the activity-dependent release of target-derived neurotrophins. Likely contributing to the loss of synapses in AD, production of several key neurotrophins is dramatically reduced early in the disease course 11. Among these, brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) in particular play critical functions in synaptic plasticity. BDNF expression and activity is especially important within the entorhinal cortex and hippocampus, two areas critically involved in learning and memory that are dramatically affected in AD. Likewise NGF produced in the cortex and hippocampus influences the survival and function of cholinergic basal forebrain neurons, another important population that influences learning and memory and undergoes dramatic degeneration in AD 12.
Interestingly, neural stem cells (NSCs) can express high levels of neurotrophins including both BDNF and NGF 13–15. Other stem cell populations including mesenchymal stem cells and embryonic stem cells can also produce several neurotrophins 16, 17. Thus, stem cells could provide a means to deliver neurotrophins to the diseased brain, potentially modulating endogenous synaptic plasticity and enhancing neuronal survival (Figure 1). Indeed a growing number of studies support this notion. We, for example previously found that NSC transplantation increases hippocampal synaptic density and improves learning and memory in transgenic models of both AD and neuronal loss 13, 18. This enhancement of synaptic growth was coupled with elevated brain levels of BDNF. In a similar study, Hampton et. al. examined NSC transplantation in a transgenic model of neurofibrillary tangle formation, finding that NSCs could reduce neuronal loss and elevate levels of glial-derived neurotrophic factor (GDNF) within the brain 19. Related to these findings, ex vivo-mediated delivery of NGF has also been shown to improve cognition both in animal models with cholinergic lesions and in a phase 1 trial in AD patients 20, 21. Delivery of NGF via NGF -secreting NSCs has also been shown to be effective at reducing cell loss in models of stroke and excitotoxicity, suggesting that stem cell-derived trophic support could be effective for a variety of conditions 22, 23.
Despite these promising data, it remains possible that stem cell-mediated effects on neurotrophin expression and neuroprotection may simply represent epiphenomena. There is considerable evidence for example that injury alone can trigger the endogenous production of neurotrophins and axonal sprouting 24, 25. Most studies include the injection of vehicle controls to account for this fact, but the use of additional cellular controls and more mechanistic examinations are clearly needed to confirm whether stem cell-mediated delivery of neurotrophins truly drives functional recovery. This problem becomes even more apparent in clinical trials, where the proposed use of control brain injections has led to considerable debate 26. To better understand the potential role of BDNF in NSC-mediated functional improvements, we have used both gain-of-function and loss-of-function approaches in our AD transgenic experiments 13. For example, we found that delivery of recombinant BDNF alone could mimic many of the effects of NSC transplantation. More importantly, when NSC BDNF expression was silenced by shRNAs, these BDNF-depleted NSCs no longer improved cognitive function in AD transgenic mice and showed diminished effects on synaptic plasticity 13. Thus, NSC-mediated delivery of neurotrophins can indeed play an important role in modulating cognition and synaptic plasticity in AD models although additional mechanistic studies are clearly needed.
Anti-inflammatory activity
Chronic inflammation plays an important role in many neurodegenerative diseases including AD 27. Interestingly, certain stem cell populations can exhibit robust anti-inflammatory properties. Mesenchymal stem cells (MSCs) in particular, have been shown to induce expression of anti-inflammatory factors such as interleukin-10 and prostaglandin E2 28, 29. In one study, MSC transplantation was found to attenuate neuroinflammation in transgenic AD mice, improving both pathology and cognitive function 30. Cord-blood derived MSCs have also recently been shown to stimulate microglial production of the Aβ-degrading enzyme neprilysin 31. Even peripheral administration of human cord blood cells has been shown to reduce AD pathology by a mechanism that appears to involve modulation of CD40 signaling 32. While there is growing evidence that MSCs can modulate the immune system, the effects of NSC transplantation on inflammation remains far less clear. Thus far one study has examined the effect of NSCs on AD-associated inflammation, finding that NSCs could reduce migrogliosis and expression of the proinflammatorry cytokine TNFα33. NSCs have also been shown to suppress inflammation in models of multiple sclerosis, although the mechanisms by which this occurs remains unknown 34, 35.
