

Abstract
Chemotherapeutic agents- and radiation therapy-induced NF-κB activation in cancer cells contributes to aggressive tumor growth and resistance to chemotherapy and ionizing radiation during cancer treatment. TAK1 has been shown to be required for genotoxic stress-induced NF-κB activation. However, whether TAK1 ubiquitination is involved in genotoxic stress-induced NF-κB activation remains unknown. Herein, we demonstrate that TAK1 ubiquitination plays an important role in the positive and negative regulation of Doxorubicin (Dox)-induced NF-κB activation. We found that TAK1 was required for Dox-induced NF-κB activation. At the early stage of Dox treatment, Dox induced Lys63-linked TAK1 polyubiquitination at lysine 158 residue. USP4 inhibited Dox-induced TAK1 Lys63-linked polyubiquitination and knockdown of USP4 enhanced Dox-induced NF-κB activation. At the late stage of Dox treatment, Dox induced Lys48-linked TAK1 polyubiquitination to promote TAK1 degradation. ITCH inhibited Dox-induced NF-κB activation by promoting Lys48-linked TAK1 polyubiquitination and its subsequent degradation. Our study indicates that TAK1 ubiquitination plays critical roles in the regulation of Dox-induced NF-κB activation. Thus, intervention of TAK1 kinase activity or TAK1 Lys63-linked polyubiquitination pathways might greatly enhance the therapeutic efficacy of Dox.
Keywords: TAK1, Ubiquitination, Doxorubicin, USP4, ITCH
Introduction
In the last decade, radiotherapy, chemotherapy or combinations of radiotherapy and chemotherapy have become the nonsurgical standard of care in many locally advanced tumors [1, 2]. However, the efficacy of currently available chemotherapeutic and radiotherapeutic agents in cancer patients is largely reduced by a number of resistance mechanisms [3–5]. Activation of transcription factor NF-κB is frequently encountered in tumor cells and is believed to be one of resistance mechanisms that contribute to aggressive tumor growth and resistance to chemotherapy and radiotherapy during cancer treatment [6–8]. Accumulating evidences over the last few years indicate that most chemotherapeutic agents and radiation therapy activate NF-κB in vitro and in vivo. However, how chemotherapeutic agents and radiation therapy activate NF-κB remains to be fully understood.
Currently, ionizing radiation (IR) and chemotherapeutic drugs, such as topoisomerase I inhibitor camptothecin (CPT), topoisomerase II inhibitor etoposide (VP16), and the DNA-intercalating agent doxorubicin (Dox) are commonly used in clinic to treat many kinds of cancer [9, 10]. Ionizing radiation and the above common chemotherapeutic drugs are DNA-damaging agents and cause genotoxic stresses. Mammalian cells have a network of highly conserved DNA-repair and cell-cycle checkpoint pathways to respond to genotoxic stresses. Double-strand breaks (DSBs), one of common DNA lesions caused by genotoxic stresses, are recognized by the Mre11-Rad50-Nbs1 (MRN) complex which transiently recruits the kinase ATM [11, 12]. DNA double-strand breaks are also sensed by the poly-(ADP)-ribosylating enzyme poly(ADP-ribose) polymerase 1 (PARP-1), which synthesizes poly-(ADP-ribose) and attaches to itself or other acceptor proteins [13–15]. Both PARP-1 and ATM are required for NF-κB activation in genotoxic stress response [16, 17]. However, how nuclear genotoxic stresses activate cytosolic IKK complex remains to be fully defined.
TAK1, a member of the MAPK kinase kinase family, is originally found to function in the transforming growth factor-β (TGF-β)-mediated MAPK activation [18]. TAK1 is critical for IKK activation in response to multiple stimuli [19–23]. Recently, several groups have reported that TAK1 mediates NF-κB activation in response to genotoxic stress [24–27]. However, the biological roles of TAK1 in response to different genotoxic stimuli are still controversial.
Ubiquitination is a reversible post-translational modification involving the covalent attachment of one or more ubiquitin monomers to a protein substrate. Ubiquitin has seven lysines (K6, K11, K27, K29, K33, K48 and K63), all of which can be conjugated to another ubiquitin to form a polyubiquitin chain through different lysine linkages to serve distinct functions in the cells [28]. Lys63-linked TAK1 polyubiquitination is required for NF-κB activation by TNFα, IL-1β and TGFβ [29–35]. Lys48-linked TAK1 polyubiquitination-mediated TAK1 degradation is critical for the termination of TNFα-induced NF-κB activation [36, 37]. Therefore, TAK1 ubiquitination plays an important role in the positive and negative regulation of TNFα-induced NF-κB activation. However, it is unclear whether TAK1 ubiquitination is also involved in genotoxic stress-induced NF-κB activation. In this study, we demonstrate that TAK1 ubiquitination plays a critical role in the regulation of Dox-induced NF-κB activation.
