

Abstract
The major event initiating atherosclerosis is hypercholesterolemia-induced disruption of vascular endothelium integrity. In settings of endothelial damage, endothelial progenitor cells (EPCs) are mobilized from bone marrow into circulation and home to sites of vascular injury where they aid endothelial regeneration. Given the beneficial effects of EPCs in vascular repair, we hypothesized that these cells play a pivotal role in atherosclerosis regression. We tested our hypothesis in the atherosclerosis-prone mouse model in which hypercholesterolemia, one of the main factors affecting EPC homeostasis, is reversible (Reversa mice). In these mice normalization of plasma lipids decreased atherosclerotic burden; however, plaque regression was incomplete. To explore whether endothelial progenitors contribute to atherosclerosis regression, bone marrow EPCs from a transgenic strain expressing green fluorescent protein under the control of endothelial cell-specific Tie2 promoter (Tie2-GFP+) were isolated. These cells were then adoptively transferred into atheroregressing Reversa recipients where they augmented plaque regression induced by reversal of hypercholesterolemia. Advanced plaque regression correlated with engraftment of Tie2-GFP+ EPCs into endothelium and resulted in an increase in atheroprotective nitric oxide and improved vascular relaxation. Similarly augmented plaque regression was also detected in regressing Reversa mice treated with the stem cell mobilizer AMD3100 which also mobilizes EPCs to peripheral blood. We conclude that correction of hypercholesterolemia in Reversa mice leads to partial plaque regression that can be augmented by AMD3100 treatment or by adoptive transfer of EPCs. This suggests that direct cell therapy or indirect progenitor cell mobilization therapy may be used in combination with statins to treat atherosclerosis.
Keywords: atherosclerosis regression, CXCR4 antagonist AMD3100, bone marrow endothelial progenitors
INTRODUCTION
Atherosclerosis and its complications remain a major cause of mortality worldwide. Deposition of low density lipoprotein (LDL) in the vascular subendothelium and the pro-inflammatory reactions of resident cells, which trigger influx of inflammatory leukocytes into the vascular wall, both lead to development of atherosclerotic plaques that can rupture, causing myocardial infarction and stroke [1]. Statins (HMG-CoA reductase inhibitors) are the primary intervention against atherosclerosis. In patients at risk or suffering from cardiovascular disease, statins effectively lower plasma cholesterol, impede atherosclerosis progression, stabilize plaques and moderately reduce adverse cardiovascular events [2]. However, recent clinical trials show that long-term administration of statins causes incomplete plaque regression, leaving patients prone to adverse cardiovascular events [3]. Thus, understanding of mechanisms that promote complete plaque resolution is important for the development of novel or improved anti-atherosclerotic therapies.
Mechanisms of atherosclerosis regression remain poorly understood. This is in part due to poor availability of animal models in which atherosclerosis risk factors such as hypercholesterolemia can be reversed. Atherosclerotic plaque regression is observed in the Reversa model (Ldlr−/−ApoB100/100Mttpfl/flMx1-Cre) [4]. Reversa mice lack low density lipoprotein receptor (Ldlr−/−) and express atherogenic apolipoprotein B 100 (ApoB100). Diet-induced hyperlipidemia causes aortic atherosclerosis reminiscent of that in apolipoprotein E-deficient (ApoE−/−) and Ldlr−/− strains. However, hypercholesterolemia is dramatically reversed upon Cre-dependent inactivation of the hepatic microsomal triglyceride transfer protein (Mttp) which assembles atherogenic LDL [5]. The Reversa model is ideal for studying regression of atherosclerosis because genetic inactivation of Mttp results in moderate lowering of plasma lipids [5] that mimics statin-induced correction of high cholesterol levels in patients. We have used the Reversa model to identify mechanisms that augment plaque regression induced by normalization of plasma lipids, with special interest paid to the contributions of endothelial progenitor cells (EPCs) to this process.
EPCs can have hematopoietic or non-hematopoietic origin. Hematopoietic EPCs, which are derived from bone marrow, comprise a heterogeneous cell population represented by colony forming EPCs, non-colony forming differentiating EPCs, myeloid EPCs and angiogenic cells. These EPCs are mainly found in circulation and are pro-vasculogenic [6–8]. Non-hematopoietic EPCs can be isolated from blood or tissue samples [9]. The origin of non-hematopoietic EPCs is unclear [10].
Bone marrow EPCs are thought to have an important role in tissue regeneration, especially in vascular repair. These cells support postnatal neovascularization by homing to and differentiating into mature CD31+ endothelial cells in damaged endothelium, promoting vasculogenesis, thereby directly contributing to endothelial regeneration [11;12]. EPCs may also produce angiogenic cytokines and growth factors that promote proliferation of existing resident endothelial cells, activate angiogenesis, and thereby indirectly contribute to the re-establishment of endothelial homeostasis [12–15].
In this report we show that treatment with either EPCs or stem cell mobilizer AMD3100 augments the partial plaque regression induced by reversal of hypercholesterolemia, demonstrating that EPCs play a pivotal role in regression of atherosclerosis.
MATERIALS AND METHODS
Experimental designs
Reversa mice (C57BL/6 background) were obtained from Jackson Laboratory (Bar Harbor, ME). Mice of both sexes were used for all experiments. Mice were separated into 3 groups immediately after weaning. All groups were fed a Western diet (21% anhydrous milkfat/butterfat, 34% sucrose, 0.2% cholesterol; Harlan Teklad) for 84 days. The atherosclerosis-resistant group was given four intraperitoneal injections of polyinosinic-polycytidylic acid (pI-pC; 500 μg/mouse; Sigma, St Louis, MO) at two-day intervals to inactivate Mttp after weaning. The atherosclerosis-prone group received no pI-pC treatment. The atheroregressing group was treated four times with pI-pC after 84 days on the Western diet. Animals were sacrificed 16, 42 or 70 days after Mttp inactivation in the atheroregressing group (Suppl. Fig. 1).
For adoptive transfer of EPCs, bone marrow Lin− cells were obtained under sterile conditions from long bones of Tie2-GFP+ donor mice (C57BL/6 background, Jackson Laboratory). Regressing Reversa recipients were not splenectomized prior to adoptive transfer because we monitored long-term effects of bone marrow EPCs on plaque regression. Recipients were injected intravenously three times (10, 20 and 30 days after reversal of hypercholesterolemia) with 1×106 donor cells and mice were sacrificed 70 days after normalization of plasma lipids.
To determine how mobilization of bone marrow progenitors affects plaque regression, Reversa mice were fed the Western Diet for 84 days, injected with pI-pC to reverse hypercholesterolemia and separated into two groups. The first group received 2.5 mg/kg of AMD3100 (Sigma) daily for a total of 70 days through an osmotic minipump implanted subcutaneously on the back, slightly posterior to the scapulae. Control mice comprising the second group received vehicle (saline) alone. Mice were sacrificed after 70 days of AMD3100 or saline treatment.
All animal care and experimental protocols were approved by the Institutional Animal Care and Use Committee of the Oklahoma Medical Research Foundation.
Atherosclerosis evaluation
Atherosclerotic plaques were evaluated by en face analysis of pinned-open aortas and by cross-sectional analysis of the proximal aorta. Aortas were excised, opened longitudinally, branching vessels were removed and aortas were pinned out flat, and stained in Oil red O solution (Newcomer Supply, Middleton, WI). Aortic images were captured with a Canon EOS-1 Mark II digital camera and analyzed using Adobe Photoshop CS. Lesional surface was measured using NIH Image J software. The size of lesions was calculated using the following formula: Oil Red+ area / aortic arch area × 100 and was expressed as percent of the total aortic arch area. Number of lesions was determined by counting atherosclerotic lesions in the whole aorta.
For cross-sectional analysis, aortas were excised and fixed, serially cryosectioned, stained with Oil Red O and counterstained with hematoxylin. Eight sections per site, collected at 40-μm intervals, were examined under the light microscope. Images were obtained with a Zeiss AxioCam MRC 12-bit color digital camera (Carl Zeiss Microimaging, Thornwood, NY). The lesional surface was measured using NIH Image J software and is expressed in μm2.
Immunofluorescence
Lesional foamy macrophages were detected with rat anti-mouse Moma 2 antibody (Ab) (clone MCA519G, Serotec, Raleigh, NC) or isotype rat anti-mouse IgG2a Ab (Abcam, Cambridge, MA) followed by staining with donkey anti-rat Alexa Fluor 488 Ab (Life Technologies, Grand Island, NY). Foamy macrophage-positive areas were determined by measuring Moma 2+ areas using the NIH Image J software and are expressed as percent of the total lesional area.
To examine incorporation of Tie2-GFP+ bone marrow EPCs into endothelium, aortic cross-sections obtained from regressing Reversa mice treated with EPCs were stained with primary rat anti-mouse CD31 (BD Biosciences, San Jose, CA) or control rat IgG2a Ab (Abcam) and secondary goat anti-rat IgG Alexa Fluor 568 Ab (Life Technologies).
To determine whether Tie2-GFP+ EPCs incorporate into vascular endothelium randomly or predominantly into areas of regressing plaques, aortic cross-sections were stained with a lipophilic stain Nile Red (Sigma). Nile Red was diluted in ethanol, which shifted its excitation and emission from 485 nm and 525 nm to 559 nm and 629 nm [16;17].
Abs and respective isotype controls to detect CXCL1, ICAM-1 and VCAM-1 in plaques of atherosclerosis-prone and atheroregressing mice treated with saline or EPCs were obtained from BD Biosciences. Aortic cross-sections were following incubation with primary Abs stained with the secondary donkey anti-rat Alexa Fluor 488 or secondary goat anti-rat IgG Alexa Fluor 568 Abs.
EPC isolation and culture
Bone marrow was flushed from long bones of Tie2-GFP+ mice with sterile Hanks’ balanced salt solution, low density cells were collected by centrifugation over Histopaque 1083. Mononuclear lineage-negative (Lin−) cells were enriched by lineage-depletion coktail containing monoclonal Abs to mouse CD3, CD11b, CD45/B220, Ly-6G and Ly-6C and TER-119 (BD Biosciences, San Jose, CA), and the Lin−GFP+ population was isolated by fluorescein-activated cell sorting (FACS) using a FACSAria cell sorter (BD Biosciences). Lin−GFP+ cells were plated on fibronectin-coated 24-well plates at a density of 5×105cells/well. They were then cultured for 4 days in endothelial cell growth medium (Lonza, Allendale, NJ) supplemented with fetal bovine serum, Gentamycin/Amphotericin, vascular endothelial growth factor (VEGF) and fibroblast growth factor. All growth factors were from Sigma. Ex vivo-expanded EPCs were subjected to flow cytometry analysis and microscopic evaluation prior to injection into recipient Reversa mice.