Although stem cell transplantation could potentially modulate AD by altering inflammation (Figure 1), it remains unclear whether suppression of the immune system represents a viable therapeutic target for this disease. Clinical trials of anti-inflammatory drugs in AD patients have for example shown no benefit 36, 37. While a few studies suggest that NSCs and MSCs can positively influence inflammation and pathogenesis in AD models, it is will be important to determine the mechanism by which this occurs and whether stem cell transplantation alters inflammation directly or simply as a result of tissue injury or xenotransplantation-associated artifacts. One approach that could help to resolve these important questions is to begin to study stem cell transplantation in immune-incompetent AD models.
Using NSCs to deliver therapeutic proteins
Although NSCs can improve cognition and modulate synaptic connectivity, they appear to have no effect on the underlying Aβ or neurofibrillary tangle pathology 13, 19. NSCs may therefore loose efficacy as pathology continues to develop unabated. To address this, combinatorial approaches may be needed. For example, supplementing NSC transplantation with Aβ-targeting therapies such as passive Aβ immunization could provide additional long-term benefit. Alternatively, NSCs could themselves be used to provide a powerful approach to deliver therapeutic Aβ-targeting proteins to the brain (Figure 1). It has been well-established that NSCs can migrate throughout the adult brain and localize to areas of injury and inflammation 18, 38. NSCs may therefore represent a promising approach to deliver therapeutic proteins specifically to those brain regions most affected by disease. NSC-mediated delivery of enzymes that degrade Aβ, such as neprilysin, would therefore likely provide considerable additional benefit (Figure 1). Such an approach would likely also prove more effective than viral gene therapy as viral vectors typically infect only a very small region (<0.5mm) of the brain 39. Similar ex vivo gene therapy approaches have been used to enhance the production of neurotrophins by NSCs. For example, NSCs modified to overexpress GDNF improve motor function and prevent neuronal loss in models of both Huntington’s disease (HD) and PD 40, 41. Thus, neural stem cells could provide a powerful tool to deliver therapeutic proteins to those damaged regions of the brain that most need them. One important caveat to this approach is the need to achieve sufficient and long-lasting NSC engraftment. In stroke models for example, the majority of newly grafted cells die, limiting potential efficacy 42. Both the survival and differentiation of NSCs appears to be influenced by the immune response and the underlying disease pathology. Survival rates in xenotransplantation models of spinal cord injury are for example dramatically higher in immune-incompetent models than immune-suppressed rodents 43. Thus far, it remains unclear whether AD-associated pathology influences NSC survival and differentiation. While we detected no difference in murine NSC differentiation rates between wild-type control and AD transgenic transplanted mice 13, transplantation of human NSCs in a different AD transgenic led to increased glial differentiation 44. Such findings are likely influenced by not only the severity of pathology but also the duration of engraftment, host immune response, and the source of NSCs examined.
Endogenous Neurogenesis and Axonal Transport
Growing evidence suggests that adult neurogenesis contributes to learning and memory 45. The generation of new-born neurons within the hippocampus is also dramatically reduced with age and appears further influenced by AD pathogenesis 46, 47. Interestingly, a variety of extrinsic factors can influence adult neurogenesis. Peripheral levels of the chemokine CCL11/eotaxin, for example, can modulate neurogenesis in an age-dependent manner 48. Transplantation of NSCs can also influence endogenous host neurogenesis. In some cases, NSC transplantation has been shown to enhance endogenous neurogenesis following stroke 49, whereas in others, neurogenesis is reduced as a result of increased microglia activation 50. It also appears that the interaction between host neurogenesis and NSC transplants can be reciprocal as changes in neurogenesis can in turn influence NSC graft survival and differentiation 51. To date, no studies have examined the effect of NSC transplantation on endogenous neurogenesis in AD models. However, findings from the stroke field suggest that such mechanisms are likely involved.