Experimental procedures
Plasmids
HA-Ub-wildtype, HA-Ub-K63-only mutant, HA-Ub-K48-only mutant, FLAG-TAK1-wildtype and TAK1-V5His expression constructs were created as previously described [29–31]. Mammalian expression vectors for USP4 wildtype was constructed as previously described [32]. The TAK1-K72R-V5His mutant was generated as previously described [36]. FLAG-ITCH was provided by Dr. Claudius Vincenz and Dr. Edward W. Harhaj.
Antibodies and reagents
The following antibodies and reagents were used: anti-phospho-IKKα/β (2078S), anti-IKKα/β (2684S), anti-phospho-p38 (9211L), anti-p38 (9212), anti-phospho-JNK (9251L), anti-JNK (9252L), anti-phospho-ERK (9106L), anti-ERK (9102), anti-TAK1(4505S), anti-mouse (7076S) and anti-Rabbit (7074S) secondary antibodies were from Cell Signaling. Anti-β-Actin (A2228) and anti-FLAG (F3165) were from Sigma. Anti-ITCH (611198) was from BD Transduction Laboratories. Anti-HA (SC-7392), anti-Ub (SC-8017) and Protein A-agarose (SC-2001) were from Santa Cruz. Anti-USP4 (A300-829A) was from Bethyl. Anti-V5 and anti-FLAG antibodies were generated by immunizing rabbits with the synthetic peptides corresponding to amino acids-GKPIPNPLLGLDST and DYKDDDDK, respectively (Genemed Synthesis, Inc., San Antonio, TX). Doxorubicin (D1515), etoposide (E1383) and camptothecin (C9911) were from Sigma.
Cell lines and cell culture
HeLa, HEK-293T and MEF cells were maintained in Dulbecco’s modified Eagle’s medium (high glucose) supplemented with 10% fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in 5% CO2. TAK1-deficient and knockin MEFs were established as previously described [29–31, 36]. USP4 stable knockdown HeLa cells were generated as previously described [36]. ITCH-deficient MEFs were provided by Lydia Matesic.
Immunoprecipitation and immunoblotting
To prepare the total cell lysates, transfected or treated cells were washed 3 times with ice-cold PBS on ice and then scraped in lysis buffer (25 mM HEPES (pH 7.7), 135 mM NaCl, 1% Triton X-100, 25 mM β-glycerophosphate, 0.1 mM sodium orthovanadate, 0.5 mM phenylmethylsulfonylfluoride, 1 mM dithiothreitol, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM Benzamidine, 20 mM disodium p-nitrophenylphosphate, 1 mM phenylmethylsulfonyl fluoride, and phosphatase inhibitor cocktail 2 and 3). Cell lysate were collected and centrifuged by 15,000 g for 15 min at 4 °C. The supernatant was transferred to a new tube and protein concentration was determined. For immunoprecipitation, primary antibodies were added to the supernatant and incubated with rotation for 3 h at 4 °C. Then the protein A-agarose beads were further incubated with rotation for 3 h at 4 °C. After that, the mixtures were centrifuged by 5,000 rpm for 4 min The precipitates were washed three times using pre-cold washing buffer (20mM HEPES (pH 7.7), 50mM NaCl, 2.5mM MgCl2, 0.1mM EDTA and 0.05% Triton X-100), then the beads were resuspended in Laemmli sample buffer and boiled for 10 min. The immunoprecipitates or the whole cell lysates were resolved by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were probed with appropriate primary antibodies and the IgG horseradish peroxidase-conjugated antibodies. The proteins were detected using the ECL-Plus Western blotting detection system (GE Health Care, Buckinghamshire, UK).
CCK-8 cell proliferation assay
Cells were seeded in 96-well plates at the concentration of 10,000 cells per well. 24 hours later, cells were checked by microscopy and treated by Dox at indicated concentrations for 48 hours. Cell viability was measured by the tetrazolium salt-based proliferation assay (CCK-8 assay, Dojindo Laboratories) following the manufacturer’s instructions.
Results
TAK1 is required for Dox-induced NF-κB activation
TAK1 has been suggested to mediate NF-κB activation in response to genotoxic stress [24–27]. To better understand the role of TAK1 in IR-, CPT-, VP16- and Dox-induced NF-κB and MAPK activation, we examined the IR-, CPT-, VP16- and Dox-induced IKKs, JNK, p38 and ERK phosphorylation in wildtype and TAK1-deficient MEFs. As expected, Dox induced IKKs, JNK, p38 and ERK phosphorylation in wildtype MEFs after one hour of stimulation. However, unlike ERK phosphorylation, Dox-induced IKKs, JNK and p38 phosphorylation were greatly impaired in TAK1-deficient MEFs (Fig. 1A). Similar results were also observed in MEFs treated with other genotoxic agents such as VP16, CPT and IR (Fig. 1C, 1D and 1E). To further confirm the role of TAK1 in Dox-induced NF-κB and MAPK activation, the control vector and TAK1 wildtype were stably introduced back into the TAK1-deficient MEF cells by a retroviral transduction system. Then vector and TAK1 wildtype reconstituted MEF cells were treated with Dox at different time points, and then the cell lysates were immunoblotted with the indicated antibodies to examine Dox-induced IKKs, JNK, p38 and ERK phosphorylation. As showed in Fig. 1B, TAK1 wildtype but not vector control rescued the Dox-induced IKKs, JNK and p38 phosphorylation in TAK1-deficient MEFs. These results suggest that TAK1 is definitely required for Dox-induced NF-κB, JNK and p38 activation.