Flow cytometry
To determine circulating EPC number, blood was collected by retro-orbital bleeding with heparin-coated capillary tubes from hypercholesterolemic controls and regressing Reversa mice treated with AMD3100 or saline. Heparinized blood was emptied into glass centrifuge tubes containing a 2% Dextran solution, mixed and the mixture was incubated to allow sedimentation of red blood cells. The upper white blood cell-enriched phase was removed. Cells were washed, resuspended in hypotonic NH4Cl solution to remove erythrocytes and stained for c-Kit (eBiosciences, San Diego, CA), Flk1 (VEGF receptor 2, VEGFR2; BD Biosciences) and CD11b (BD Biosciences) Abs. Gate was set on CD11b− cells and c-Kit+Flk1+ cell count was determined by flow cytometry. Dead cells were excluded by propidium iodide staining (Life Technologies, Eugene, OR).
Alternative analysis to determine circulating EPCs was also performed by staining cells with monoclonal anti-CD34 (BD Biosciences), anti-Flk1 and anti-CD133 (BioLegend, San Diego, CA) Abs. Gate was set on Flk1+ cells and the number of viable CD34+CD133+ cells was determined by flow cytometry. The contaminant mature TER-119+ and B220+ white blood cells were excluded from analysis.
To determine the phenotype of ex-vivo expanded Lin−Tie2-GFP+ EPCs prior to adoptive transfer, cells were confirmed to express EPC markers by staining with anti–CD133 (BioLegend), anti–Flk1 or anti–Sca-1 (BD Biosciences) Abs. EPC identity was also confirmed by testing cells for the uptake of TRITC-Lectin (Sigma) and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylin-docarbocyanine-labelled acetylated LDL (Dil-Ac-LDL; Life Technologies).
Flow cytometry was performed on a BD LSRII (BD Biosciences), correcting for nonspecific staining with isotype Ab controls. FlowJo software (Tree Star, San Carlos, CA) was used for data analysis.
Plasma analysis
Plasma samples were collected after an overnight fast. Total cholesterol, LDL, HDL and triglycerides were determined by colormetric assays (BioVision Research Products, Mountain View, CA). Plasma nitrite, a reservoir of atheroprotective NO [18;19], and plasma nitrate, a measure of NO production [20;21], were determined using a colorimetric NO metabolite detection kit (Cayman Chemical, Ann Arbor, MI).
Vascular relaxation
Aortas were removed from hypercholesterolemic and regressing Reversa mice that received Tie2-GFP+ EPCs, cut into 3 mm-wide rings and pre-contracted with 30 nmol/L U45519 in organ chambers (PowerLab ADInstruments, Colorado Springs, CO). Endothelium-dependent and -independent vasodilation responses were monitored in the presence of acetylcholine (ACh) and sodium nitroprusside (SNP) respectively [22].
Statistical analysis
Experiments were repeated at least three times using 4–6 mice per each group. Dataare presented as median ± SEM. The statistical significance of differences between two groups was tested by the non-parametric Mann-Whitney U test using the GraphPad Prism 5.04 program (GraphPad Software, La Jolla, CA). Values of p<0.05 were considered statistically significant. In the Figure 3B the alternative flow cytometry evaluation of circulating EPCs was performed once with a minimal number of n=2 mice per group.
Figure 3. AMD3100 markedly increases circulating EPC availability.
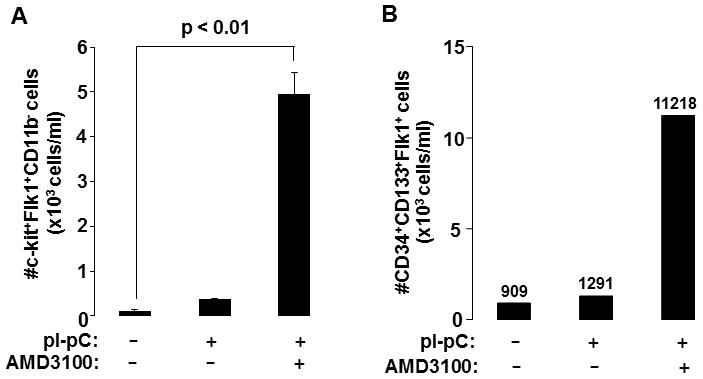
Reversa mice fed the Western diet for 84 days were left hypercholesterolemic (−pI-pC) or were injected with pI-pC and sacrificed 16 days post Mttp inactivation. Atheroregressing mice (+pI-pC) were treated with saline or AMD3100. Peripheral blood was collected by the retro-orbital bleed. Circulating mononuclear cells were separated, immunostained and absolute numbers of (A) c-kit+Flk1+ in the CD11b− population or the absolute number of (B) CD34+CD133+ in the Flk1+ population were determined. Numerical values in B indicate the average absolute number of CD34+CD133+ Flk1+ cells in each group.
RESULTS
Normalization of plasma lipids reduces atherosclerosis burden in Reversa mice
Feig and colleagues described use of Reversa mice to study regression of advanced atherosclerotic plaques [4]. However, the existence, cellular composition, and features of advanced plaques in atherosclerosis-prone mouse models remain controversial [23]. To overcome this problem, we developed a new experimental protocol to monitor plaque regression in these mice. Our experimental approach involved feeding Reversa mice with the atherosclerosis-inducing Western diet (21% anhydrous milkfat/butterfat, 34% sucrose, 0.2% cholesterol) for 84 days. With this feeding protocol, mice developed plaques that contain predominantly foamy macrophages (Suppl. Fig. 2). These plaques are, in content and features, similar to lesions in well-characterized atherosclerosis-prone ApoE−/− and Ldlr−/− strains [5].
Mice of both genders were grouped into atherosclerosis-resistant, atherosclerosis-prone and atheroregressing groups as illustrated in Suppl. Fig. 1. Immediately after weaning, the atherosclerosis-resistant group was given four intraperitoneal injections of pI-pC to inactivate Mttp. The atherosclerosis-prone group was not injected with pI-pC. After 84 days on the Western diet, Mttp was inactivated in the atheroregressing group. We assessed plasma lipids and atherosclerosis burden in all three groups at 16, 42 and 70 days after Mttp inactivation in the atheroregressing group.
Consistent with previous findings [5], we observed that pI-pC injections beginning just after weaning rendered mice resistant to Western diet-induced hypercholeresterolemia. Failure to inactivate the Mttp gene in this way resulted in hypercholesterolemic animals. We found that hypercholesterolemia in the atheroregressing group was reversed within 16 days of pI-pC treatment (Fig. 1). All subsequent experiments involved comparison of disease-prone and reversed groups of mice.
Figure 1. Hyperlipidemia is reversed following Mttp inactivation.
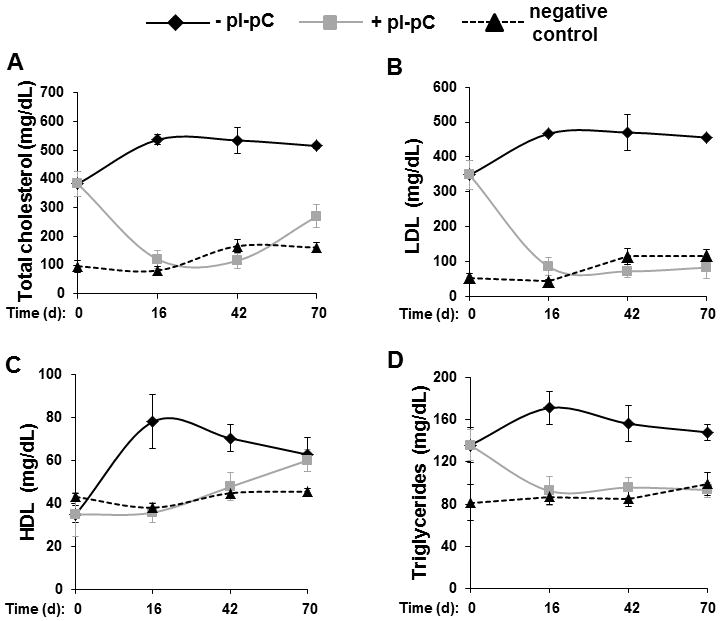
Reversa mice were treated with pI-pC as shown in Suppl. Fig. 1. and sacrificed immediately after Mttp inactivation (day 0) or 16, 42 or 70 days after cessation of hepatic lipoprotein production in the atheroregressing group (+pI-pC). Fasting total plasma cholesterol (A) and fasting plasma levels of LDL (B), HDL (C) and triglycerides (D) were measured in atherosclerosis-prone (−pI-pC), atheroregressing and atherosclerosis-resistant (negative control) mice using a colorimetric Cholesterol quantification kit. d, days.
Atherosclerosis-prone mice developed aortic plaques within 84 days that further progressed if animals were maintained on the Western diet (Fig. 2) for an additional 70 days. In contrast, the atheroregressing group displayed a substantial regression of atherosclerosis (Fig. 2A). Regressing animals had 49%, 60% and 65% smaller lesions at the aortic valve level than did the hypercholesterolemic mice at 16, 42 and 70 days after Mttp inactivation, respectively (Figs. 2B and 2C and Suppl. Fig. 3). Furthermore, we were able to quantify reversal of hypercholesterolemia-induced regression in terms of area and number of plaques (Figs. 2C and 2D). Numbers of Moma 2+ foamy macrophages in regressing lesions also declined (Fig. 2E), a change reaching the greatest significance 70 days after Mttp inactivation (Fig. 2F). We conclude that reversal of hypercholesterolemia results in significant but incomplete plaque regression. However, the Reversa mouse model is appropriate for observations of atherosclerosis regression involving our experimental protocol, which monitors time-dependent changes in atherosclerosis burden up to 70 days post Mttp inactivation.
Figure 2. Reversal of hypercholesterolemia triggers regression of plaques and reduces Moma 2+ macrophages in lesions.
