

Abstract
Investigation of the endemic Madagascan plant Sterculia tavia Baill (Malvaceae) for antiproliferative activity against the A2780 ovarian cancer cell line led to the isolation of two new bioactive calamenene-type sesquiterpenoids, named tavinin A (2) and epi-tavinin A (3) together with the known sesquiterpenoid mansonone G (1). The structures of the two new compounds were elucidated based on analysis of their 1D and 2D NMR spectra and mass spectrometric data, and were confirmed by de novo synthesis. The three isolated sesquiterpenoids (1–3) had modest antiproliferative activities against the A2780 ovarian cancer cell line, with IC50 values of 10.2, 5.5 and 6.7 µM, respectively.
Keywords: Calamenene-type sesquiterpenoid, Antiproliferative activity, Sterculia tavia, Synthesis, Nuclear Magnetic Resonance
1. Introduction
Discovering antiproliferative natural products from both tropical dry forests and rainforests of Madagascar has been one of the objectives of our group’s involvement for more than 15 years in the International Cooperative Biodiversity Group (ICBG) program.1 As a part of this research, an EtOH extract from the bark of Sterculia tavia Arènes (Malvaceae) from the rain forest of northern Madagascar was found to exhibit antiproliferative activity against the A2780 human ovarian cancer cell line, with an IC50 value of 14 µg/mL. The Malvaceae family is a family of flowering plants containing over 200 genera with close to 2,300 species.2 Sterculia, one of the largest genera of this family, is a rich source of alkaloids, saponins and flavonoid glycosides, which are well-known for their broad range of bioactivity, including antimicrobial, antifungal, insecticidal, cytotoxic, antioxidant, and anti-inflammatory activities among others.3 Although the genus Sterculia has been well investigated, the present species is endemic to Madagascar and has not been explored for its phytochemical composition and biological activity. The EtOH extract of S. tavia was thus selected for bioassay-guided fractionation to isolate its antiproliferative components.
2. Results and Discussion
The EtOH extract of the bark part of the S. tavia was subjected to liquid-liquid partitioning to give an active dichloromethane fraction with an IC50 value of 9.6 µg/mL. Bioassay-guided separation, including LH-20 size-exclusion, normal-phase silica gel chromatography, and C-18 reverse-phase HPLC, was used to obtain the known cadinane-type sesquiterpenoid mansonone (1), and the two new calamenene-type sesquiterpenoids designated tavinin A (2) and epi-tavinin A (3) as the major antiproliferative constituents of the extract. Herein, we report the structural elucidation and synthesis of the two new compounds.
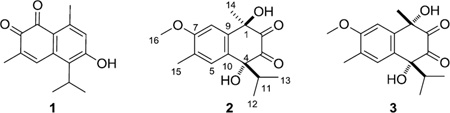
Compound 1 was identified as the cadinane-type sesquiterpenoid mansonone G (6-hydroxy-5-isopropyl-3,8-dimethyl-naphthalene-1,2-dione) by comparison of its chemical and spectroscopic data with those reported in the literature.4
Tavinin A (2), [α] 21D +15° (c 1.2, MeOH), was isolated as a light yellow oil. Its positive ion HRESIMS revealed quasi-molecular ion peaks at m/z 293.1399 [M+H]+ and 315.1194 [M+Na]+, corresponding to a molecular formula of C16H20O5, with seven degrees of unsaturation. Based on the similarity of the NMR spectral data of 2 and those of compound 1, compound 2 was identified as a sesquiterpenoid with a similar carbon skeleton. The six aromatic 13C NMR resonances: δ 162.9, 145.3, 127.8, 123.1, 116.7 and 107.4, as well as the two singlet signals (Table 1) observed in the aromatic region of its 1H NMR spectrum (δ 7.64 and 7.37), revealed the presence of a tetrasubstituted benzene ring with two protons located in the para-position.5 The presence of two deshielded carbon signals at δ 204.0 and 196.7, as well as the compound’s IR absorption (νmax = 1659 cm−1), indicated the presence of two hydrogen-bonded carbonyl groups in the structure. Since the benzene rings and two carbonyl groups contribute four and two degrees of unsaturation, respectively, the remaining one was assigned to a ring. The above data indicated that the basic skeleton of 2 was that of an oxygenated calamenene-type sesquiterpenoid.6 The methyl proton signal located at δ 2.27 and the methoxy proton at δ 4.04 (both singlets) were assignable to a C-6 methyl carbon and to the carbon of a C-7 methoxyl group of the benzene ring.