Figure 1. TAK1 is required for doxorubicin (Dox) induced NF-κB, p38 and JNK activation.

(A) TAK1+/+ and TAK1−/− MEFs were treated with Dox at the indicated time points or left untreated, protein extracts were subjected to SDS-PAGE and immunoblotted with antibodies indicated. (B) TAK1-deficient and TAK1 wildtype reconstituted MEF cells were treated with Dox at the indicated time points or left untreated, protein extracts were subjected to SDS-PAGE and immunoblotted with antibodies indicated. (C) TAK1+/+ and TAK1−/− MEFs were treated with VP-16 at the indicated time points or left untreated, protein extracts were subjected to SDS-PAGE and immunoblotted with antibodies indicated. (D) TAK1+/+ and TAK1−/− MEFs were treated with CPT at the indicated time points or left untreated, protein extracts were subjected to SDS-PAGE and immunoblotted with antibodies indicated. (E) TAK1+/+ and TAK1−/− MEFs were treated with ionizing radiation (IR) at the indicated dose or left untreated, protein extracts were subjected to SDS-PAGE and immunoblotted with antibodies indicated. β-actin was detected as a loading control for whole cell extracts.
Lys63-linked TAK1 polyubiquitination at Lys-158 is required for Dox-induced NF-κB activation
Lys63-linked TAK1 polyubiquitination is required for TNFα- and IL-1β-induced IKK-NF-κB activation [29–31]. However, whether Lys63-linked TAK1 polyubiquitination is also involved in Dox-induced NF-κB activation remains unknown. To answer this question, we first tested whether Dox could induce Lys63-linked TAK1 polyubiquitination. In this assay, we co-transfected FLAG-TAK1 into HEK-293T cells along with HA-ubiquitin Lys63-only (HA-Ub-K63) mutant. Transfected cells were treated with or without Dox for the time points indicated. FLAG-TAK1 was then immunoprecipitated from cell lysates and blotted with anti-HA antibodies to detect ubiquitinated TAK1. As showed in Fig. 2A, Dox induced Lys63-linked TAK1 polyubiquitination. We further confirmed this result by using a FLAG-TAK1 stably expressed HeLa cell line (Fig. 2B).
Figure 2. Lys63-linked TAK1 polyubiquitination at Lys-158 is required for Dox-induced NF-κB activation.

(A) Expression vectors encoding HA-Ub-K63 and FLAG-TAK1 were co-transfected into HEK-293T cells. Thirty six hours after transfection, cells were treated with Dox at the indicated time points or left untreated. FLAG-TAK1 proteins in the cell lysates were immunoprecipitated with anti-FLAG antibodies and immunoblotted with anti-HA antibodies to detect the presence of polyubiquitinated FLAG-TAK1. (B) HeLa cells with stable expression of FLAG-TAK1 were treated with Dox at the indicated time points or left untreated. Then, FLAG-TAK1 proteins in the cell lysates were immunoprecipitated with anti-FLAG antibodies and immunoblotted with anti-ubiquitin antibodies to detect the presence of polyubiquitinated FLAG-TAK1. (C) TAK1 wildtype and TAK1 K158R mutant reconstituted MEF cells were treated with Dox at the indicated time points or left untreated, cell extracts were subjected to SDS-PAGE and immunoblotted with antibodies indicated. (D) pBabe vector, TAK1 wildtype and TAK1 K158R mutant reconstituted TAK1-deficient MEFs were seeded into 96-well plates at the concentration of 10,000 cells per well. 24 hours later, cells were treated by Dox at the indicated concentrations for 48 hours. Cell viability was measured by the CCK-8 assay following the manufacturer’s instructions.
We previously showed that Lys63-linked TAK1 polyubiquitination occurs at Lys-158 and this modification is critical for NF-κB activation induced by many stimuli [29–31]. Thus, we tested whether Lys63-linked TAK1 polyubiquitination at Lys-158 is also required for Dox-induced NF-κB activation. TAK1 wildtype and K158R mutant reconstituted MEF cells were treated with Dox at different time points, and then the cell lysates were immunoblotted with the indicated antibodies to examine Dox-induced IKKs, JNK, and p38 phosphorylation. As showed in Fig. 2C, Dox-induced IKKs, JNK, and p38 phosphorylation were greatly impaired in MEFs reconstituted with TAK1 K158R mutant compared to ones with TAK1 wildtype. Consistently, TAK1-deficient and K158R mutant reconstituted MEF cells were more sensitive to Dox-induced cell death compared to MEFs with TAK1 wildtype (Fig. 2D). These data suggest that Lys63-linked TAK1 polyubiquitination at Lys-158 is required for Dox-induced NF-κB activation.