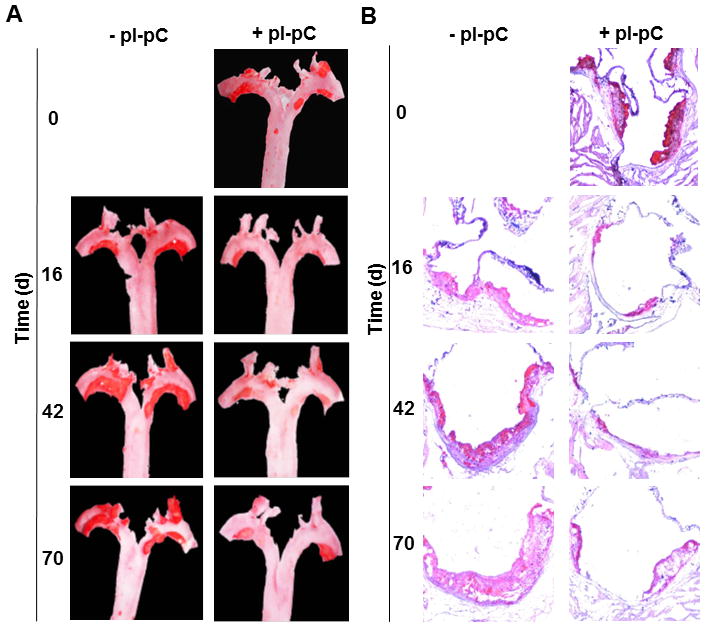
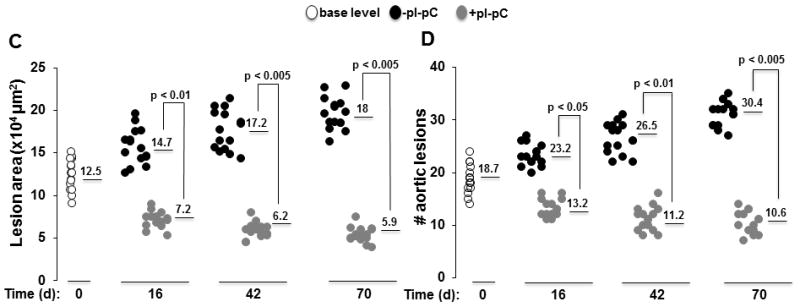
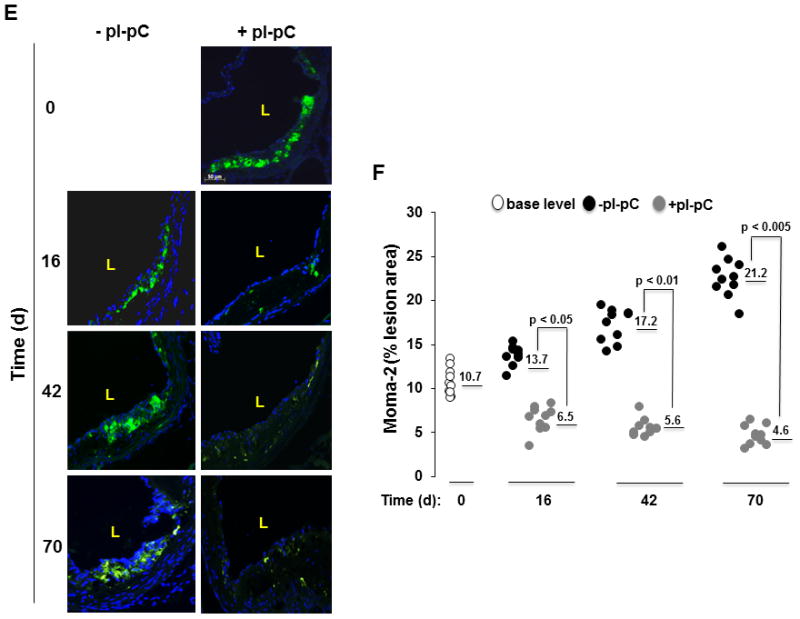
Reversa mice fed the Western diet were sacrificed immediately after the last pI-pC injection (day 0, base level) or 16, 42 and 70 days after Mttp inactivation. Aortas were removed and stained en face or fixed and embedded into OCT compound for cross-sectional analysis to determine location, size and number of plaques. Aortic roots were fixed and cryosections were stained with Moma 2-directed monoclonal Ab (green). A, Representative en face staining of the aorta from atherosclerosis-prone (−pI-pC) or atheroregressing (+pI-pC) mice. B, Representative images of Oil Red O staining of aortic root cross-sections. C, Quantification of lesion size at the aortic root cross-sections. Numerical values indicate the average lesion surface in each group, expressed in μm2. D, Quantification of lesion number in the whole aorta. Numerical values indicate the average number of aortic lesions in each group. E, Representative images of Moma 2+ atherosclerotic lesions in aortic roots of +pI-pC and −pI-pC mice. F, Quantification of Moma 2+ areas in plaques. Numerical values indicate foamy macrophage-positive areas in each group and are expressed as percent of the total lesion area. #, number; L, vessel lumen; d, days.
Mobilization of endothelial progenitors with the stem cell mobilizer AMD3100 augments plaque regression
Hypercholesterolemia and vascular inflammation negatively affect both EPC number and function and may deplete the EPC bone marrow pool [24–27]. In contrast, nonpharmacologic and statin-induced correction of hyperlipidemia in cardiovascular patients correlate with increased EPCs in peripheral blood [28;29] and their homing to sites of vascular injury [30]. Furthermore, increased EPC availability promoted by the endothelial progenitor cell-based therapy was demonstrated to stimulate endothelial repair in animal models of atherosclerosis and ischemic limb [31;32]. Since EPCs are thought to stimulate vascular repair, we tested whether endothelial progenitor availability is rate limiting in atherosclerotic plaque regression.
Although there are no surface markers specific for EPCs, the minimal antigenic profile for mouse circulating EPCs includes at least one marker of immaturity such as CD34, CD133, c-kit or Sca-1 plus one marker of endothelial commitment such as VEGFR2 (Flk1) [33;34].
We tested whether reversal of hypercholesterolemia increases circulating EPCs in atheroregressing Reversa mice. We found that regardless of which antigenic profile was used to identify circulating EPCs, the absolute number of c-kit+Flk1+CD11b− and CD34+CD133+Flk1+ cells in peripheral blood remained low after normalization of plasma lipids (Fig. 3 and Suppl. Fig. 5A).
To increase EPCs availability, we mobilized progenitors from bone marrow stores. A potent agent for inducing rapid deployment of bone marrow EPCs into peripheral blood in mice is AMD3100 (plerixafor, FDA-approved Mozobil) [35–38], a highly selective antagonist of the chemokine receptor CXCR4. AMD3100 mobilizes EPCs by disrupting the CXCL12-CXCR4 axis which is central to the retention of stem cells and progenitors within the bone marrow [39]. Reversa mice in which plasma lipids were normalized after 84 days on the Western diet were treated with AMD3100 or vehicle for 70 days and atherosclerosis burden was monitored.
As expected for this compound [39], AMD3100 triggered mild neutrophilia and leukocytosis in atheroregressing mice (Suppl. Fig. 4A). However, AMD3100 did not alter plasma cholesterol or triglyceride levels (Suppl. Fig. 4B). A marked increase in circulating c-kit+Flk1+CD11b− and CD34+CD133+Flk1+ cells was observed in AMD3100-treated group after 16 days of treatment (Fig. 3). Interestingly, a 70-day treatment with AMD3100 significantly reduced plaque burden (Fig. 4A). We recorded a 51% decrease in the lesion surface in the aortic arch (Fig. 4B) and a 83% decrease in the thoracoabdominal aorta (Fig. 4C). Moreover, AMD3100 decreased lesion number 2.3-fold (Fig. 4D). These data suggest that the availability of circulating EPCs may be rate limiting in regression of atherosclerotic lesions.
Figure 4. AMD3100 accelerates atherosclerosis regression.
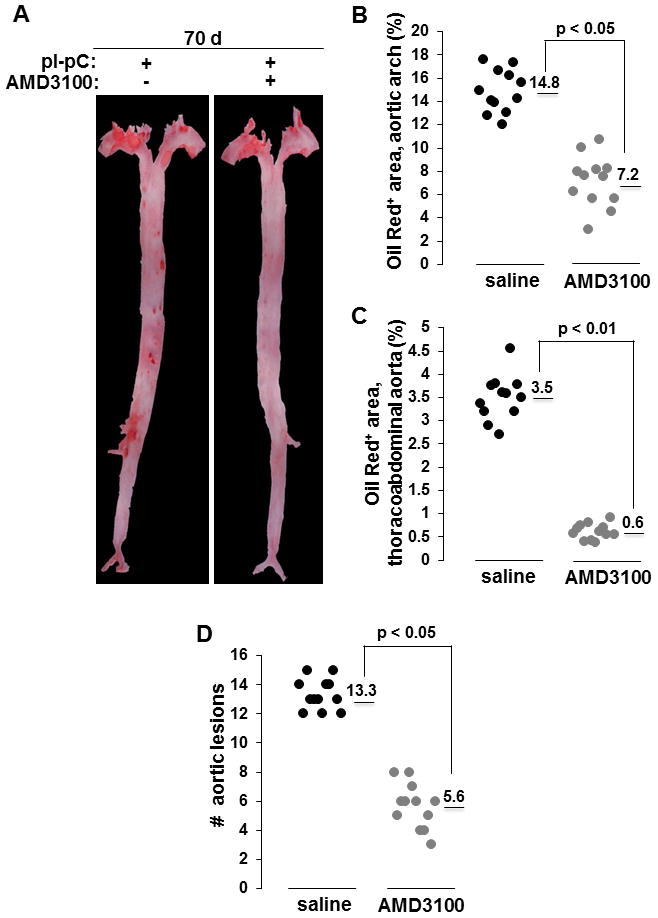
Hypercholesterolemia was reversed after 84 days on the Western diet and atheroregressing Reversa mice were treated with AMD3100 or saline for a total of 70 days. Location, size and number of atherosclerotic lesions on excised aortas were determined. A, Representative aortas. B, Quantification of lesion size in the aortic arch. Numerical values indicate the average lesional surface in each group, expressed as percent of the total aortic arch area. C, Quantification of lesion size in thoracoabdominal aorta. Numerical values indicate the average lesional surface in each group, expressed as percent of the total thoracoabdominal aorta. D, Quantification of lesion number. Numerical values indicate the average number of aortic lesions in each group. Representative experiment is shown in B, C and D. d, days; #, number.