Recent data from the stroke field also implicates another mechanism by which NSC transplantation may modulate AD pathogenesis. Following a stroke, NSC transplantation can improve ischemia-induced axonal transport deficits 52. Although a similar analysis in AD models has not yet been performed, there is clear evidence of axonal transport deficits in AD 53. Thus, similar mechanisms could potentially influence AD pathogenesis and cognitive function, although additional studies in AD models are clearly needed.
Hurdles in the development of a stem cell-based approach to treat AD
The preclinical studies discussed above suggest that stem cell based therapies could possibly be developed to treat AD. However, considerable work and appropriate caution is needed before these findings can be translated to clinical trials. Identification of appropriate good manufacturing process (GMP)-compliant human NSC lines that exhibit similar efficacy and safety profiles is clearly needed. Validation of findings using such GMP-compatible lines is also a prerequisite to clinical testing. As AD represents a protracted and progressive disease, examination of efficacy and safety at longer timepoints is also critically needed. It may also be important to further assess the impact of NSC transplantation on AD-associated neuronal loss. Unfortunately, most AD models show very limited neuronal loss 54. Alternative approaches such as neuronal ablation or aggressive AD models such as 5xfAD transgenic mice may therefore need to be used 18, 55.
A major hurdle in achieving these important preclinical studies is the need to establish AD models that allow prolonged human NSC xenotransplantation. Long-term use of common immunosuppressants such as Cyclosporin A of FK506 cause toxicity, especially in aged mice, and can also alter AD-related pathology, complicating interpretation 56, 57. An alternative approach is to utilize AD transgenic models with immune incompetent backgrounds. Unfortunately, no such model has been published to date and thus important questions remain as to whether altering the adaptive immune response will influence AD-associated pathology or cognitive dysfunction.
Investigating AD with human stem cells
The role of AD-associated genes in neural development
The idea of studying AD with pluripotent stem cells initially seems counterintuitive. Taking a cell type that mimics the earliest stages of human development and expecting these cells to inform one about a disease that typically afflicts people over the age of 65, does initially seem at odds with our basic understanding of AD. However, a growing number of studies have shown that many of the genes implicated in AD play critical roles in neural development. Among these, considerable data suggests that amyloid precursor protein (APP), the protein that gives rise to Aβ, is involved in both neurogenesis and neural migration. Combined knockout of APP and its homologue APLP2, for example cause perinatal lethality and cortical dysplasia that closely resembles human lissencephaly 58. Array studies have also shown that both APP and APLP2 single knockouts exhibit significant dysregulation of neurogenenic transcriptional pathways 59. Roles for APP and its derivatives in neurite outgrowth, adult neurogenesis, NSC differentiation, and human ESC differentiation have also recently been shown (Figure 2) 47, 60–62. Thus, APP appears to play important roles in neural development and neurogenesis that can be modeled and examined using human stem cells.
Figure 2. Several key genes implicated in AD play important roles in neural development and neurogenesis.
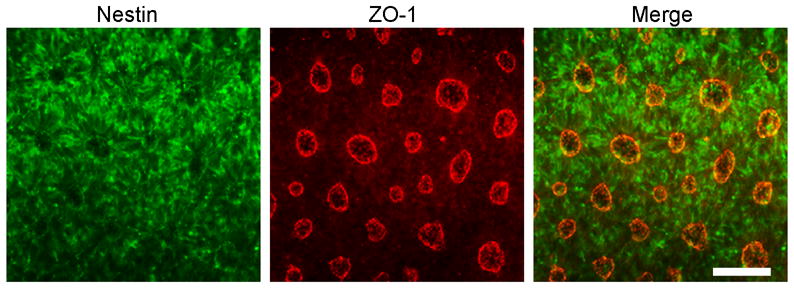
We previously showed that 2-fold overexpression of APP or treatment with recombinant sAPPα/β drives rapid neural differentiation of human ESCs 60. As shown, APP-transgenic human ESCs rapidly differentiate into nestin-positive neural progenitors (green) within just 5 days of manual passaging. Co-labeling for the gap-junction marker zonula occludens-1 (ZO-1, red) and nestin reveals the characteristic formation of neural rosette-like structures. Scale Bar = 120μm. Other studies of embryonic and neural stem cells also show that AD-associated genes mediate important development functions. By studying human stem cells and patient-derived induced pluripotent stem cells we will likely enhance our understanding of not only the pathogenesis of AD but also the development of the human nervous system.