USP4 deubiquitinates TAK1 with Lys63-linked polyubiquitination and inhibits Dox-induced NF-κB activation
USP4 inhibits TNFα-induced NF-κB activation by acting as a TAK1 deubiquitinase [32]. Therefore, we want to test whether USP4 also acts as a TAK1 deubiquitinase to inhibit Dox-induced NF-κB activation. We overexpressed FLAG-TAK1 and MYC-USP4 with HA-Ub-K63 mutant in HEK-293T cells and then treated cells with Dox for the time points indicated. In this assay, we found that Dox-induced TAK1 Lys63-linked polyubiquitination was inhibited by USP4 (Fig. 3A). To determine whether USP4 is involved in the negative regulation of Dox-induced NF-κB activation, we generated USP4 stable knockdown HeLa cell line using a retroviral transduction system (Fig. 3B). We then analyzed the effect of USP4 knockdown on Dox-induced IKKs phosphorylation. In this assay, we found that Dox induced a higher level of IKKs phosphorylation in the USP4-knockdown cells compared with the control cells (Fig. 3B). Furthermore, knockdown of USP4 enhances Dox-induced JNK and p38 phosphorylation (Fig. 3B). Therefore, USP4 acts as a TAK1 deubiquitinase to inhibit Dox-induced NF-κB activation.
Figure 3. USP4 deubiquitinates Lys63-linked TAK1 polyubiquitination and inhibits Dox-induced NF-κB activation.

(A) Expression vectors encoding HA-Ub-K63 and FLAG-TAK1 were co-transfected into HEK-293T cells with control vector or expression vectors encoding MYC-USP4. Thirty six hours after transfection, cells were treated with Dox at the indicated time points or left untreated. FLAG-TAK1 proteins in the transfected cells were immunoprecipitated with anti-FLAG antibodies and immunoblotted with anti-HA antibodies to detect the presence of polyubiquitinated FLAG-TAK1. (B) USP4 stable knockdown and sh-Control HeLa cells were treated with Dox at the indicated time points or left untreated. Then, protein extracts were subjected to SDS-PAGE and immunoblotted with antibodies indicated.
Dox induces Lys48-linked TAK1 polyubiquitination and degradation
Lys48-linked TAK1 polyubiquitination mediated TAK1 degradation plays a critical role in the termination of TNFα-induced NF-κB activation [36, 37]. We then tested whether Dox could induce Lys48-linked TAK1 polyubiquitination and degradation. In this assay, we co-transfected FLAG-TAK1 into HEK-293T cells with HA-Ub-K48 mutant. Transfected cells were treated with MG132 along with or without Dox for the time points indicated. FLAG-TAK1 was then immunoprecipitated from cell lysates and blotted with anti-HA antibodies to detect TAK1 with Lys48-linked polyubiquitination. We found that Dox induced Lys48-linked TAK1 polyubiquitination in the presence of MG132, while without Dox stimulation, MG132 alone was not sufficient to cause Lys48-linked TAK1 polyubiquitination (Fig. 4A). Dox-induced Lys48-linked TAK1 polyubiquitination was also confirmed in FLAG-TAK1 stably expressed HeLa cell line (Fig. 4B). Next we examined whether Dox could induce TAK1 degradation in HeLa cells. We first incubated the cells with cycloheximide (CHX) to block protein synthesis. Consistent with the observation that Dox could induce Lys48-linked TAK1 polyubiquitination, Dox promoted degradation of a significant portion of TAK1 protein after sixteen hours of treatment (Fig. 4C and 4D).
Figure 4. Dox induces Lys48-linked TAK1 polyubiquitination and degradation.

(A) Expression vectors encoding HA-Ub-K48 and FLAG-TAK1 were co-transfected into HEK-293T cells. Thirty six hours after transfection, cells were treated with Dox and MG132 at the indicated time points or left untreated. FLAG-TAK1 proteins in the cell lysates were immunoprecipitated with anti-FLAG antibodies and immunoblotted with anti-HA antibodies to detect the presence of polyubiquitinated FLAG-TAK1. (B) Stable expression of FLAG-TAK1 or control HeLa cells were treated with Dox and MG132 at the indicated time points or left untreated. FLAG-TAK1 proteins in the cell lysates were immunoprecipitated with anti-FLAG antibodies and immunoblotted with anti-ubiquitin antibodies to detect the presence of ubiquitinated FLAG-TAK1. (C) HeLa cells were treated with Dox and cycloheximide (CHX) at the indicated time points or left untreated. TAK1 proteins in the cell lysates were detected by anti-TAK1 antibodies. (D) Quantification of C.