Adoptive transfer of bone marrow-derived EPCs accelerates atherosclerosis regression
AMD3100-induced inhibition of the CXCL12-CXCR4 axis in the bone marrow indiscriminately mobilizes EPCs, hematopoietic stem cells and hematopoietic progenitors to the periphery [40]. Consequently, AMD3100 is used for autologous hematopoietic stem cell mobilization and transplantation in patients with non-Hodgkin lymphoma or multiple myeloma [41]. Although AMD3100 treatment of regressing Reversa mice increased circulating c-kit+Flk1+CD11b− and CD34+CD133+Flk1+ cells (Fig. 3) that correlated with advanced plaque regression (Fig. 4), these data do not provide evidence for a direct role of EPCs in plaque regression. Thus, to determine whether EPCs directly augment atherosclerosis regression, a pure population of bone marrow EPCs from Tie2-GFP+ donor mice expanded ex vivo was adoptively transferred into reversing recipients and plaque regression was monitored.
Tie2 is a receptor for angiopoietins that is expressed specifically in endothelial cells throughout development and in adults [42]. In transgenic Tie2-GFP+ mice, the Tie2 promoter drives GFP expression specifically in vascular endothelial cells [43]. Tie2-GFP+ mice were used as bone marrow EPC donors so that the fate of injected cells could be followed.
The purity of EPC populations adoptively transferred into reversing recipients after 4 days in culture was high since at least 95% of Tie2-GFP+ bone marrow EPCs expressed CD133, Sca-1or Flk1 (Suppl. Fig. 5B). Furthermore, Tie2-GFP+ cells also bound TRITC-lectin and internalized Dil-AcLDL (Suppl. Fig. 5C), confirming that Tie2-GFP+ bone marrow cells grown ex vivo have the EPC phenotype.
Atheroregressing Reversa mice received intravenous adoptive transfer of Tie2-GFP+ EPCs at 10, 20 and 30 days after Mttp inactivation. When assessed 70 days later, plaque surface was reduced up to 43% (Figs. 5A–5D and Suppl. Fig. 6) while the lesion number was reduced up to 2.9-fold relative to saline-treated controls receiving no adoptive transfer (Fig. 5E). Moreover, atherosclerosis burden in the EPC-treated group was reduced to a magnitude comparable to that achieved with AMD3100 (Fig. 5). Enhanced plaque regression also correlated with successful engraftment and differentiation of Tie2-GFP+ EPCs into CD31+ endothelial cells in the vascular wall (Fig. 6A). Importantly, Tie2-GFP+ EPCs mostly incorporated into endothelium covering Nile red+ areas of lipid deposits in the vessel wall (Fig. 6B), suggesting that these cells engraft the vascular wall predominantly in the areas of regressing plaques.
Figure 5. Adoptive transfer of bone marrow EPCs augments plaque resolution.
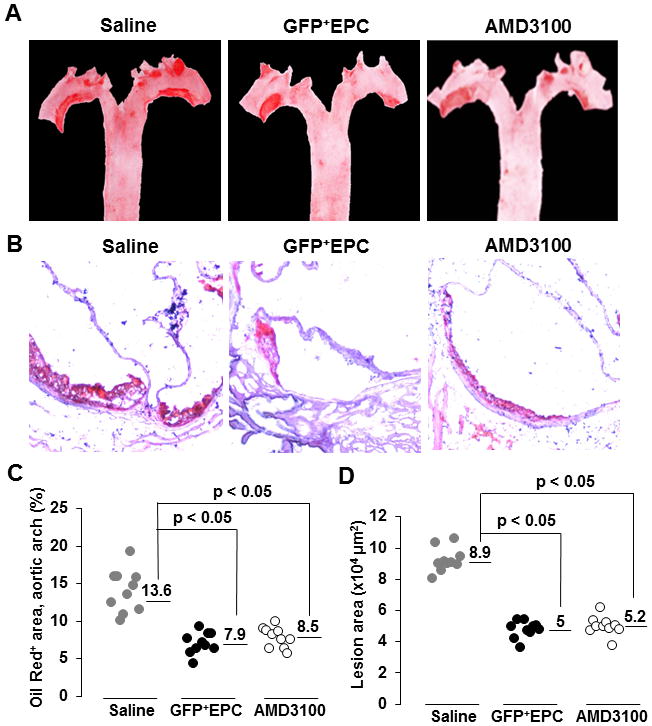
Reversa mice fed the Western diet for 84 days in which hypercholesterolemia was reversed were either treated with AMD3100 or saline, or received adoptive transfer of Tie2-GFP+ bone marrow EPCs (GFP+EPCs) three times, 10, 20 and 30 days after Mttp inactivation. Mice in all three groups were sacrificed 70 days after the last pI-pC injection and atherosclerosis burden was assessed. A, Representative en face staining of aortas. B, Representative Oil Red O staining of aortic root cross-sections. C, Quantification of lesion size in the aortic arch. Numerical values indicate the average lesional surface in each group expressed as percent of the total aortic arch area. D, Quantification of lesion size. Numerical values indicate the average lesional surface in each group, expressed in μm2. E, Quantification of lesion number in aorta. Numerical values indicate the average number of aortic lesions in each group. Representative experiments are shown in C, D and E. #, number.
Figure 6. Adoptively transferred Tie2-GFP+ bone marrow EPCs incorporate into vascular endothelium and differentiate into CD31+ endothelial cells.
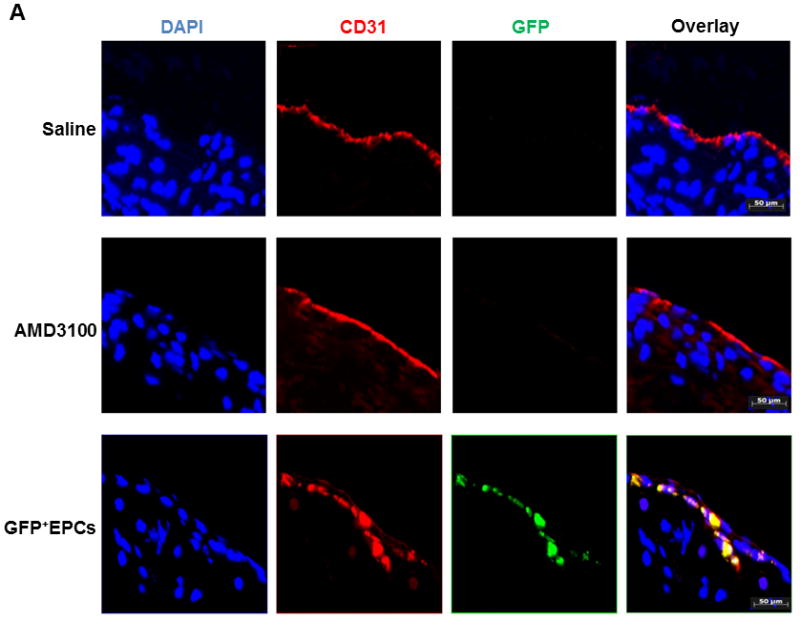
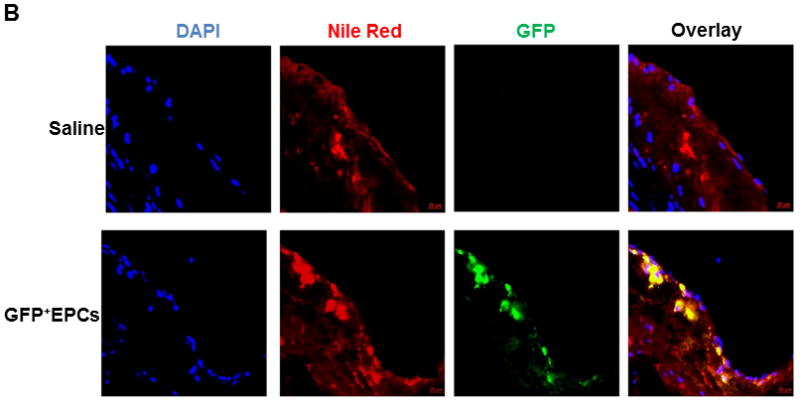
Atheroregressing Reversa mice treated with either saline or AMD3100, or with Tie2-GFP+ bone marrow EPCs were sacrificed 70 days after Mttp inactivation and the location and efficiency of GFP+ cell incorporation into endothelium were evaluated. A, Aortic roots cryosections were stained with primary rat anti-mouse CD31 and secondary goat anti-rat IgG Alexa Fluor 568 antibody to identify differentiated endothelial cells and mounted in 4’,6-diamidino-2-phenylindole (DAPI)-containing medium. Representative images. DAPI (blue), GFP (green), CD31 (red), red + green emission overlap = yellow. B, Cryosections of aortic roots in A were stained with Nile Red and mounted in DAPI-containing medium. Representative images. DAPI (blue), GFP (green), Nile red (red), red + green emission overlap = yellow.
To gain the initial perspective on mechanisms supporting homing of Tie2-GFP+ EPCs into regressing plaques, we evaluated lesions of atherosclerosis-prone and atheroregressing Reversa mice that received saline or EPC treatment for expression of chemokines including CXCL12 and CXCL1 and adhesion molecules ICAM-1 and VCAM-1. All these molecules have critical roles in homing of EPCs to sites of vascular repair [44–46]. Correction of hypercholesterolemia or EPC treatment of regressing mice upregulated CXCL1 (Suppl. Fig. 7). Only very low expression of CXCL12 was detected in plaques and normalization of plasma lipids did not upregulate this chemokine (data not shown). Reversal of hypercholesterolemia also had no effect on expression of VCAM-1 or on ICAM-1. However, expression of both adhesion molecules was significant at all times (Suppl. Fig. 7). This observation suggests that CXCL1 may promote bone marrow Tie2-GFP+ EPC recruitment to regressing plaques whereas expression of VCAM-1 and ICAM-1 may support adhesion of these cells to endothelium and/or in the vascular wall.
Targeting of EPCs to the vascular wall represents a challenge because these cells translocate into non-target organs through vessels, especially if EPCs are injected intravenously. Thus, we examined whether donor Tie2-GFP+ EPCs in addition to the vessel wall also populate non-target organs such as spleen, lungs and liver. We detected GFP+ cells in spleen but not in lungs and liver (Suppl. Fig. 8).