Table 1.
NMR Spectroscopic Data for 2 and 3 in acetone-d6 (600 MHz)
 | ||||
|---|---|---|---|---|
| 2 | 3 | |||
| position | δH(J in Hz) | δCa type | δH(J in Hz) | δCa, type |
| 1 | - | 73.8, C | - | 74.7, C |
| 2 | - | 204.0, C | - | 208.2, C |
| 3 | - | 196.7, C | - | 200.7, C |
| 4 | - | 88.9, C | - | 91.2, C |
| 5 | 7.64 s | 127.8, CH | 7.68 s | 128.1, CH |
| 6 | - | 116.7, C | - | 116.9, C |
| 7 | - | 162.9, C | - | 163.2, C |
| 8 | 7.37 s | 107.4, CH | 7.33 s | 108.0, C |
| 9 | - | 145.3, C | - | 144.8, C |
| 10 | - | 123.1, C | - | 122.8, C |
| 11 | 2.52 sept | 36.1, CH | 2.35 sept | 35.6, CH |
| 12 | 0.67 d (J= 6.7 Hz) | 15.4, CH3 | 0.81 d (J= 6.7 Hz) | 15.6, CH3 |
| 13 | 0.88 d (J= 6.7 Hz) | 15.9, CH3 | 0.90 d (J= 6.7 Hz) | 16.2, CH3 |
| 14 | 1.62 s | 28.1, CH3 | 1.86 s | 27.4, CH3 |
| 15 | 2.27 s | 15.1, CH3 | 2.27 s | 15.1, CH3 |
| 16 | 4.04 s | 55.5, CH3 | 4.05 s | 55.5, CH3 |
The 13C-NMR spectral data were generated from gHSQC and gHMBC spectra.
This assignment was supported by HMBC correlations (Fig. 1a) between H-15 (δ 2.27) and C-7 (δ 162.9) and the C-5 methine carbon (δ 127.8), and between H-16 (δ 4.04) and C-7. An oxygen-bearing quaternary carbon (δ 88.9, C-4) was determined to be attached to C-10 of the benzene ring (δ 123.1) due the presence of a 3J HMBC crosspeak between the methine proton at H-5 and C-4. The 1H NMR spectrum exhibited resonances at δ 0.67 (d, J = 6.7 Hz, H-12) and δ 0.88 (d, J= 6.7 Hz, H-13) for the two methyl groups of an isopropyl group, and these showed COSY correlations with a methine proton at δ 2.52 (sept, J = 6.8 Hz, H-11). The 3J HMBC correlation between the protons of one of the doublet methyl groups (H-12/H-13) and the carbon at δ 88.9 allowed us to determine the connection of the isopropyl unit to be at C-4. Moreover, the 2J HMBC correlation observed between the protons at δ 1.62 (H-14) and C-1, the 3J HMBC crosspeaks between H-14 and C-9 (δ 145.3) and between H-14 and one carbonyl carbon at δ 204.0 (C-2), as well as the 3J HMBC correlations between the septet methine proton (H-11) and the carbon at δ 196.7 (C-3), allowed us to determine the location of a second oxygen-bearing quaternary carbon (δ 73.8) and the two carbonyl carbons to be at C-1, C-2 and C-3, respectively. The relative configuration of 2 (Fig.1a) as well as the assignment of the aromatic methyl and methoxy groups to C-6 and C-7 respectively, rather than the reverse, was deduced by the interpretation of the results of the 1D NOE-difference experiment. When the H-14 protons (δ 1.62) were irradiated, only the signal at δ 7.37 (H-8) was amplified (1.23%). Similarly, irradiation of H-11 (δ 2.52) only enhanced the aromatic signals at δ 7.64 (H-5) by 3.18%.
Figure 1.