ITCH promotes Lys48-linked TAK1 polyubiquitination and inhibits Dox-induced NF-κB activation
Recently, ITCH is suggested to be a TAK1 E3 ligase to mediate TAK1 Lys48-linked polyubiquitination and negatively regulates TNFα-induced NF-κB activation [37]. To test whether ITCH is a TAK1 E3 ligase to mediate Dox-induced Lys48-linked polyubiquitination of TAK1, we expressed TAK1-V5His and FLAG-ITCH along with HA-Ub-wildtype, HA-Ub-K48 or HA-Ub-K63. We observed more polyubiquitinated forms of TAK1 when ITCH and TAK1 were expressed together compared to TAK1 alone (Fig. 5A). ITCH catalyzed TAK1 polyubiquitination mainly through Ub-K48 (Fig. 5A). TAK1 Lys-72 has been suggested to mediate TNFα-induced TAK1 polyubiquitination with Lys48 linkage type [36, 37]. Here we examined whether ITCH mediated Lys48-linked TAK1 polyubiquitination through Lys-72 site. To test this, we expressed FLAG-ITCH and HA-tagged Ub K48 mutant along with TAK1 wildtype or K72R mutant. As shown in Fig. 5B, ITCH dramatically promoted Lys48-linked polyubiquitination of TAK1 wildtype but not K72R mutant. This result suggests that ITCH mediates Lys48-linked TAK1 polyubiquitination mainly through TAK1 Lys-72 residue.
Figure 5. ITCH promotes Lys48-linked TAK1 polyubiquitination and inhibits Dox-induced NF-κB activation.

(A) Expression vectors encoding HA-Ub-WT, HA-Ub-K63, HA-Ub-K48 and TAK1-V5His were co-transfected into HEK-293T cells with control vector or expression vectors encoding FLAG-ITCH. Thirty six hours after transfection, TAK1-V5His proteins in the cell lysates were immunoprecipitated with anti-V5 antibodies and immunoblotted with anti-HA antibodies to detect the presence of polyubiquitinated TAK1-V5His. (B) Expression vectors encoding HA-Ub-K48 and TAK1-wildtype-V5His or TAK1-K72R-V5His were co-transfected into HEK-293T cells with control vector or expression vectors encoding FLAG-ITCH. Thirty six hours after transfection, TAK1-V5His proteins in the cell lysates were immunoprecipitated with anti-V5 antibodies and immunoblotted with anti-HA antibodies to detect the presence of polyubiquitinated TAK1-V5His. (C) ITCH+/+ and ITCH−/− MEFs were treated with CHX and Dox at the indicated time points or left untreated, protein extracts were subjected to SDS-PAGE and immunoblotted with antibodies indicated. (D) ITCH+/+ and ITCH−/− MEFs were treated with Dox at the indicated time points or left untreated, protein extracts were subjected to SDS-PAGE and immunoblotted with antibodies indicated.
Because ITCH was a TAK1 E3 ligase to mediate TAK1 Lys48-linked polyubiquitination, we reasoned that TAK1 should be more stable in ITCH-deficient MEFs than in wildtype MEFs. Therefore, we examined TAK1 protein level and Dox-induced TAK1 degradation in wildtype and ITCH-deficient MEFs. As expected, the TAK1 protein level is much higher in ITCH-deficient MEFs than in wildtype MEFs (Fig. 5C). Futhermore, in ITCH-deficient MEFs, TAK1 was resistant to Dox-induced degradation when compared to its level in wildtype MEFs (Fig. 5C). Furthermore, we investigated the effect of ITCH deficiency on Dox-induced NF-κB activation. As shown in Fig. 5D, Dox induced a dramatically higher and prolonger IKKs, JNK and p38 phosphorylation in ITCH-deficient MEFs when compared to wildtype MEFs. These data collectively suggest that ITCH negatively regulates Dox-induced NF-κB activation by acting as a TAK1 E3 ligase.
Discussion
The efficacy of currently available chemotherapeutic and radiotherapeutic agents in cancer patients is largely dampened by a number of resistance mechanisms. Activation of transcription factor NF-κB is one of resistance mechanisms that contribute to resistance to chemotherapy and ionizing radiation during cancer treatment. Therefore, understanding the molecular mechanisms of how NF-κB activation is induced by chemotherapy and radiotherapy is of pivotal importance. Here, we provide convincing genetic and biochemical evidences to prove that TAK1 polyubiquitination plays an important role not only in Dox-induced NF-κB activation but also in the termination of Dox-induced NF-κB activation. TAK1 positively and negatively regulates its downstream signaling events through its dynamic polyubiquitination modification. At the early stage of Dox treatment, TAK1 is modified by Lys63-linked polyubiquitination at Lys-158 that is required for Dox-induced NF-κB activation. Subsequently, activated TAK1 will be modified by Lys48-linked polyubiquitination at Lys-72 that results in TAK1 degradation to terminate Dox-induced NF-κB activation. To obtain an optional NF-κB activation, cells have evolved a precise ubiquitination regulatory mechanism to control TAK1 activity. Given the pivotal role of TAK1 and TAK1 ubiquitination in Dox-induced NF-κB activation, we reasoned that inhibition of TAK1 kinase activity or intervention of TAK1 ubiquitination pathways might greatly enhance the therapeutic efficacy of Dox.
The human genome encodes about 95 putative deubiquitinating enzymes (DUBs) that are divided into five subclasses based on their Ub-protease domains [38]. USP4 is a member of the Ub-specific peptidases (USPs) family that represents the largest subclass of DUBs. USP4 is previously reported to act as a TAK1 deubiquitinase to inhibit TNFα- and IL-1β-induced NF-κB activation [32]. Here, we provide evidence to show that USP4 also acts as a TAK1 deubiquitinase to inhibit Dox-induced NF-κB activation. High expression of USP4 in cancer cells might be beneficial to chemotherapy-induced cancer cell death. Moreover, USP4 is recently showed to enhance EMT and metastasis by stabilizing TβRI [39]. It is very possible that USP4 has diverse or even opposite roles in tumor initiation, tumor development, metastasis and chemotherapy response by targeting on different substrates.