Since adoptive transfer of highly pure bone marrow EPCs advanced atherosclerosis regression induced by reversal of hypercholesterolemia, we conclude that EPCs directly contribute to regression of plaques in the Reversa model.
EPC treatment increases atheroprotective nitric oxide and improves vascular relaxation
Vascular endothelial cells are major producers of nitric oxide (NO) detected in blood. Endothelial NO, which is synthesized by the endothelial nitric oxide synthase (eNOS), modulates vascular tone by controlling a cascade of events that ultimately trigger relaxation of vascular smooth muscle in response to vasoactive substances. Thus, endothelial NO acts as a major vasodilator regulating vessel wall relaxation. Endothelial NO is also atheroprotective, maintaining anti-inflammatory and anti-thrombogenic phenotypes in vascular endothelium. Disturbances in either production or availability of NO can cause endothelial dysfunction and contribute to atherosclerosis [47]. Lipid lowering with statins upregulates and activates eNOS, increasing NO production and thereby improving vascular relaxation [48;49].
Rapid metabolism and short half-life of NO represent a considerable obstacle in measuring its concentrations in blood. The major pathway for NO metabolism is a stepwise oxidation to nitrate (NO2−) and nitrite (NO3−), which are both stable end-metabolites and can be used as an index of NO production. Thus, the sum of NO2− and NO3− plasma levels, NOx, can be used as a measure of NO in blood and in plasma [18–21].
We hypothesized that atheroregressing conditions increase plasma NO and improve vasorelaxation. Thus, we determined plasma NOx levels before and immediately after reversal of hypercholesterolemia. We have also evaluated long-term (70 days) effects of AMD3100 or EPC treatment of regressing Reversa mice on plasma NOx and vascular relaxation. We found that reversal of hypercholesterolemia increases plasma NOx in Reversa mice as early as 16 days after normalization of plasma lipids (Fig 7A). We also observed that either AMD3100 or EPC treatment markedly elevated plasma NOx (Fig. 7B). Furthermore, plasma NOx in these mice were above even those of aged-matched C57BL/6 controls fed the regular chow diet and displaying no hypercholesterolemia or atherosclerosis (Fig. 7B). Importantly, EPC treatment of atheroregressing mice improved endothelium-dependent ACh-induced vascular relaxation (Fig. 7C) but had no effect on endothelium-independent SNP-induced vasorelaxation (Fig. 7D).
Figure 7. Reversal of hypercholesterolemia and EPC treatment correlate with an increase in plasma nitric oxide and improved vascular relaxation.
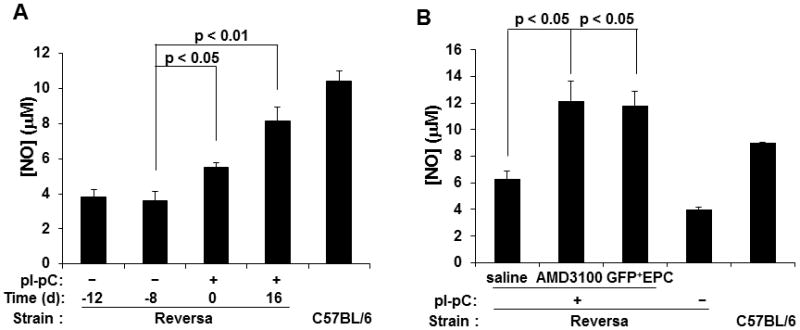
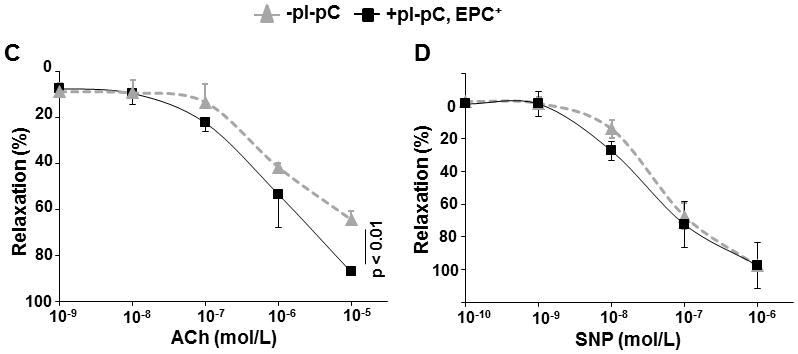
A, Peripheral blood was collected from Reversa mice 12 and 8 days before Mttp inactivation as well as 16 days after reversal of hypercholesterolemia, and assayed for NOx as described in Materials and Methods. B, NOx levels were also determined in plasma from atheroregressing Reversa mice sacrificed 70 days after Mttp inactivation that were treated with either saline or AMD3100, or received Tie2-GFP+ bone marrow EPCs. C and D, 3-mm wide rings were prepared from aortas removed from atherosclerosis-prone (−pI-pC) and atheroregressing mice that received EPC treatment (+pI-pC, EPC+). Acetylcholine (ACh)- or sodium nitroprusside (SNP)-specific vascular relaxation was determined.
We conclude that treatment with bone marrow EPCs in regressing conditions increases available NO and increases endothelium-dependent vessel relaxation. This observation also suggests that the restoration of endothelial function may be necessary for efficient regression of plaques.
DISCUSSION
Statin treatment reduces atherogenic plasma lipoproteins and vascular inflammation, thereby limiting atherosclerosis progression. However, these lipid-lowering drugs facilitate incomplete plaque regression that leaves patients at risk for adverse cardiovascular events [3]. Thus, it is significant that we have identified the mechanism and found a means to augment lesion regression. We used a mouse model in which reversal of hypercholesterolemia reduces atherosclerosis burden. Plaque regression in Reversa mice in which hypercholesterolemia was reversed was markedly improved by treatment with EPC mobilizing agent AMD3100 or by adoptive transfer of EPCs that in turn were incorporated into damaged arteries, possibly supporting endothelial repair. Indeed, increased plasma NOx, indicating improved production and availability of atheroprotective NO, suggest that endothelial repair was achieved by both manipulations.
Atherosclerosis decreases contractility of smooth muscle cells in vascular subendothelium [50]. Thus, an additional important line of evidence exposed by our investigation is that EPC treatment of regressing Reversa mice significantly improved vascular relaxation. These data further support the idea that endothelial regeneration is necessary for atherosclerosis regression, a process that was thus far defined by shrinkage of plaques and may from now on also be associated with improved vascular tone.
EPCs normally reside in bone marrow niches that are characterized by low oxygen tension and high levels of CXCL12. They may exit that environment in response to EPC-activation factors produced in peripheral tissues [51]. Circulating EPC numbers are a surrogate marker for cumulative cardiovascular risk. In fact, low circulating EPC numbers that accompany hyperlipidemia and vascular inflammation are an independent predictor of endothelial dysfunction and atherosclerosis [52;53]. Reciprocally, correction of hyperlipidemia in cardiovascular patients correlates with an increase in circulating EPCs [28;29] and improved cardiovascular risk profiles [54]. However, the exact role of EPCs in atherosclerosis remains controversial.
Treatment with EPC mobilizing agents such as granulocyte colony stimulating factor (G-CSF) prevents progression of atherosclerosis and improves symptoms of intractable atherosclerotic peripheral artery disease in patients [55]. However, accelerated plaque development was observed following administration of EPC mobilizer VEGF into hypercholesterolemic ApoE−/− mice [56].
Investigations measuring impact of cell-based therapy on plaque development, especially infusion of a pure EPC population, have also generated inconsistent results. Chronic treatment with bone marrow EPCs from nonatherosclerotic young ApoE−/− mice prevented atherosclerosis progression in adult ApoE−/− recipients despite persistent hypercholesterolemia [57]. In contrast, administration of spleen-derived EPCs resulted in an increase in atherosclerosis burden in ApoE−/− mice [58]. Thus, EPCs seem to exert a variety of effects in an atherosclerotic milieu. A plausible explanation for these ambiguous results could be that cardiovascular risk factors change during progression of atherosclerosis and therefore differentially affect EPC function and survival. While it is possible that EPCs could prevent development of early stage lesions or ameliorate endothelial dysfunction, their beneficial effects are not favored in the local pro-thrombotic environment of advanced atherosclerotic disease. Furthermore, it is also possible that EPCs isolated from spleen and bone marrow differentially contribute to plaque development because they derive from different sources and thus have different functional properties. Because of complications limiting investigations into the roles of EPCs in plaque development, the understanding of mechanisms by which EPCs may regenerate damaged vascular endothelium remains very limited.
Compared to atherosclerosis-prone models in which studying effects of EPCs atherosclerosis progression is difficult, the Reversa model has an advantage because cardiovascular risk factors affecting homeostasis of EPCs, especially hypercholesterolemia, are reversible. We confirmed that lowering of plasma lipids reduces atherosclerosis burden in these mice. Importantly, as adoptively transferred EPCs advanced plaque resolution, incorporated into the endothelium and survived in regressing Reversa recipients for up to 70 days, this model can be manipulated to study short- and long-term effects of EPCs on plaque regression and repair of vascular endothelium. Furthermore, AMD3100 treatment and adoptive transfer of EPCs similarly improved plaque regression indicating that consistent results are obtained by two different approaches, both supporting the conclusion that EPCs play a pivotal role in atherosclerosis resolution. Overall, our study indicates that Reversa mice present a novel and a reliable model for studying mechanisms by which EPCs and possibly other cell types in the vascular wall contribute to atherosclerosis regression and/or vascular remodeling supporting reductions in atherosclerosis burden.
Recruitment of EPCs to sites of neovascularization shares common features with the homing of leukocytes to sites of inflammation [51] in which chemokines and adhesion molecules play pivotal roles. We have observed that the most potent chemokine ligand for the chemokine receptor CXCR2, CXCL1, is upregulated in the vascular wall in regressing versus hypercholesterolemic conditions. Furthermore, previous studies have reported that mouse and human EPCs express CXCR2 [44]. This chemokine receptor was shown to recruit human circulating EPCs to sites of acute arterial injury in the athymic nude mice [59]. Furthermore, CXCR2 is also the main receptor mediating homing of mouse bone marrow EPCs to the peribronchial blood vessel endothelium during chronic allergic inflammation in lungs [44]. The in vivo homing of bone marrow EPCs to ischemic tissues also depends upon interactions of β2 integrins expressed on EPCs and endothelial ICAM-1[46] as well as on EPC α4β1 integrin (VLA-4) interaction with VCAM-1 in the vessel wall [45]. Interestingly, regressing plaques express ICAM-1 and VCAM-1. Thus, the recruitment of bone marrow EPCs into regressing plaques in our model may therefore require cooperative action of chemotactic and adhesive mechanisms mediated by the CXCL1-CXCR2 axis and ICAM-1 and VCAM-1. However, detailed evaluation of all homing cues produced in the vascular wall following reversal of hypercholesterolemia is mandatory for a clear understanding of how EPCs migrate to sites of vascular damage to augment plaque resolution.