a. HMBC, COSY, and NOESY correlations of 2; b. HMBC, COSY and NOESY correlations of 3
The second compound isolated, epi-tavinin A (3), [α] 21D +8° (c 1.2, MeOH) had the same molecular formula (C16H20O5) as 2 by interpretation of its HRESIMS data. The NMR spectroscopic data of compound 3 were very similar to those of 2 except for the 1H chemical shifts of H-11, H-12, H-13 and H-14. Compounds 2 and 3 were thus assigned as a pair of diastereomers differing in one of their two stereogenic centers (C-1 and C-4). The relative configuration of 3 was determined in the same manner as that of 2. Irradiation of H-14 in a 1D NOE-difference experiment enhanced the signals of both H-8 and H-11 by 1.32% and 1.35%, respectively, indicating a syn relationship between the methyl and isopropyl groups in 3. Based on these observations, the relative configurations of C-1 and C-4 of compound 2 were assigned as R* and R* while those of C-1 and C-4 of 3 were assigned as S* and R*.
The structures of compounds 2 and 3 were confirmed by synthesis (Scheme 1). The synthesis initially followed McCormick's method for the synthesis of lacinilene C methyl ether (10) from 2-methylanisole.7 The literature procedure was modified by running the Grignard reaction to convert the known ketoester 4 to 5-isopropyl-5-(4-methoxy-3-methylphenyl)-dihydrofuran-2-one (6) at low temperature, which increased the yield of 6 from 20% to 42% and of the reduction byproduct 5 from 23% to 30%. Dihydrofuranone 6 was then converted to cycloalkene 7, diol 8, and ketone 9 as previously described,7 except that conversion of 8 to 9 was carried out using the sulfur trioxide pyridine complex instead of the Swern oxidation originally used.7 Oxidation of the carbon-carbon double bond of compound 10 to triol 13 could not be accomplished directly with osmium tetroxide in the presence of N-methylmorpholine N-oxide because of the electron deficient nature of the αβ-unsaturated double bond. The ketone 10 was thus reduced to alcohol 118 by Luche reduction9 in a yield of 90.3%. The product appeared to consist of a single compound as judged by HPLC and by the presence of only one singlet for H-2 in its 1H NMR spectrum, presumably because the orientation of the hydride ion attack was governed by complexation of the borohydride with the adjacent C-1 hydroxyl group. Oxidation of 11 with osmium tetroxide in the presence of N-methylmorpholine N-oxide afforded tetraol 12 as a mixture of stereoisomers. Without separation of this mixture, the tetraols 12 were further oxidized by the sulfur trioxide pyridine complex to give a mixture of four diones as two pairs of enantiomers in the relatively low yield of 43.2%. The diones were separated by HPLC to afford synthetic racemic 2 and synthetic racemic 3. The structures of the natural products isolated were confirmed by comparison of their 1H NMR, UV spectrometric and HRESIMS data with those of their synthetic counterparts.
Scheme 1.

Conditions: a. Isopropylmagnesium chloride, THF, −15 °C, 1h, 42.1%; b. 2% Osmium tetroxide, N-Methyl-morpholine-N-oxide (NMO), dichloromethane, RT, 48h, 75.4%; c. Sulfur trioxide-pyridine complex, dichloromethane, 0 °C, 2h, 91.9%; d. 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ), benzene, reflux, 24h, 58%; e. NaBH4, CeCl3.7H2O, rt, 5min, 90.3%. f. 2% Osmium tetroxide, N-Methyl-morpholine-N-oxide (NMO), dichloromethane, RT, 48 h; g. Sulfur trioxide-pyridine complex, dichloromethane, 0°C, 2 h, 13% (rac-2) 10% (rac-3).
Synthesis of compound 2 and 3
3. Bioassay data
The isolated tavinin A (2) and epi-tavinin A (3), the synthetic compounds rac-2 and rac-3 as well as the intermediates involved in the synthetic route were tested for antiproliferative activity against the A2780 ovarian cancer. As listed in Table 2, both natural products 2 and 3 showed weak inhibition of the A2780 ovarian cancer cells, with IC50 values of 5.5 and 6.7 µM, respectively. The synthetic compounds rac-2 and rac-3 had identical values within experimental error, indicating that bioactivity is not dependent on stereochemistry in this series. Among the synthetic intermediates, only lacinilene C methyl ether (10, IC50 value of 4.0 µM) was of comparable potency to the natural products; the other calamenene-type sesquiterpenoid intermediates were less active or inactive. The cytotoxic activity of compound 10 may be due to the presence of an α,β-unsaturated ketone function in the molecule; such groups are electrophilic and are able to bind to receptors, leading to facile reactions with protein thiols, and ultimately to induction of apoptosis.10
Table 2.