ITCH (AIP4) is an E3 ubiquitin ligase that was originally identified through genetic studies aimed to examine the agouti locus, whose mutation results in coat color alterations in mice [40]. After that there are an emerging number of Itch protein targets that have been implicated in tumorigenesis and chemosensitivity [41, 42]. Recently, ITCH is shown to inhibit lung cancer growth by acting as a TAK1 Lys48-specific E3 ligase to promote TAK1 degradation [37]. Consistent with their reports, we found that ITCH catalyzed Dox-induced Lys48-linked TAK1 polyubiquitination at TAK1 Lys-72 residue and inhibited Dox-induced NF-κB activation by promoting TAK1 degradation. Therefore, loss of ITCH may play a role in the resistance to chemotherapy. Moreover, ITCH also stabilizes p63 and p73 to promote DNA damage-induced cell death [43–47]. Together, the role of ITCH in the resistance of chemotherapy is worth further study.
In summary, here we demonstrate that Lys63-linked TAK1 polyubiquitination at Lys-158 is required for Dox-induced NF-κB activation. USP4 acts as a TAK1 Lys63-specific deubiquitinase to deubiquitinate TAK1 and inhibit Dox-induced NF-κB activation. Forthermore, Dox also induces Lys48-linked TAK1 polyubiquitination at the late stage which is critical for TAK1-mediated NF-κB termination. ITCH acts as a TAK1 Lys48-specific E3 ligase to promote Lys48-linked TAK1 polyubiquitination mainly at Lys-72 site and terminate Dox-induced NF-κB activation. In view of the data presented here and previous reports, we proposed a working model to show how TAK1 ubiquitination regulates Dox-induced NF-κB activation and termination. Dox causes double-stranded breaks (DSBs) that activate PARP-1 and ATM. PARP-1 signalosome formation promotes NEMO SUMOylation. The coupled export of ATM and sumoylated NEMO translocates to the cytoplasm and assembles a large complex containing TRAF6, cIAP and ELKS. Lys63-linked polyubiquitination of TRAF6 or ELKS recruits TAB2/TAK1 complex and TRAF6 to catalyze Lys63-linked TAK1 polyubiquitination at Lys-158 site. TAK1 with Lys63-linked polyubiquitination recruits IKKs complex and results in IKK-NF-κB activation. After TAK1 is activated with Lys63-linked polyubiquitination by Dox, USP4 deubiquitinates TAK1 and inhibits TAK1-mediated signaling. Meanwhile, E3 ligase ITCH catalyzes Lys48-linked TAK1 polyubiquitination and promotes TAK1 degradation to terminate TAK1-mediated signal transduction (Fig. 6).
Figure 6. Working model.
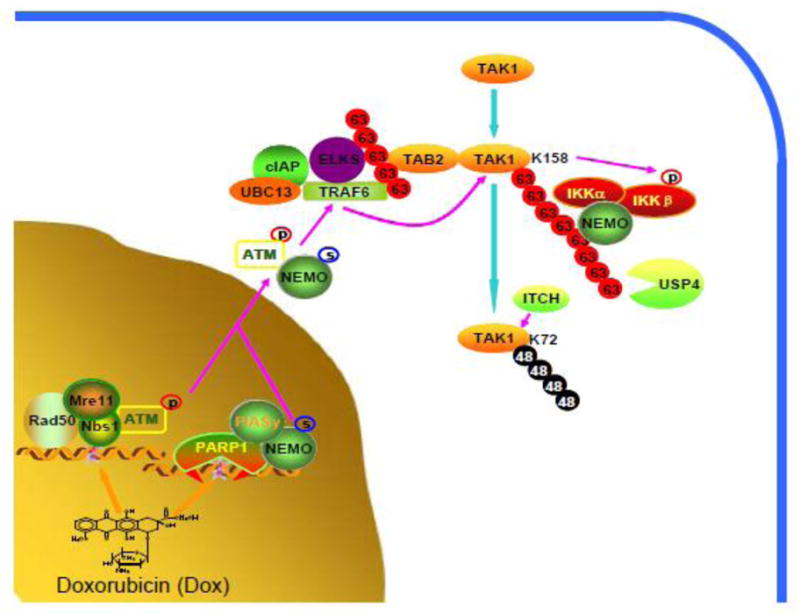
Dox-induced double-stranded breaks (DSBs) activate PARP-1 and ATM. PARP-1 signalosome formation promotes NEMO SUMOylation. The coupled export of ATM and sumoylated NEMO translocates to the cytoplasm and assembles a large complex containing TRAF6, cIAP, ELKS. Lys63-linked polyubiquitination of TRAF6 or ELKS recruits TAB2/TAK1 complex and TRAF6 further promotes Lys63-linked polyubiquitination of TAK1 at Lys-158 site. TAK1 with Lys63-linked polyubiquitination recruits IKKs complex and activates IKKs and NF-κB. After TAK1 activation, USP4 deubiquitinates TAK1 with Lys63-linked polyubiquitination and inhibits TAK1-mediated downstream signal transduction. Meanwhile, E3 ligase ITCH catalyzes Lys48-linked polyubiquitination of TAK1 and promotes TAK1 degradation to terminate downstream signal transduction.