Postnatal neovascularization is supported by angiogenesis and by vasculogenesis [11–15]. Our work suggests that adoptively transferred Tie2-GFP+CD133+Sca-1+Flk1+ EPCs augment plaque resolution by facilitating vasculogenesis, since bone marrow progenitors were observed to incorporate into regenerating vasculature in vivo and differentiate into mature endothelial cells. However, EPCs may also promote new vessel formation via production and secretion of paracrine or justacrine factors that trigger in situ proliferation and migration of pre-existing endothelial cells.
Direct endothelial progenitor cell therapy and indirect EPC mobilization therapy have been proposed for the prevention or treatment chronic ischemic heart disease and myocardial infarction [31]. However, stem cells and progenitors including EPCs have not been evaluated for their potential to support regression of atherosclerosis. The main obstacle in using circulating EPCs to facilitate atheroregression would be the exceedingly low number of these cells in peripheral blood. Accordingly, an intervention to increase circulating EPCs may be to reduce chronic vascular inflammation by dietary supplements [60], exercise [61] or statin therapy [29]. Since reducing inflammation may not profoundly increase their numbers, administration of autologous EPCs harvested from peripheral blood by leukapheresis [62] may be tested. Instead of administering EPC, another therapeutic intervention could be to mobilize EPCs from bone marrow. G-CSF has been used clinically for mobilization of hematopoietic stem cells (HSC) for more than a decade. It was found that in addition to mobilizing HSC, G-CSF also mobilizes EPC [63]. G-CSF has been approved for long-term use in patients with severe chronic neutropenia. Toxic and adverse events have been catalogued but are not clinically troublesome and, aside from occasional adjustment of scheduling and dosing, seldom necessitate stopping therapy [64;65]. Reports indicate that AMD3100 (Mozobil) is a more potent stem cell mobilizer than G-CSF [62]. However, Mozobil is clinically approved only for acute and not chronic use [41]. Since the benefits far outweigh the risks in long-term administration of G-CSF [65], the use of G-CSF rather than Mozobil may be necessary for this intervention to reach fruition.
In conclusion, our investigation based on the use of EPC mobilizer AMD3100 and administration of pure endothelial progenitors suggests application of EPCs in combination with statins in treatment of atherosclerosis, making regression of atherosclerotic plaques more efficient than lipid-lowering treatment alone.
CONCLUSION
Current therapies for atherosclerosis do not completely reverse the pathology of this disease. New approaches facilitating complete plaque resolution may offer novel avenues to reduce the disease burden. We demonstrate that both EPC mobilization with AMD3100 and adoptive transfer of bone marrow EPCs significantly improve regression of plaques in an atherosclerosis-prone mouse model with reversible hypercholesterolemia. Our data indicate that bone marrow EPCs play a pivotal role in atherosclerosis regression. Although further investigations are needed to determine how EPCs recruit to regressing plaques and how they augment resolution of atherosclerosis, results from this study provide the basis to explore the use of EPC-based regenerative therapy in treatment of atherosclerosis.
Supplementary Material
Female and male Reversa mice were weaned at 21 days of age and separated into three groups. All three groups were fed a Western diet for 84 days. The first group, atherosclerosis-resistant (negative control), was given 4 intraperitoneal injections of pI-pC immediately after weaning. In the second group, the atherosclerosis-prone group (−pI-pC), hepatic lipoprotein production remained unaltered as these mice were not injected with pI-pC. In the third group Mttp was inactivated after 84 days on the Western diet. We named this latter group of mice the atheroregressing group (+pI-pC). Atherosclerosis burden was monitored in all three groups immediately after the last (4th) pI-pC injection (day 0) or 16, 42 or 70 days after Cre-dependent Mttp inactivation in the atheroregressing group. Yellow block area, mice fed the Western diet; Atheroregression, in the +pI-pC group only; d, days.
Mice were fed the Western diet for 84 days and sacrificed. Aortic cross-sections were stained with Oil Red O and counterstained with hematoxylin (left). Sections were also stained with Moma 2-directed monoclonal Ab to identify lesional foamy macrophages (green, right).
Reversa mice fed the Western diet were sacrificed immediately after the last pI-pC injection (day 0, base level) or 16, 42 and 70 days after Mttp inactivation. Aortas were removed, fixed and sectioned. Representative Oil Red O staining demonstrating the entire aortic root cross-sections of aortas from atherosclerosis-prone (−pI-pC) or atheroregressing (+pI-pC) mice.
Hypercholesterolemia was reversed after 84 days on the Western diet and atheroregressing mice were treated with AMD3100 or saline for a total of 70 days. Peripheral blood was collected and total leukocyte and neutrophil numbers and plasma cholesterol levels were examined. A, Leukocyte and neutrophil numbers were determined with Hemavet. B, Fasting total plasma cholesterol and fasting plasma levels of LDL, HDL and triglycerides were measured using the colorimetric Cholesterol quantification kit.
A, Circulating mononuclear cells obtained from atherosclerosis-prone (−pI-pC) or atheroregressing mouse treated with saline (+pI-pC) or AMD3100 (+pI-pC + AMD3100) were stained and analyzed as detailed in Materials and Methods. Fluorescence staining profiles of Flk1+ cells (left) and expression of CD34 and CD133 in the Flk1+ populations (right) are shown. Numbers in the upper right corner indicate percent of cells with the indicated phenotype. B, Bone marrow was collected from long bones of Tie2-GFP+ donor mice and EPCs isolated by negative selection and sorting were cultured ex vivo for 4 days. A, FACS plot shows representative Lin−GFP+ cells (GFP) co-stained with CD133, Sca-1-, Flk1-specific monoclonal antibodies, or with isotype rat IgG2b antibody. C, Representative images of Lin−GFP+ bone marrow cells (GFP, green) that were prior to adoptive transfer labeled with TRITC-Lectin (red) or evaluated for the uptake of Dil-AcLDL (red), and stained with DAPI (blue). Original magnification ×200.
Reversa mice fed the Western diet for 84 days in which hypercholesterolemia was reversed were either treated with AMD3100 or saline, or received adoptive transfer of Tie2-GFP+ bone marrow EPCs (GFP+EPCs) three times, 10, 20 and 30 days after Mttp inactivation. Mice in all three groups were sacrificed 70 days after the last pI-pC injection and atherosclerosis burden was assessed. Representative Oil Red O staining showing the entire aortic root cross-sections.
Reversa mice fed the Western diet for 84 days in which hypercholesterolemia was reversed were either treated with AMD3100 or saline, or received adoptive transfer of Tie2-GFP+ bone marrow EPCs (GFP+EPCs) three times, 10, 20 and 30 days after Mttp inactivation. Mice in all three groups were sacrificed 70 days after the last pI-pC injection and atherosclerosis burden was assessed. Representative Oil Red O staining showing the entire aortic root cross-sections.
Atherosclerosis-prone (−pI-pC) or atheroregressing (+pI-pC) mice treated with saline or with Tie2-GFP+ bone marrow EPCs were sacrificed 70 days after Mttp inactivation. Aortas were removed, aortic roots were excised and frozen, and cryosections were stained with CXCL1, ICAM-1 or VCAM Ab and mounted in DAPI-containing medium. Representative images of the aortic cross-sections from each group are shown.
Atheroregressing Reversa mice treated with either saline or AMD3100, or with Tie2-GFP+ bone marrow EPCs were sacrificed 70 days after Mttp inactivation and lungs, liver and spleen were collected, frozen, and cryosections were stained with primary rat anti-mouse CD31 or control rat IgG2a antibody and secondary goat anti-rat IgG Alexa Fluor 568 antibody to identify differentiated endothelial cells and mounted in DAPI-containing medium. The only organ that Tie2-GFP+ bone marrow EPCs were detected in addition to the vasculature was spleen. Representative images of the spleen section from each group. DAPI (blue), GFP (green), CD31 (red), red + green emission overlap = yellow.
Acknowledgments
We thank Drs. Kevin Moore, Lijun Xia, Rodger McEver and Paul Kincade for helpful discussions and critical review of the manuscript. We also thank Dr. John Knight and Mia Pederson-Rambo for editing the manuscript.
Sources of funding: this work was supported by grants from the National Center for Research Resources (5 P20 RR018758-08) and the National Institute of General Medical Sciences (8 P20 GM103441-08), from the Oklahoma Center for the Advancement of Science and Technology (HR10-099) and the Oklahoma Center for Adult Stem Cell Research (4340-03-06).
Footnotes
Author contributions: L.Y.: Conception and design, collection and/or assembly of data; data analysis and interpretation; J.H.B.: Collection and/or assembly of data; O.H.P.: Collection and/or assembly of data, data analysis and interpretation; R.I.: Collection and/or assembly of data; Q.W.: Collection and/or assembly of data; M-H.Z.: Data analysis and interpretation; J.B.D.: Conception and design, data analysis and interpretation, manuscript writing, and final approval of manuscript.
DISCLOSURES
None.