Antiproliferative activity of compounds 1–3 and synthetic intermediates 7–12 against A2780 ovarian cancer cells.
| Compound | IC50 (µ M) |
|---|---|
| 1 | 10.2 ± 0.9 |
| 2 | 5.5 ± 0.9 |
| rac-2 | 5.9 ± 0.7 |
| 3 | 6.7 ± 0.3 |
| rac-3 | 6.3 ± 0.4 |
| 7 | >50 |
| 8 | >50 |
| 9 | 33.9 ± 1.5 |
| 10 | 4.0 ± 0.5 |
| 11 | 22.1 ± 3.4 |
| 12 | 35.5 ± 2.7 |
| Paclitaxel | 0.028± 0.003 |
4. Experimental Section
4.1. General Experimental Procedures
Optical rotations were recorded on a JASCO P-2000 polarimeter. IR spectroscopic data were measured on a MIDAC M-series FTIR spectrophotometer. NMR spectra were recorded in acetone-d6 or CDCl3 on Bruker Avance 500 or Bruker Avance 600 spectrometers. The chemical shifts are given in δ (ppm), and coupling constants (J) are reported in Hz. Mass spectra were obtained on an Agilent 6220 LC-TOF-MS in the positive ion mode.
4.2. Antiproliferative Bioassays
Antiproliferative activities were obtained at Virginia Tech against the drug-sensitive A2780 human ovarian cancer cell line11 as previously described.1c
4.3. Plant Material
A sample of the bark parts of Sterculia tavia Baill was collected in October 2005. The sample was collected from a tree 24 meters high, with yellowish seeds and light brown fruits. The collection was made in the rainforest 8 km from Ankijabe, in the Daraina region of Madagascar, at coordinates 13°16'12"S 049°36'40"E and elevation 617 m. Voucher specimens have been deposited at the Parc Botanique and Zoologique de Tsimbazaza (TAN), at the Centre National d’Application des Recherches Pharmaceutiques in Antananarivo, Madagascar (CNARP), the Missouri Botanical Garden in St. Louis, Missouri (MO), and the Muséum National d’Histoire Naturelle in Paris, France (P), voucher Sennen Randrianasolo et al. 533.
4.4. Extraction and Isolation
Dried bark parts of S. tavia (252 g) were ground in a hammer mill, then extracted with EtOH by percolation for 24 h at room temperature to give the crude extract MG 3532 (5.2 g), of which 1.42 g was shipped to Virginia Tech for bioassay-guided isolation. A 1.4 g sample of MG 3532 (IC50 14 µg/mL) was suspended in aqueous MeOH (MeOH-H2O 9:1, 100 mL), and extracted with hexane (3 × 100 mL portions). The aqueous layer was then diluted to 60% MeOH (v/v) with H2O and extracted with dichloromethane (DCM) (3 150 mL portions). The hexane fraction was evaporated in vacuo to leave 497.5 mg of material with IC50 = 15µg/mL. The residue from the DCM fraction (213.5 mg) had an IC50 of 9.6 µg/mL, and the remaining aqueous MeOH fraction had an IC50 > 20 µg/mL. LH-20 size exclusion open column chromatography (DCM: MeOH=1:1) of the DCM fraction was used to obtain six fractions, of which the two most active fractions F3 (67.7 mg) and F4 (51.4 mg) had IC50 values of 5.9 and 4.8 µg/mL, respectively. Fraction F4 was then applied to a silica gel column with elution by hexane: EtOAc, 7:3 to give seventeen fractions, of which fraction 4 (2.3 mg) was the most active (IC50 3.7 µg/mL), and yielded compound 1 (1.5 mg, IC50 2.5 µg/mL) on chromatography on C-18 HPLC with elution by 85% MeOH in water, with the retention time of 14.9 minute. Fraction F3 was also applied to a silica gel column with elution by hexane: EtOAC = 4:1 to give six fractions, of which fraction 5 (26.2 mg) was the most active (IC50 2.7 µg/mL). Compounds 2 (0.32 mg, IC50 1.7 µg/mL) and 3 (0.28 mg, IC50 1.9 µg/mL), with retention times of 14.7 and 15.2 minutes, respectively, were obtained by using C-18 HPLC eluted by 70% MeOH in water to purify fraction 5.