As the catalytic component of the gamma-secretase complex, the AD-associated proteins presenilin-1 and -2 also play a critical role in development by cleaving Notch. It follows that mutations or deletion of presenilins dramatically alter neurogenesis. PS1−/− knockout mice for example, have diminished progenitor populations, a thinning of the ventricular zone, and impaired embryonic neurogenesis 63. Adult neurogenesis is also reduced in PS-1 mutant mice 64. A fourth gene implicated in AD, Apoliprotein E (APOE), also plays a role in neurogenesis, influencing the maintenance of the dentate gyrus neuroprogenitor pool 65. Whereas APOE knockout or knockin of the more pathogenic ApoE4 allele impairs neurogenesis, the human ApoE3 allele does not 66. Thus, it appears that many of the genes implicated in AD play important roles in neural development. Whether these functions contribute to the development or pathogenesis of AD remains unknown but it is intriguing to speculate that altered neurogenesis may influence the development or progression of AD.
Studying APP with human ESCs
APP, presenilin, and ApoE transgenic and knockout mouse models have clearly taught us a great deal about the function of these genes and their role in AD pathogenesis. However, it is reasonable to conclude that genetic differences between humans and mice have also contributed to our difficulties in accurately modeling AD and generating clinically predictive data. Human stem cell based models may therefore provide an alternative approach to clarify both the normal and pathogenic roles of AD-associated genes. Along those lines, we recently generated hESC clones that overexpress wild-type or mutant forms of human APP 60. Surprisingly, we found that all of the resulting APP hES clones rapidly and spontaneously differentiated toward a neural lineage (Figure 2). Despite maintenance in standard hESC media, up to 80% of cells expressed the neural stem cell marker nestin and 65% exhibited the more mature neural marker TUJ1 within just 5 days of manual passaging. In comparison, standard protocols typically require > 3 weeks to achieve similar amounts of neural differentiation. To understand the mechanism by which this occurred we examined the various cleavage products that result from APP proteolysis, revealing that soluble secreted N-terminal fragments of APP (sAPPα and sAPPβ) were critical for this effect 60. Thus, our findings further support the notion that AD associated genes may play important roles in neural development.
Modeling AD with induced pluripotent stem cells (iPSCs)
Genetic studies of early-onset AD identified mutations in APP, Presenilin 1, and Presenilin 2 as driving the development of familial AD. In contrast, sporadic AD does not result from a single highly penetrant genetic cause. Nevertheless, twin studies reveal that the heritability of sporadic AD is as high as 79% 67. Polymorphisms in the Apolipoprotein E gene clearly provide the greatest single genetic influence on sporadic AD risk 68, 69. However, recent genome-wide association studies (GWAS) have identified several other genes that can influence the development of sporadic AD 70, 71. Thus, genetics clearly plays an important albeit complex role in sporadic AD that could potentially be modeled with induced pluripotent stem cells (iPSCs).
Patient-derived iPSCs have been used to model a growing list of human genetic disorders 72. Most recently, two groups have reported the establishment and investigation of AD iPSCs 73, 74. In one study, Yagi et. al., generated iPSCs from patients carrying familial mutations in PS1 or PS2 74. The patient-derived iPSCs recapitulated an important aspect of familial AD; altered generation of Aβ42 versus Aβ40. This report also gave an example of the potential use of AD iPSCs for testing drug efficacy. In a second study, Israel et al., generated iPSCs from not only fAD patients but also 2 cases of sporadic AD and 2 unaffected controls 73. Interestingly, neurons derived from one of the sporadic AD cases mimicked some of the findings from fAD cases; showing increased Aβ40 generation and tau phosphorylation, activation of glycogen synthase kinase-3β, and accumulation of enlarged early endosomes. Importantly, these disease-associated phenotypes were not detected in fibroblast cultures from this case, demonstrating the importance of studying these phenotypes in iPSC-derived neurons. Taken together these studies represent critical first steps in assessing the potential of AD iPSCs to model AD. As Israel and colleagues point out, generation of many additional sporadic AD iPSC lines will of course be needed to fully establish the ability of this approach to guide drug development and enhance our understanding of AD. The generation of directly reprogrammed induced NSCs (iNSCs) from AD patients may also help to accelerate such research.