Highlights.
TAK1 is required for doxorubicin (Dox)-induced NF-κB activation.
Lys63-linked TAK1 polyubiquitination at lysine 158 residue is required for Dox-induced NF-κB activation.
USP4 inhibits Dox-induced Lys63-linked TAK1 polyubiquitination and NF-κB activation.
Dox induces Lys48-linked TAK1 polyubiquitination to promote TAK1 degradation.
ITCH inhibits Dox-induced NF-κB activation by promoting Lys48-linked TAK1 polyubiquitination and its subsequent degradation.
Acknowledgments
This work was supported, in whole or in part, by the NIH/NINDS grant 1R01NS072420-01 (to J.Y.), the state Ministry of Health of China grant 201002009 (to L.L.). Jin Cheng and Wei Jia are the recipients of China Scholarship Council training grants.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Riesterer O, Milas L, Ang KK. J Clin Oncol. 2007;25:4075. doi: 10.1200/JCO.2007.11.8497. [DOI] [PubMed] [Google Scholar]
- 2.Nakayama A, Alladin KP, Igbokwe O, White JD. Cancer Invest. 2011;29:655. doi: 10.3109/07357907.2011.626479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morrison R, Schleicher SM, Sun Y, Niermann KJ, Kim S, Spratt DE, Chung CH, Lu B. J Oncol. 2011:941876. doi: 10.1155/2011/941876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frosina G. Mol Cancer Res. 2009;7:989. doi: 10.1158/1541-7786.MCR-09-0030. [DOI] [PubMed] [Google Scholar]
- 5.Gilbert LA, Hemann MT. Cancer Res. 2011;71:5062. doi: 10.1158/0008-5472.CAN-11-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li F, Sethi G. Biochim Biophys Acta. 2010;1805:167. doi: 10.1016/j.bbcan.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Aggarwal BB, Sung B. Cancer Discov. 2011;1:469. doi: 10.1158/2159-8290.CD-11-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaturvedi MM, Sung B, Yadav VR, Kannappan R, Aggarwal BB. Oncogene. 2011;30:1615. doi: 10.1038/onc.2010.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gabizon A, Shmeeda H, Grenader T. Eur J Pharm Sci. 2011;45:388. doi: 10.1016/j.ejps.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 10.Duggan ST, Keating GM. Drugs. 2011;71:2531. doi: 10.2165/11207510-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 11.Harper JW, Elledge SJ. Mol Cell. 2007;28:739. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 12.Lee JH, Paull TT. Oncogene. 2007;26:7741. doi: 10.1038/sj.onc.1210872. [DOI] [PubMed] [Google Scholar]
- 13.Kim MY, Zhang T, Kraus WL. Genes Dev. 2005;19:1951. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- 14.Mortusewicz O, Ame JC, Schreiber V, Leonhardt H. Nucleic Acids Res. 2007;35:7665. doi: 10.1093/nar/gkm933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schreiber V, Dantzer F, Ame JC, Murcia G. Nat Rev Mol Cell Biol. 2006;7:517. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 16.Janssens S, Tschopp J. Cell Death Differ. 2006;13:773. doi: 10.1038/sj.cdd.4401843. [DOI] [PubMed] [Google Scholar]
- 17.Wu ZH, Miyamoto S. J Mol Med. 2007;85:1187. doi: 10.1007/s00109-007-0227-9. [DOI] [PubMed] [Google Scholar]
- 18.Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. Science. 1995;270:2008. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 19.Chen ZJ, Bhoj V, Seth RB. Cell Death Differ. 2006;13:687. doi: 10.1038/sj.cdd.4401869. [DOI] [PubMed] [Google Scholar]
- 20.Hayden MS, Ghosh S. Cell. 2008;132:344. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 21.Sun SC, Ley SC. Trends Immunol. 2008;29:469. doi: 10.1016/j.it.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, Akira S. Nat Immunol. 2005;6:1087. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- 23.Shim JH, Xiao C, Paschal AE, Bailey ST, Rao P, Hayden MS, Lee KY, Bussey C, Steckel M, Tanaka N, Yamada G, Akira S, Matsumoto K, Ghosh S. Genes Dev. 2005;19:2668. doi: 10.1101/gad.1360605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu ZH, Wong ET, Shi Y, Niu J, Chen Z, Miyamoto S, Tergaonkar V. Mol Cell. 2010;40:75. doi: 10.1016/j.molcel.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y, Xia F, Hermance N, Mabb A, Simonson S, Morrissey S, Gandhi P, Munson M, Miyamoto S, Kelliher MA. Mol Cell Biol. 2011;31:2774. doi: 10.1128/MCB.01139-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinz M, Stilmann M, Arslan SC, Khanna KK, Dittmar G, Scheidereit C. Mol Cell. 2010;40:63. doi: 10.1016/j.molcel.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 27.Jin HS, Lee DH, Kim DH, Chung HH, Lee SJ, Lee TH. Cancer Res. 2009;69:1782. doi: 10.1158/0008-5472.CAN-08-2256. [DOI] [PubMed] [Google Scholar]