4.5. (1R,4R)-1,4-dihydroxy-1-isopropyl-6-methoxy-4,7-dimethylnaphthalene-2,3(1H,4H)-dione (2, Tavinin A)
Light yellow oil; [α]D21 +15 (c 1.2, MeOH); UV (MeOH) λmax (ε) 207 (3.20), 232 (2.35), 280 (1.62); IR νmax cm−1: 3418, 2926, 1659, 1463, 1052 cm−1. 1H NMR (600 MHz, acetone-d6), and 13C NMR (125 MHz, acetone-d6), see Table 1; HRESIMS m/z 315.1194 [M+Na]+ (calcd for C16H20NaO5, 315.1208).
4.6. (1R,4S)-1,4-dihydroxy-1-isopropyl-6-methoxy-4,7-dimethylnaphthalene-2,3(1H,4H)-dione (3, Epi-Tavinin A)
Light yellow oil; [α]D21 +8 (c 1.2, MeOH); UV (MeOH) λmax (ε) 207 (3.20), 232 (2.35), 280 (1.62); IR νmax cm−1: 3432, 2932, 1671, 1459, 1078 cm−1. 1H NMR (600 MHz, acetone-d6), and 13C NMR (125 MHz, acetone-d6), see Table 1; HRESIMS m/z 315.1201 [M+Na]+ (calcd for C16H20NaO5, 315.1208).
4.7. Synthesis of 4-hydroxy-4-(4-methoxy-3-methylphenyl)-5-methyl-hexanoic acid lactone (6)
Ketoester 412 (500 mg) was dissolved in 50 mL of THF, and the solution cooled to −15 °C. Isopropyl magnesium chloride (1.5 mL of a 2.0 M solution in THF) was added during a 15 min period. After stirring for 2h, the mixture was dried under reduced pressure, and 50 mL of saturated NH4Cl solution was added. The aqueous solution was extracted with 50 mL Et2O three times, and the extracts were washed with saturated NaCl solution, dried over Na2SO4 and concentrated to afford 468 mg brown oil, which was refluxed together with 500 mg KOH in 100 mL 95% EtOH for 4 h. The EtOH was removed from the mixture under reduced pressure, 100 mL of water was added, and the resulting mixture was extracted three times with 50 mL Et2O. The aqueous portion was acidified to pH 1 with HCl and stirred at rt for 2 h. The acidified solution was extracted three times by another 50 mL of Et2O, and the ethereal potion was washed with saturated Na2CO3 and NaCl solution, respectively, dried by Na2SO4 and concentrated under reduced pressure to afford 313 mg of a yellow oil. Chromatographic purification of the crude lactone (50 g of silica gel; eluted with EtOAc:hexane, 3:7) afforded 167 mg of compound 6 (Rf 0.56, silica gel TLC, EtOAc:hexane, 3:7) and 140 mg of compound 5 (Rf 0.40). The NMR and mass spectrometric data were consistent with the data reported.7
4.8. 1,2,3,4-Tetrahydro-1,2-dihydroxy-4-isopropyl-7-methoxy-1,6-dimethylnaphthalenes (8)
This compound was prepared from 4-hydroxy-4-(4-methoxy-3-methylphenyl)-5-methylhexanoic acid lactone (6) in four steps as previously described; its spectroscopic data matched the literature data.7
4.9. l-Hydroxy-4-isopropyl-7-methoxy-1,6-dimethyl-2-naphthalenone (10)
Diol mixture 8 (50 mg) was dissolved in 50 mL of CH2Cl2 and the solution cooled to 0 °C. Sulfur trioxide pyridine complex (150.6 mg) was added to the solution during a 5 min period. After stirring for 2 h, 50 mL of saturated Na2CO3 solution was added, and the layers were separated. The aqueous layer was extracted with 50 mL of CH2Cl2 three times, and the combined organic layers were washed with saturated NaCl solution, dried over Na2SO4, and concentrated under reduced pressure to afford 45.3 mg of ketone 9 as a yellow oil. The crude ketone and 56.6 mg dichlorodicyanobenzoquinone were stirred in 50 mL of benzene for 24 h at room temperature, after which the solvent was evaporated under reduced pressure. The resulting oil was purified by column chromatography (50 g of silica gel; eluted with EtOAc:hexane, 2:3) to give 28.9 mg (58%) of compound 10 (Rf 0.40). The NMR and mass spectrometric data were consistent with the reported data.7
4.10. 4-Isopropyl-7-methoxy-1,6-dimethyl-1,2-dihydronaphthalene-1,2-diol (11)