Conclusions
In this review, we have discussed many of the current studies that examine the use of stem cells to treat and model AD. A growing amount of evidence suggests that stem cell based-therapies could prove beneficial in AD, albeit via indirect mechanisms rather than cellular replacement (Figure 1). Studies of embryonic, neural, and iPSCs are also beginning to unravel the normal and pathogenic function of AD-associated genes and may provide powerful new approaches to model this disorder (Figure 2). Future work will hopefully clarify the potential of stem cells to treat AD and decipher the complex genetic differences that predispose a person to developing this devastating disease.
Acknowledgments
This work was supported by NIH grant AG029378 and AG16573 (MBJ). Many thanks to Dr. Wayne Poon for his critical review of the manuscript.
Footnotes
Competing interests:
The authors declare that they have no competing interests.
References
- 1.Prince MJJ, Ferri CP, Sousa R, Albanese E, Ribeiro WS, Honyashiki M, editors. World Alzheimer Report. Alzheimer’s Disease International; 2009. [Google Scholar]
- 2.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 3.Cruts M, Van Broeckhoven C. Molecular genetics of Alzheimer’s disease. Ann Med. 1998;30:560–565. doi: 10.3109/07853899809002605. [DOI] [PubMed] [Google Scholar]
- 4.Blurton-Jones M, Laferla FM. Pathways by which Aβ facilitates tau pathology. Curr Alzheimer Res. 2006;3:437–448. doi: 10.2174/156720506779025242. [DOI] [PubMed] [Google Scholar]
- 5.Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 6.Golde TE, Schneider LS, Koo EH. Anti-abeta therapeutics in Alzheimer’s disease: the need for a paradigm shift. Neuron. 2011;69:203–213. doi: 10.1016/j.neuron.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Selkoe DJ. Resolving controversies on the path to Alzheimer’s therapeutics. Nat Med. 2011;17:1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- 8.Lindvall O, Rehncrona S, Gustavii B, et al. Fetal dopamine-rich mesencephalic grafts in Parkinson’s disease. Lancet. 1988;2:1483–1484. doi: 10.1016/s0140-6736(88)90950-6. [DOI] [PubMed] [Google Scholar]
- 9.Morizane A, Li JY, Brundin P. From bench to bed: the potential of stem cells for the treatment of Parkinson’s disease. Cell Tissue Res. 2008;331:323–336. doi: 10.1007/s00441-007-0541-0. [DOI] [PubMed] [Google Scholar]
- 10.Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 11.Arancio O, Chao MV. Neurotrophins, synaptic plasticity and dementia. Curr Opin Neurobiol. 2007;17:325–330. doi: 10.1016/j.conb.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 12.Cuello AC, Bruno MA, Bell KF. NGF-cholinergic dependency in brain aging, MCI and Alzheimer’s disease. Curr Alzheimer Res. 2007;4:351–358. doi: 10.2174/156720507781788774. [DOI] [PubMed] [Google Scholar]
- 13.Blurton-Jones M, Kitazawa M, Martinez-Coria H, et al. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A. 2009;106:13594–13599. doi: 10.1073/pnas.0901402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kamei N, Tanaka N, Oishi Y, et al. BDNF, NT-3, and NGF released from transplanted neural progenitor cells promote corticospinal axon growth in organotypic cocultures. Spine (Phila Pa 1976) 2007;32:1272–1278. doi: 10.1097/BRS.0b013e318059afab. [DOI] [PubMed] [Google Scholar]
- 15.Lu P, Jones LL, Snyder EY, et al. Neural stem cells constitutively secrete neurotrophic factors and promote extensive host axonal growth after spinal cord injury. Exp Neurol. 2003;181:115–129. doi: 10.1016/s0014-4886(03)00037-2. [DOI] [PubMed] [Google Scholar]
- 16.Kim HJ, Lee JH, Kim SH. Therapeutic effects of human mesenchymal stem cells on traumatic brain injury in rats: secretion of neurotrophic factors and inhibition of apoptosis. J Neurotrauma. 2009;27:131–138. doi: 10.1089/neu.2008.0818. [DOI] [PubMed] [Google Scholar]