References
- 1.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 2.Liao JK. Effects of statins on 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition beyond low-density lipoprotein cholesterol. Am J Cardiol. 2005;96:24–33. doi: 10.1016/j.amjcard.2005.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicholls SJ, Ballantyne CM, Barter PJ, et al. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. 2011;365:2078–2087. doi: 10.1056/NEJMoa1110874. [DOI] [PubMed] [Google Scholar]
- 4.Feig JE, Parathath S, Rong JX, et al. Reversal of hyperlipidemia with a genetic switch favorably affects the content and inflammatory state of macrophages in atherosclerotic plaques. Circulation. 2011;123:989–998. doi: 10.1161/CIRCULATIONAHA.110.984146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lieu HD, Withycombe SK, Walker Q, et al. Eliminating atherogenesis in mice by switching off hepatic lipoprotein secretion. Circulation. 2003;107:1315–1321. doi: 10.1161/01.cir.0000054781.50889.0c. [DOI] [PubMed] [Google Scholar]
- 6.Bailey AS, Jiang S, Afentoulis M, et al. Transplanted adult hematopoietic stems cells differentiate into functional endothelial cells. Blood. 2004;103:13–19. doi: 10.1182/blood-2003-05-1684. [DOI] [PubMed] [Google Scholar]
- 7.Pelosi E, Valtieri M, Coppola S, et al. Identification of the hemangioblast in postnatal life. Blood. 2002;100:3203–3208. doi: 10.1182/blood-2002-05-1511. [DOI] [PubMed] [Google Scholar]
- 8.Grant MB, May WS, Caballero S, et al. Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization. Nat Med. 2002;8:607–612. doi: 10.1038/nm0602-607. [DOI] [PubMed] [Google Scholar]
- 9.Ingram DA, Caplice NM, Yoder MC. Unresolved questions, changing definitions, and novel paradigms for defining endothelial progenitor cells. Blood. 2005;106:1525–1531. doi: 10.1182/blood-2005-04-1509. [DOI] [PubMed] [Google Scholar]
- 10.Timmermans F, Van Hauwermeiren F, De Smedt M, et al. Endothelial outgrowth cells are not derived from CD133+ Cells or CD45+ hematopoietic precursors. Arterioscler Thromb Vasc Biol. 2007;27:1572–1579. doi: 10.1161/ATVBAHA.107.144972. [DOI] [PubMed] [Google Scholar]
- 11.Asahara T, Masuda H, Takahashi T, et al. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85:221–228. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 12.Isner JM, Asahara T. Angiogenesis and vasculogenesis as therapeutic strategies for postnatal neovascularization. J Clin Invest. 1999;103:1231–1236. doi: 10.1172/JCI6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267:10931–10934. [PubMed] [Google Scholar]
- 14.Urbich C, Aicher A, Heeschen C, et al. Soluble factors released by endothelial progenitor cells promote migration of endothelial cells and cardiac resident progenitor cells. J Mol Cell Cardiol. 2005;39:733–742. doi: 10.1016/j.yjmcc.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Shantsila E, Watson T, Tse HF, et al. New insights on endothelial progenitor cell subpopulations and their angiogenic properties. J Am Coll Cardiol. 2008;51:669–671. doi: 10.1016/j.jacc.2007.09.057. [DOI] [PubMed] [Google Scholar]
- 16.Greenspan P, Fowler SD. Spectrofluorometric studies of the lipid probe, nile red. J Lipid Res. 1985;26:781–789. [PubMed] [Google Scholar]
- 17.Greenspan P, Mayer EP, Fowler SD. Nile red: a selective fluorescent stain for intracellular lipid droplets. J Cell Biol. 1985;100:965–973. doi: 10.1083/jcb.100.3.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gladwin MT, Raat NJH, Shiva S, et al. Nitrite as a vascular endocrine nitric oxide reservoir that contributes to hypoxic signaling, cytoprotection, and vasodilation. Am J Physiol Heart Circ Physiol. 2006;291:H2026–H2035. doi: 10.1152/ajpheart.00407.2006. [DOI] [PubMed] [Google Scholar]
- 19.Kelm M, Preik-Steinhoff H, Preik M, et al. Serum nitrite sensitively reflects endothelial NO formation in human forearm vasculature: evidence for biochemical assessment of the endothelial l-arginine-NO pathway. Cardiovasc Res. 1999;41:765–772. doi: 10.1016/s0008-6363(98)00259-4. [DOI] [PubMed] [Google Scholar]
- 20.Zeballos GA, Bernstein RD, Thompson CI, et al. Pharmacodynamics of plasma nitrate/nitrite as an indication of nitric oxide formation in conscious dogs. Circulation. 1995;91:2982–2988. doi: 10.1161/01.cir.91.12.2982. [DOI] [PubMed] [Google Scholar]
- 21.Malte K. Nitric oxide metabolism and breakdown. Biochim Biophys Acta - Bioenergetics. 1999;1411:273–289. doi: 10.1016/s0005-2728(99)00020-1. [DOI] [PubMed] [Google Scholar]
- 22.Russell A, Watts S. Vascular reactivity of isolated thoracic aorta of the C57BL/6J mouse. J Pharmacol Exp Ther. 2000;294:598–604. [PubMed] [Google Scholar]
- 23.Rosenfeld ME, Averill MM, Bennett BJ, et al. Progression and disruption of advanced atherosclerotic plaques in murine models. Curr Drug Targets. 2008;9:210–216. doi: 10.2174/138945008783755575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siddique A, Shantsila E, Lip G, et al. Endothelial progenitor cells: what use for the cardiologist? J Angiogenes Res. 2010;2:6. doi: 10.1186/2040-2384-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Umemura T, Higashi Y. Endothelial progenitor cells: therapeutic target for cardiovascular diseases. J Pharmacol Sci. 2008;108:1–6. doi: 10.1254/jphs.08r01cp. [DOI] [PubMed] [Google Scholar]
- 26.Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004;95:343–353. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- 27.Gomes AL, Carvalho T, Serpa J, et al. Hypercholesterolemia promotes bone marrow cell mobilization by perturbing the SDF-1:CXCR4 axis. Blood. 2010;115:3886–3894. doi: 10.1182/blood-2009-08-240580. [DOI] [PubMed] [Google Scholar]
- 28.Croce G, Passacquale G, Necozione S, et al. Nonpharmacological treatment of hypercholesterolemia increases circulating endothelial progenitor cell population in adults. Arterioscler Thromb Vasc Biol. 2006;26:e38–e39. doi: 10.1161/01.ATV.0000218504.71680.b5. [DOI] [PubMed] [Google Scholar]
- 29.Vasa M, Fichtlscherer S, Adler K, et al. Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation. 2001;103:2885–2890. doi: 10.1161/hc2401.092816. [DOI] [PubMed] [Google Scholar]
- 30.Walter DH, Rittig K, Bahlmann FH, et al. Statin therapy accelerates reendothelialization: a novel effect involving mobilization and incorporation of bone marrow-derived endothelial progenitor cells. Circulation. 2002;105:3017–3024. doi: 10.1161/01.cir.0000018166.84319.55. [DOI] [PubMed] [Google Scholar]
- 31.Kalka C, Masuda H, Takahashi T, et al. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc Natl Acad Sci U S A. 2000;97:3422–3427. doi: 10.1073/pnas.070046397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sprengers RW, Lips DJ, Moll FL, et al. Progenitor cell therapy in patients with critical limb ischemia without surgical options. Ann Surg. 2008;247:411–420. doi: 10.1097/SLA.0b013e318153fdcb. [DOI] [PubMed] [Google Scholar]
- 33.Hirschi KK, Ingram DA, Yoder MC. Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28:1584–1595. doi: 10.1161/ATVBAHA.107.155960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoder MC, Ingram DA. Endothelial progenitor cell: ongoing controversy for defining these cells and their role in neoangiogenesis in the murine system. Curr Opin Hematol. 2009;16:269–273. doi: 10.1097/MOH.0b013e32832bbcab. [DOI] [PubMed] [Google Scholar]
- 35.Broxmeyer HE, Orschell CM, Clapp DW, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005;201:1307–1318. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin Y, Huang L, Zhao X, et al. AMD3100 mobilizes endothelial progenitor cells in mice, but inhibits its biological functions by blocking an autocrine/paracrine regulatory loop of stromal cell derived factor-1 in vitro. J Cardiovasc Pharmacol. 2007;50:61–67. doi: 10.1097/FJC.0b013e3180587e4d. [DOI] [PubMed] [Google Scholar]
- 37.Capoccia BJ, Shepherd RM, Link DC. G-CSF and AMD3100 mobilize monocytes into the blood that stimulate angiogenesis in vivo through a paracrine mechanism. Blood. 2006;108:2438–2445. doi: 10.1182/blood-2006-04-013755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jujo K, Hamada H, Iwakura A, et al. CXCR4 blockade augments bone marrow progenitor cell recruitment to the neovasculature and reduces mortality after myocardial infarction. Proc Natl Acad Sci U S A. 107:11008–11013. doi: 10.1073/pnas.0914248107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Clercq E. Recent advances on the use of the CXCR4 antagonist plerixafor (AMD3100, Mozobil) and potential of other CXCR4 antagonists as stem cell mobilizers. Pharmacol Ther. 2010;128:509–518. doi: 10.1016/j.pharmthera.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 40.Liles WC, Broxmeyer HE, Rodger E, et al. Mobilization of hematopoietic progenitor cells in healthy volunteers by AMD3100, a CXCR4 antagonist. Blood. 2003;102:2728–2730. doi: 10.1182/blood-2003-02-0663. [DOI] [PubMed] [Google Scholar]
- 41.Flomenberg N, Comenzo RL, Badel K, et al. Plerixafor (Mozobil) alone to mobilize hematopoietic stem cells from multiple myeloma patients for autologous transplantation. Biol Blood Marrow Transplant. 2010;16:695–700. doi: 10.1016/j.bbmt.2009.12.538. [DOI] [PubMed] [Google Scholar]
- 42.Schlaeger TM, Bartunkova S, Lawitts JA, et al. Uniform vascular-endothelial-cell-specific gene expression in both embryonic and adult transgenic mice. Proc Natl Acad Sci U S A. 1997;94:3058–3063. doi: 10.1073/pnas.94.7.3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Motoike T, Loughna S, Perens E, et al. Universal GFP reporter for the study of vascular development. Genesis. 2000;28:75–81. doi: 10.1002/1526-968x(200010)28:2<75::aid-gene50>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 44.Jones CP, Pitchford SC, Lloyd CM, et al. CXCR2 mediates the recruitment of endothelial progenitor cells during allergic airways remodeling. Stem Cells. 2009;27:3074–3081. doi: 10.1002/stem.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qin G, Ii M, Silver M, et al. Functional disruption of alpha4 integrin mobilizes bone marrow-derived endothelial progenitors and augments ischemic neovascularization. J Exp Med. 2006;203:153–163. doi: 10.1084/jem.20050459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jin H, Aiyer A, Su J, et al. A homing mechanism for bone marrow-derived progenitor cell recruitment to the neovasculature. J Clin Invest. 2006;116:652–662. doi: 10.1172/JCI24751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Forstermann U. Oxidative stress in vascular disease: causes, defense mechanisms and potential therapies. Nat Clin Pract Cardiovasc Med. 2008;5:338–349. doi: 10.1038/ncpcardio1211. [DOI] [PubMed] [Google Scholar]