- 28.Adhikari A, Chen ZJ. Dev Cell. 2009;16:485. doi: 10.1016/j.devcel.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 29.Fan YH, Yu Y, Shi Y, Sun W, Xie M, Ge N, Mao R, Chang A, Xu G, Schneider MD, Zhang H, Fu S, Qin J, Yang JJ. J Biol Chem. 2010;285:5347. doi: 10.1074/jbc.M109.076976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mao R, Fan Y, Mou Y, Zhang H, Fu S, Yang J. Cell Signal. 2011;23:222. doi: 10.1016/j.cellsig.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan Y, Yu Y, Mao R, Zhang H, Yang J. Cell Signal. 2011;23:660. doi: 10.1016/j.cellsig.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan YH, Yu Y, Mao RF, Tan XJ, Xu GF, Zhang H, Lu XB, Fu SB, Yang J. Cell Death Differ. 2011;18:1547. doi: 10.1038/cdd.2011.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamazaki K, Gohda J, Kanayama A, Miyamoto Y, Sakurai H, Yamamoto M, Akira S, Hayashi H, Su B, Inoue J. Sci Signal. 2009;2:ra66. doi: 10.1126/scisignal.2000387. [DOI] [PubMed] [Google Scholar]
- 34.Sorrentino A, Thakur N, Grimsby S, Marcusson A, Bulow V, Schuster N, Zhang S, Heldin CH, Landstrom M. Nat Cell Biol. 2008;10:1199. doi: 10.1038/ncb1780. [DOI] [PubMed] [Google Scholar]
- 35.Li Q, Yan J, Mao AP, Li C, Ran Y, Shu HB, Wang YY. Proc Natl Acad Sci U S A. 2011;108:19341. doi: 10.1073/pnas.1110946108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fan Y, Shi Y, Liu S, Mao R, An L, Zhao Y, Zhang H, Zhang F, Xu G, Qin J, Yang J. Cell Signal. 2012;24:1381. doi: 10.1016/j.cellsig.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahmed N, Zeng M, Sinha I, Polin L, Wei WZ, Rathinam C, Flavell R, Massoumi R, Venuprasad K. Nat Immunol. 2011;12:1176. doi: 10.1038/ni.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R. Cell. 2005;123:773. doi: 10.1016/j.cell.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 39.Zhang L, Zhou F, Drabsch Y, Gao R, Snaar-Jagalska BE, Mickanin C, Huang H, Sheppard KA, Porter JA, Lu CX, Ten P. Nat Cell Biol. 2012;14:717. doi: 10.1038/ncb2522. [DOI] [PubMed] [Google Scholar]
- 40.Perry WL, Hustad CM, Swing DA, O’Sullivan TN, Jenkins NA, Copeland NG. Nat Genet. 1998;18:143. doi: 10.1038/ng0298-143. [DOI] [PubMed] [Google Scholar]
- 41.Bernassola F, Karin M, Ciechanover A, Melino G. Cancer Cell. 2008;14:10. doi: 10.1016/j.ccr.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 42.Melino G, Gallagher E, Aqeilan RI, Knight R, Peschiaroli A, Rossi M, Scialpi F, Malatesta M, Zocchi L, Browne G, Ciechanover A, Bernassola F. Cell Death Differ. 2008;15:1103. doi: 10.1038/cdd.2008.60. [DOI] [PubMed] [Google Scholar]
- 43.Rossi M, Simone M, Pollice A, Santoro R, Mantia G, Guerrini L, Calabro V. Cell Cycle 2006. 2006;5:1816. doi: 10.4161/cc.5.16.2861. [DOI] [PubMed] [Google Scholar]
- 44.Rossi M, Aqeilan RI, Neale M, Candi E, Salomoni P, Knight RA, Croce CM, Melino G. Proc Natl Acad Sci U S A. 2006;103:12753. doi: 10.1073/pnas.0603449103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rossi M, Laurenzi V, Munarriz E, Green DR, Liu YC, Vousden KH, Cesareni G, Melino G. Embo J. 2005;24:836. doi: 10.1038/sj.emboj.7600444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oberst A, Rossi M, Salomoni P, Pandolfi PP, Oren M, Melino G, Bernassola F. Biochem Biophys Res Commun. 2005;331:707. doi: 10.1016/j.bbrc.2005.03.158. [DOI] [PubMed] [Google Scholar]
- 47.Munarriz E, Bano D, Sayan AE, Rossi M, Melino G, Nicotera P. Biochem Biophys Res Commun. 2005;333:954. doi: 10.1016/j.bbrc.2005.05.188. [DOI] [PubMed] [Google Scholar]