- 17.Bentz K, Molcanyi M, Riess P, et al. Embryonic stem cells produce neurotrophins in response to cerebral tissue extract: Cell line-dependent differences. J Neurosci Res. 2007;85:1057–1064. doi: 10.1002/jnr.21219. [DOI] [PubMed] [Google Scholar]
- 18.Yamasaki TR, Blurton-Jones M, Morrissette DA, et al. Neural stem cells improve memory in an inducible mouse model of neuronal loss. J Neurosci. 2007;27:11925–11933. doi: 10.1523/JNEUROSCI.1627-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hampton DW, Webber DJ, Bilican B, et al. Cell-mediated neuroprotection in a mouse model of human tauopathy. J Neurosci. 2010;30:9973–9983. doi: 10.1523/JNEUROSCI.0834-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tuszynski MH, Roberts J, Senut MC, et al. Gene therapy in the adult primate brain: intraparenchymal grafts of cells genetically modified to produce nerve growth factor prevent cholinergic neuronal degeneration. Gene Ther. 1996;3:305–314. [PubMed] [Google Scholar]
- 21.Tuszynski MH, Thal L, Pay M, et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med. 2005;11:551–555. doi: 10.1038/nm1239. [DOI] [PubMed] [Google Scholar]
- 22.Andsberg G, Kokaia Z, Bjorklund A, et al. Amelioration of ischaemia-induced neuronal death in the rat striatum by NGF-secreting neural stem cells. Eur J Neurosci. 1998;10:2026–2036. doi: 10.1046/j.1460-9568.1998.00214.x. [DOI] [PubMed] [Google Scholar]
- 23.Martinez-Serrano A, Bjorklund A. Protection of the neostriatum against excitotoxic damage by neurotrophin-producing, genetically modified neural stem cells. J Neurosci. 1996;16:4604–4616. doi: 10.1523/JNEUROSCI.16-15-04604.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steward O, Cotman CW, Lynch GS. Growth of a new fiber projection in the brain of adult rats: Re-innervation of the dentate gyrus by the contralateral entorhinal cortex following ipsilateral entorhinal lesions. Exp Brain Res. 1974;20:45–66. doi: 10.1007/BF00239017. [DOI] [PubMed] [Google Scholar]
- 25.Cafferty WB, McGee AW, Strittmatter SM. Axonal growth therapeutics: regeneration or sprouting or plasticity? Trends Neurosci. 2008;31:215–220. doi: 10.1016/j.tins.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galpern WR, Corrigan-Curay J, Lang AE, et al. Sham neurosurgical procedures in clinical trials for neurodegenerative diseases: scientific and ethical considerations. Lancet Neurol. 2012;11:643–650. doi: 10.1016/S1474-4422(12)70064-9. [DOI] [PubMed] [Google Scholar]
- 27.Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Y, Yuan J, Zhou B, et al. The therapeutic efficacy of human adipose tissue-derived mesenchymal stem cells on experimental autoimmune hearing loss in mice. Immunology. 2011;133:133–140. doi: 10.1111/j.1365-2567.2011.03421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ylostalo JH, Bartosh TJ, Coble K, et al. Human Mesenchymal Stem/Stromal Cells (hMSCs) Cultured as Spheroids are Self-activated to Produce Prostaglandin E2 (PGE2) that Directs Stimulated Macrophages into an Anti-inflammatory Phenotype. Stem Cells. 2012 doi: 10.1002/stem.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee HJ, Lee JK, Lee H, et al. Human umbilical cord blood-derived mesenchymal stem cells improve neuropathology and cognitive impairment in an Alzheimer’s disease mouse model through modulation of neuroinflammation. Neurobiol Aging. 2010;33:588–602. doi: 10.1016/j.neurobiolaging.2010.03.024. [DOI] [PubMed] [Google Scholar]