- 48.Walter DH, Zeiher AM, Dimmeler S. Effects of statins on endothelium and their contribution to neovascularization by mobilization of endothelial progenitor cells. Coron Artery Dis. 2004;15:235–242. doi: 10.1097/01.mca.0000131572.14521.8a. [DOI] [PubMed] [Google Scholar]
- 49.Rikitake Y, Liao JK. Rho GTPases, Statins, and Nitric Oxide. Circ Res. 2005;97:1232–1235. doi: 10.1161/01.RES.0000196564.18314.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bian K, Doursout MF, Murad F. Vascular system: role of nitric oxide in cardiovascular diseases. J Clin Hypertens. 2008;10:304–310. doi: 10.1111/j.1751-7176.2008.06632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chavakis E, Dimmeler S. Homing of progenitor cells to ischemic tissues. Antioxid Redox Signal. 2011;15:967–980. doi: 10.1089/ars.2010.3582. [DOI] [PubMed] [Google Scholar]
- 52.Schmidt-Lucke C, Rossig L, Fichtlscherer S, et al. Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events. Circulation. 2005;111:2981–2987. doi: 10.1161/CIRCULATIONAHA.104.504340. [DOI] [PubMed] [Google Scholar]
- 53.Sen S, McDonald SP, Coates PT, et al. Endothelial progenitor cells: novel biomarker and promising cell therapy for cardiovascular disease. Clin Sci (Lond) 2011;120:263–283. doi: 10.1042/CS20100429. [DOI] [PubMed] [Google Scholar]
- 54.Shantsila E, Watson T, Lip GYH. Endothelial progenitor cells in cardiovascular disorders. J Am Coll Cardiol. 2007;49:741–752. doi: 10.1016/j.jacc.2006.09.050. [DOI] [PubMed] [Google Scholar]
- 55.Arai M, Misao Y, Nagai H, et al. Granulocyte colony-stimulating factor a noninvasive regeneration therapy for treating atherosclerotic peripheral artery disease. Circ J. 2006;70:1093–1098. doi: 10.1253/circj.70.1093. [DOI] [PubMed] [Google Scholar]
- 56.Lucerna M, Zernecke A, de Nooijer R, et al. Vascular endothelial growth factor-A induces plaque expansion in ApoE knock-out mice by promoting de novo leukocyte recruitment. Blood. 2007;109:122–129. doi: 10.1182/blood-2006-07-031773. [DOI] [PubMed] [Google Scholar]
- 57.Rauscher FM, Goldschmidt-Clermont PJ, Davis BH, et al. Aging, progenitor cell exhaustion, and atherosclerosis. Circulation. 2003;108:457–463. doi: 10.1161/01.CIR.0000082924.75945.48. [DOI] [PubMed] [Google Scholar]
- 58.George J, Afek A, Abashidze A, et al. Transfer of endothelial progenitor and bone marrow cells influences atherosclerotic plaque size and composition in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2005:2636–2641. doi: 10.1161/01.ATV.0000188554.49745.9e. [DOI] [PubMed] [Google Scholar]
- 59.Hristov M, Zernecke A, Bidzhekov K, et al. Importance of CXC chemokine receptor 2 in the homing of human peripheral blood endothelial progenitor cells to sites of arterial injury. Circ Res. 2007;100:590–597. doi: 10.1161/01.RES.0000259043.42571.68. [DOI] [PubMed] [Google Scholar]
- 60.Phillips T, Childs AC, Dreon DM, Phinney S, Leeuwenburgh C. A dietary supplement attenuates IL-6 and CRP after eccentric exercise in untrained males. Med Sci Sports Exerc. 2003;35:2032–2037. doi: 10.1249/01.MSS.0000099112.32342.10. [DOI] [PubMed] [Google Scholar]
- 61.Colbert LH, Visser M, Simonsick EM, et al. Physical activity, exercise, and inflammatory markers in older adults: findings from the health, aging and body composition study. J Am Geriatr Soc. 2004;52:1098–1104. doi: 10.1111/j.1532-5415.2004.52307.x. [DOI] [PubMed] [Google Scholar]
- 62.Shepherd RM, Capoccia BJ, Devine SM, et al. Angiogenic cells can be rapidly mobilized and efficiently harvested from the blood following treatment with AMD3100. Blood. 2006;108:3662–3667. doi: 10.1182/blood-2006-06-030577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Körbling M, Reuben J, Gao H, et al. Recombinant human granulocyte colony-stimulating factor-mobilized and apheresis-collected endothelial progenitor cells: a novel blood cell component for therapeutic vasculogenesis. Transfusion. 2006;46:1795–1802. doi: 10.1111/j.1537-2995.2006.00985.x. [DOI] [PubMed] [Google Scholar]
- 64.Cottle TE, Fier CJ, Donadieu J, et al. Risk and benefit of treatment of severe chronic neutropenia with granulocyte colony-stimulating factor. Semin Hematol. 2002;39:134–140. doi: 10.1053/shem.2002.31914. [DOI] [PubMed] [Google Scholar]
- 65.Mueller MM, Bialleck H, Bomke B, et al. Safety and efficacy of healthy volunteer stem cell mobilization with filgrastim G-CSF and mobilized stem cell apheresis: results of a prospective longitudinal 5-year follow-up study. Vox Sang. 2012 doi: 10.1111/j.1423-0410.2012.01632.x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Female and male Reversa mice were weaned at 21 days of age and separated into three groups. All three groups were fed a Western diet for 84 days. The first group, atherosclerosis-resistant (negative control), was given 4 intraperitoneal injections of pI-pC immediately after weaning. In the second group, the atherosclerosis-prone group (−pI-pC), hepatic lipoprotein production remained unaltered as these mice were not injected with pI-pC. In the third group Mttp was inactivated after 84 days on the Western diet. We named this latter group of mice the atheroregressing group (+pI-pC). Atherosclerosis burden was monitored in all three groups immediately after the last (4th) pI-pC injection (day 0) or 16, 42 or 70 days after Cre-dependent Mttp inactivation in the atheroregressing group. Yellow block area, mice fed the Western diet; Atheroregression, in the +pI-pC group only; d, days.
Mice were fed the Western diet for 84 days and sacrificed. Aortic cross-sections were stained with Oil Red O and counterstained with hematoxylin (left). Sections were also stained with Moma 2-directed monoclonal Ab to identify lesional foamy macrophages (green, right).
Reversa mice fed the Western diet were sacrificed immediately after the last pI-pC injection (day 0, base level) or 16, 42 and 70 days after Mttp inactivation. Aortas were removed, fixed and sectioned. Representative Oil Red O staining demonstrating the entire aortic root cross-sections of aortas from atherosclerosis-prone (−pI-pC) or atheroregressing (+pI-pC) mice.
Hypercholesterolemia was reversed after 84 days on the Western diet and atheroregressing mice were treated with AMD3100 or saline for a total of 70 days. Peripheral blood was collected and total leukocyte and neutrophil numbers and plasma cholesterol levels were examined. A, Leukocyte and neutrophil numbers were determined with Hemavet. B, Fasting total plasma cholesterol and fasting plasma levels of LDL, HDL and triglycerides were measured using the colorimetric Cholesterol quantification kit.
A, Circulating mononuclear cells obtained from atherosclerosis-prone (−pI-pC) or atheroregressing mouse treated with saline (+pI-pC) or AMD3100 (+pI-pC + AMD3100) were stained and analyzed as detailed in Materials and Methods. Fluorescence staining profiles of Flk1+ cells (left) and expression of CD34 and CD133 in the Flk1+ populations (right) are shown. Numbers in the upper right corner indicate percent of cells with the indicated phenotype. B, Bone marrow was collected from long bones of Tie2-GFP+ donor mice and EPCs isolated by negative selection and sorting were cultured ex vivo for 4 days. A, FACS plot shows representative Lin−GFP+ cells (GFP) co-stained with CD133, Sca-1-, Flk1-specific monoclonal antibodies, or with isotype rat IgG2b antibody. C, Representative images of Lin−GFP+ bone marrow cells (GFP, green) that were prior to adoptive transfer labeled with TRITC-Lectin (red) or evaluated for the uptake of Dil-AcLDL (red), and stained with DAPI (blue). Original magnification ×200.
Reversa mice fed the Western diet for 84 days in which hypercholesterolemia was reversed were either treated with AMD3100 or saline, or received adoptive transfer of Tie2-GFP+ bone marrow EPCs (GFP+EPCs) three times, 10, 20 and 30 days after Mttp inactivation. Mice in all three groups were sacrificed 70 days after the last pI-pC injection and atherosclerosis burden was assessed. Representative Oil Red O staining showing the entire aortic root cross-sections.
Reversa mice fed the Western diet for 84 days in which hypercholesterolemia was reversed were either treated with AMD3100 or saline, or received adoptive transfer of Tie2-GFP+ bone marrow EPCs (GFP+EPCs) three times, 10, 20 and 30 days after Mttp inactivation. Mice in all three groups were sacrificed 70 days after the last pI-pC injection and atherosclerosis burden was assessed. Representative Oil Red O staining showing the entire aortic root cross-sections.
Atherosclerosis-prone (−pI-pC) or atheroregressing (+pI-pC) mice treated with saline or with Tie2-GFP+ bone marrow EPCs were sacrificed 70 days after Mttp inactivation. Aortas were removed, aortic roots were excised and frozen, and cryosections were stained with CXCL1, ICAM-1 or VCAM Ab and mounted in DAPI-containing medium. Representative images of the aortic cross-sections from each group are shown.
Atheroregressing Reversa mice treated with either saline or AMD3100, or with Tie2-GFP+ bone marrow EPCs were sacrificed 70 days after Mttp inactivation and lungs, liver and spleen were collected, frozen, and cryosections were stained with primary rat anti-mouse CD31 or control rat IgG2a antibody and secondary goat anti-rat IgG Alexa Fluor 568 antibody to identify differentiated endothelial cells and mounted in DAPI-containing medium. The only organ that Tie2-GFP+ bone marrow EPCs were detected in addition to the vasculature was spleen. Representative images of the spleen section from each group. DAPI (blue), GFP (green), CD31 (red), red + green emission overlap = yellow.