Compound 10 (26.0 mg) was dissolved in 25 mL of 0.4 mM methanolic CeCl3.7H2O at room temperature, and 1 equivalent of NaBH4 (3.8 mg) was added to the solution. After stirring for 5 minutes, the mixture was dried under reduced pressure, redissolved in 25 mL of water and extracted three times with 25 mL of dichloromethane. The organic portion was washed with saturated NaCl solution, dried over Na2SO4 and concentrated under reduced pressure. The resulting light yellow oil was purified by column chromatography (50 g of silica gel; eluted with EtOAc:hexane, 2:3) to afford 23.7 mg (90%) of diol 11. The product appeared to consist of a single compound as judged by HPLC and by the presence of only one singlet for H-2 in its 1H NMR spectrum, presumably because the orientation of the hydride ion attack was governed by complications of the borohydride with the adjacent C-1 hydroxyl group. m/z 262.1658; 1H-NMR (CDCl3) δ 7.12 (H-8, s, 1), 7.06 (H-5, s, 1), 5.56 (H-3, br s, 1), 4.48 (H-2, br s, 1), 3.86 (H-16, s, 3), 2.87 (H-11, sept, 1, J = 6.65), 2.19 (H-15, s, 3), 1.30 (H-14, s, 3), 1.19 (H-12, d, 3, J = 6.68), 1.06 (H-13, d, 3, J = 6.68); 13C-NMR (CDCl3) δ 157.5 (C-7, C), 142.6 (C-4, C), 142.2 (C-9, C), 126.2 (C-5, CH), 125.2 (C-10, C), 125.2 (C-6, C), 121.7 (C-3, CH), 105.7 (C-8, CH), 77.1 (C-2, CH), 77.0 (C-1, C), 55.7 (C-16, CH3), 28.2 (C-11, CH); 22.7 (C-14, CH3), 21.6 (C-12, CH3), 20.8 (C-13, CH3), 16.4 (C-15, CH3).
4.11. 1,4-Dihydroxy-1-isopropyl-6-methoxy-4,7-dimethylnaphthalene-2,3-diones rac-2 and rac-3
Diol 11 (22.0 mg) was dissolved in 6 mL of acetone and 2.5 mL of water and the solution cooled to −25 °C. N-methylmorpholine N-oxide (0.05 mL of a 50% solution in water) and 0.1 mL of 2% osmium tetroxide aqueous solution were then added under nitrogen. The mixture was allowed to warm to room temperature and stirred for an additional 48 h, and then 50 mg of Na2S2O4 was added. The acetone was removed under reduced pressure, the pH was adjusted to 2 by HCl, and the solution was extracted with 20 mL dichloromethane three times. The combined extracts were washed with saturated NaCl solution, dried by Na2SO4 and concentrated under reduced pressure to afford product 12 as a light brown oil The crude product was oxidized directly with sulfur trioxide pyridine complex as described in the synthesis of compound 10 above. The resulting 6.6 mg of light brown oil was applied to C-18 HPLC and eluted by 68% MeOH in water to afford 3.2 mg of light yellow oil (rac-2) and 2.5 mg of light yellow oil (rac-3), at retention times of 19.5 and 20.7 min, respectively. The NMR and mass spectrometric data of both compounds were identical to those of the corresponding natural products.
Supplementary Material
Acknowledgments
This project was supported by the Fogarty International Center, the National Cancer Institute, the National Science Foundation, the National Heart, Lung and Blood Institute, the National Institute of Mental Health, the Office of Dietary Supplements, and the Office of the Director of NIH, under Cooperative Agreement U01 TW000313 with the International Cooperative Biodiversity Groups. This project was also supported by the National Research Initiative of the Cooperative State Research, Education and Extension Service, USDA, Grant #2008-35621-04732. These supports are gratefully acknowledged. This work was also supported by the National Science Foundation under Grant no. CHE-0619382 for purchase of the Bruker Avance 500 NMR spectrometer and Grant no. CHE-0722638 for the purchase of the Agilent 6220 mass spectrometer. We thank Mr. Bill Bebout for obtaining the mass spectra and Dr. Hugo Azurmendi for assistance with the NMR spectra. Field work essential for this project was conducted under a collaborative agreement between the Missouri Botanical Garden and the Parc Botanique et Zoologique de Tsimbazaza and a multilateral agreement between the ICBG partners, including the Centre National d’Applications des Recherches Pharmaceutiques. We gratefully acknowledge courtesies extended by the Government of Madagascar (Ministère des Eaux et Forêts).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Biodiversity Conservation and Drug Discovery in Madagascar, Part 53. For Part 52, see Ref. 1b.
Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:XXX
References and notes
- 1.a) Zhou BN, Baj NJ, Glass TE, Malone S, Werkhoven MCM, van Troon F, David M, Wisse JH, Kingston DGI. J. Nat. Prod. 1997;60:1287. doi: 10.1021/np970233c. [DOI] [PubMed] [Google Scholar]; b) Dai Y, Harinantenaina L, Brodie PJ, Birkinshaw C, Randrianasolo S, Ratsimbason M, Rasamison VE, Shen Y, TenDyke K, Kingston DGI. Chem. Biodivers. 2012 doi: 10.1002/cbdv.201200156. accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Pan E, Harinantenaina L, Brodie PJ, Callmander M, Rakotonandrasana S, Rakotobe E, Rasamison VE, TenDyke K, Shen Y, Suh EM, Kingston DGI. Bioorg. Med. Chem. 2011;19:422. doi: 10.1016/j.bmc.2010.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Pan E, Gorka AP, Alumasa JN, Slebodnick C, Harinantenaina L, Brodie PJ, Roepe PD, Randrianaivo R, Birkinshaw C, Kingston DGI. J. Nat. Prod. 2011;74:2174. doi: 10.1021/np200499d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Judd W, Manchester S. Brittonia. 1997;49:384. [Google Scholar]
- 3.a) Yang H, Protiva P, Cui B, Ma C, Baggett S, Hequet V, Mori S, Weinstein IB, Kennelly EJ. J. Nat. Prod. 2003;66:1501. doi: 10.1021/np034002j. [DOI] [PubMed] [Google Scholar]; b) Reid KA, Jäger AK, Light ME, Mulholland DA, Staden JV. J. Ethnopharmacol. 2005;97:285. doi: 10.1016/j.jep.2004.11.010. [DOI] [PubMed] [Google Scholar]; c) Rani P, Rajasekharreddy P. J. Pest Sci. 2010;83:273. [Google Scholar]; d) Naik DG, Mujumdar AM, Waghole RJ, Misar AV, Bligh SW, Bashall A, Crowder J. Planta Med. 2004;70:68. doi: 10.1055/s-2004-815459. [DOI] [PubMed] [Google Scholar]
- 4.a) Tiew P, Puntumchai A, Kokpol U, Chavasiri W. Phytochemistry. 2002;60:773. doi: 10.1016/s0031-9422(02)00194-2. [DOI] [PubMed] [Google Scholar]; b) Tanaka N, Yasue M, Imamura H. Tetrahedron Lett. 1966;7:2767. [Google Scholar]
- 5.Nabeta K, Katayama K, Nakagawara S, Katoh K. Phytochemistry. 1992;32:117. [Google Scholar]
- 6.Kashman Y. Tetrahedron. 1979;35:263. [Google Scholar]
- 7.McCormick JP, Shinmyozu T, Pachlatko JP, Schafer TR, Gardner JW, Stipanovic RD. J. Org. Chem. 1984;49:34. [Google Scholar]
- 8.Jeffs PW, Lynn DG. J. Org. Chem. 1975;40:2958. [Google Scholar]
- 9.Gemal AL, Luche JL. J. Am. Chem. Soc. 1981;103:5454. [Google Scholar]
- 10.Nakayachi T, Yasumoto E, Nakano K, Morshed SR, Hashimoto K, Kikuchi H, Nishikawa H, Kawase M, Sakagami H. Anticancer Res. 2004;24:737. [PubMed] [Google Scholar]
- 11.Louie KG, Behrens BC, Kinsella TJ, Hamilton TC, Grotzinger KR, McKoy WM, Winker MA, Ozols RF. Cancer Res. 1985;45:2110. [PubMed] [Google Scholar]
- 12.Bell AA, Stipanovic RD, Zhang J, Mace ME, Reibenspies JH. Phytochemistry. 1998;49:431. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.