- 31.Kim JY, Kim DH, Kim JH, et al. Soluble intracellular adhesion molecule-1 secreted by human umbilical cord blood-derived mesenchymal stem cell reduces amyloid-beta plaques. Cell Death Differ. 2011;19:680–691. doi: 10.1038/cdd.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nikolic WV, Hou H, Town T, et al. Peripherally administered human umbilical cord blood cells reduce parenchymal and vascular beta-amyloid deposits in Alzheimer mice. Stem Cells Dev. 2008;17:423–439. doi: 10.1089/scd.2008.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ryu JK, Cho T, Wang YT, et al. Neural progenitor cells attenuate inflammatory reactivity and neuronal loss in an animal model of inflamed AD brain. J Neuroinflammation. 2009;6:39. doi: 10.1186/1742-2094-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fainstein N, Vaknin I, Einstein O, et al. Neural precursor cells inhibit multiple inflammatory signals. Mol Cell Neurosci. 2008;39:335–341. doi: 10.1016/j.mcn.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 35.Giannakopoulou A, Grigoriadis N, Polyzoidou E, et al. Inflammatory changes induced by transplanted neural precursor cells in a multiple sclerosis model. Neuroreport. 2011;22:68–72. doi: 10.1097/WNR.0b013e32834272eb. [DOI] [PubMed] [Google Scholar]
- 36.Green RC, Schneider LS, Amato DA, et al. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA. 2009;302:2557–2564. doi: 10.1001/jama.2009.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bentham P, Gray R, Sellwood E, et al. Aspirin in Alzheimer’s disease (AD2000): a randomised open-label trial. Lancet Neurol. 2008;7:41–49. doi: 10.1016/S1474-4422(07)70293-4. [DOI] [PubMed] [Google Scholar]
- 38.Muller FJ, Snyder EY, Loring JF. Gene therapy: can neural stem cells deliver? Nat Rev Neurosci. 2006;7:75–84. doi: 10.1038/nrn1829. [DOI] [PubMed] [Google Scholar]
- 39.de Backer MW, Brans MA, Luijendijk MC, et al. Optimization of adeno-associated viral vector-mediated gene delivery to the hypothalamus. Hum Gene Ther. 2010;21:673–682. doi: 10.1089/hum.2009.169. [DOI] [PubMed] [Google Scholar]
- 40.Ebert AD, Barber AE, Heins BM, et al. Ex vivo delivery of GDNF maintains motor function and prevents neuronal loss in a transgenic mouse model of Huntington’s disease. Exp Neurol. 2010;224:155–162. doi: 10.1016/j.expneurol.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 41.Emborg ME, Ebert AD, Moirano J, et al. GDNF-secreting human neural progenitor cells increase tyrosine hydroxylase and VMAT2 expression in MPTP-treated cynomolgus monkeys. Cell Transplant. 2008;17:383–395. [PubMed] [Google Scholar]
- 42.Lindvall O, Kokaia Z. Recovery and rehabilitation in stroke: stem cells. Stroke. 2004;35:2691–2694. doi: 10.1161/01.STR.0000143323.84008.f4. [DOI] [PubMed] [Google Scholar]
- 43.Anderson AJ, Haus DL, Hooshmand MJ, et al. Achieving stable human stem cell engraftment and survival in the CNS: is the future of regenerative medicine immunodeficient? Regen Med. 2011;6:367–406. doi: 10.2217/rme.11.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marutle A, Ohmitsu M, Nilbratt M, et al. Modulation of human neural stem cell differentiation in Alzheimer (APP23) transgenic mice by phenserine. Proc Natl Acad Sci U S A. 2007;104:12506–12511. doi: 10.1073/pnas.0705346104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deng W, Aimone JB, Gage FH. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci. 2010;11:339–350. doi: 10.1038/nrn2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;16:2027–2033. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lazarov O, Mattson MP, Peterson DA, et al. When neurogenesis encounters aging and disease. Trends Neurosci. 2010;33:569–579. doi: 10.1016/j.tins.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Villeda SA, Luo J, Mosher KI, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477:90–94. doi: 10.1038/nature10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jin K, Xie L, Mao X, et al. Effect of human neural precursor cell transplantation on endogenous neurogenesis after focal cerebral ischemia in the rat. Brain Res. 2011;1374:56–62. doi: 10.1016/j.brainres.2010.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Minnerup J, Kim JB, Schmidt A, et al. Effects of neural progenitor cells on sensorimotor recovery and endogenous repair mechanisms after photothrombotic stroke. Stroke. 2011;42:1757–1763. doi: 10.1161/STROKEAHA.110.599282. [DOI] [PubMed] [Google Scholar]
- 51.Madhavan L, Daley BF, Sortwell CE, et al. Endogenous neural precursors influence grafted neural stem cells and contribute to neuroprotection in the parkinsonian rat. Eur J Neurosci. 2012;35:883–895. doi: 10.1111/j.1460-9568.2012.08019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Andres RH, Horie N, Slikker W, et al. Human neural stem cells enhance structural plasticity and axonal transport in the ischaemic brain. Brain. 2011;134:1777–1789. doi: 10.1093/brain/awr094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stokin GB, Lillo C, Falzone TL, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- 54.Zahs KR, Ashe KH. ‘Too much good news’ - are Alzheimer mouse models trying to tell us how to prevent, not cure, Alzheimer’s disease? Trends Neurosci. 2010;33:381–389. doi: 10.1016/j.tins.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 55.Oakley H, Cole SL, Logan S, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 57.Mollison KW, Fey TA, Krause RA, et al. Nephrotoxicity studies of the immunosuppressants tacrolimus (FK506) and ascomycin in rat models. Toxicology. 1998;125:169–181. doi: 10.1016/s0300-483x(97)00167-4. [DOI] [PubMed] [Google Scholar]
- 58.Herms J, Anliker B, Heber S, et al. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 2004;23:4106–4115. doi: 10.1038/sj.emboj.7600390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aydin D, Filippov MA, Tschape JA, et al. Comparative transcriptome profiling of amyloid precursor protein family members in the adult cortex. BMC Genomics. 2011;12:160. doi: 10.1186/1471-2164-12-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Freude KK, Penjwini M, Davis JL, et al. Soluble amyloid precursor protein induces rapid neural differentiation of human embryonic stem cells. J Biol Chem. 2011;286:24264–24274. doi: 10.1074/jbc.M111.227421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Young-Pearse TL, Chen AC, Chang R, et al. Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin beta1. Neural Dev. 2008;3:15. doi: 10.1186/1749-8104-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kwak YD, Brannen CL, Qu T, et al. Amyloid precursor protein regulates differentiation of human neural stem cells. Stem Cells Dev. 2006;15:381–389. doi: 10.1089/scd.2006.15.381. [DOI] [PubMed] [Google Scholar]
- 63.Shen J, Bronson RT, Chen DF, et al. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- 64.Wen PH, Hof PR, Chen X, et al. The presenilin-1 familial Alzheimer disease mutant P117L impairs neurogenesis in the hippocampus of adult mice. Exp Neurol. 2004;188:224–237. doi: 10.1016/j.expneurol.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 65.Yang CP, Gilley JA, Zhang G, et al. ApoE is required for maintenance of the dentate gyrus neural progenitor pool. Development. 2011;138:4351–4362. doi: 10.1242/dev.065540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li G, Bien-Ly N, Andrews-Zwilling Y, et al. GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell. 2009;5:634–645. doi: 10.1016/j.stem.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168–174. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- 68.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 69.Strittmatter WJ, Saunders AM, Schmechel D, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 72.Grskovic M, Javaherian A, Strulovici B, et al. Induced pluripotent stem cells--opportunities for disease modelling and drug discovery. Nat Rev Drug Discov. 2011;10:915–929. doi: 10.1038/nrd3577. [DOI] [PubMed] [Google Scholar]
- 73.Israel MA, Yuan SH, Bardy C, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yagi T, Ito D, Okada Y, et al. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20:4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]