

Abstract
Parkinson's disease (PD) is a progressive neurodegenerative disease caused by the interaction of genetic and environmental factors. However, the etiology of PD remains largely unknown. Macroautophagy is known to play an essential role in the degradation of abnormal proteins and organelles. Furthermore, the loss of autophagy-related (Atg) genes results in neurodegeneration and abnormal protein accumulation. Since these are also pathologic features of Parkinson's disease, the conditional impairment of autophagy may lead to improved animal models for the study of PD. Using transgenic mice expressing Cre recombinase under the control of either the dopamine transporter or the engrailed-1 promoters, we generated mice with the conditional deletion of Atg7 in the dopamine neurons of the substantia nigra pars compacta, other regions of the midbrain, and also the hindbrain. This conditional impairment of autophagy results in the age-related loss of dopaminergic neurons and corresponding loss of striatal dopamine, the accumulation of low-molecular-weight α-synuclein, and the presence of ubiquitinated protein aggregates, recapitulating many of the pathologic features of PD. These conditional knock-out animals provide insight into the process of autophagy in Parkinson's disease pathology.
Introduction
Parkinson's disease (PD) is a progressive neurodegenerative disorder characterized pathologically by the extensive loss of dopamine neurons in the substantia nigra pars compacta (SNpc) and the presence of dystrophic neurites and Lewy bodies in the surviving neurons (Savitt et al., 2006). Lewy bodies are proteinaceous intracellular inclusions containing ubiquitin and α-synuclein among many other components. Although incompletely understood, the etiology of PD is thought to involve both genetic and environmental factors. Further research, including the development of a more accurate animal model of PD, is necessary to elucidate the molecular pathways involved in this disease.
There is growing evidence that the development of PD pathology involves macroautophagy (henceforth termed autophagy; Levine and Kroemer, 2008; Cheung and Ip, 2009; Jaeger and Wyss-Coray, 2009; Geisler et al., 2010). For example, autophagic vacuoles are present in the substantia nigra of Parkinson's disease patients (Anglade et al., 1997), suggesting abnormal activation or impaired function of autophagy in PD. In addition, α-synuclein is cleared in part through autophagy (Vogiatzi et al., 2008), and the protein Parkin, mutated in the most common cause of recessive Parkinsonism, may mediate the clearance of abnormal mitochondria through autophagy (Narendra et al., 2008). Finally, mutations in the LRRK2 gene, the most common monogenetic cause of PD, can induce pathology in neurites by an autophagy-dependent process (Plowey et al., 2008).
Autophagy is a bulk lysosomal degradation pathway essential for the turnover of long-lived, misfolded or aggregated proteins, as well as damaged or excess organelles (Levine and Kroemer, 2008). The autophagy-related protein Atg7 is a necessary component of this process and, among several cellular functions, mediates the conversion of LC3-I to the phosphatidylethanolamine-conjugated form, LC3-II. Previous research described widespread neurodegeneration in mice with CNS conditional deletion of Atg7 (Komatsu et al., 2006), suggesting that basal levels of autophagy may play a protective role against neurodegeneration. The effect of autophagy impairment in the SNpc has not been reported. Therefore we sought to examine the effect of Atg7 deletion and subsequent impairment of autophagy specifically in midbrain dopamine neurons. We have generated a novel mouse model of PD in which the selective impairment of autophagy leads to a progressive loss of dopamine neurons, an accompanying reduction in striatal dopamine levels, an accumulation of low-molecular-weight α-synuclein, and the presence of aggregates that contain K48-linked polyubiquitin and the ubiquitin binding protein p62.
Materials and Methods
Animals.
Mice of either sex were housed and cared for in accordance with the NIH Health Guide for the Care and Use of Laboratory Animals. Animals were housed in a pathogen-free facility with a 12 h light/dark cycle and provided food and water ad libitum unless otherwise indicated. En-Cre (Kimmel et al., 2000), Dat-Cre (Zhuang et al., 2005), and Atg7flox/flox mice (Komatsu et al., 2006) were genotyped using published protocols. Genotyping was accomplished using a three-primer PCR method that includes ATG7flox forward, ATG7mid reverse, and ATG7flox reverse primers. Sequences are 5′-TGGCTGCTACTTCTGCAATGATGTG-3′, 5′-CTGTTCGGGCACGGGACGGG-3′, and 5′-GTCCTGTCTCTGGTAGTCGAGGTG-3′, respectively. The reaction yields a wild-type band at 350 bp and a floxed allele band at 600 bp. ROSA26 lacZ mice were genotyped and stained per protocol (Jackson Laboratories).
Western blot analysis.
Atg7 flox/flox; En-Cre and Atg7 flox/flox control mice were killed via pentobarbital injection. Atg7 protein expression was monitored from indicated brain regions that were homogenized (2 min) in RIPA (radioimmunoprecipitation assay) lysis buffer (Sigma) containing protease inhibitor (BioVision, Inc), incubated (30 min) on ice, and centrifuged at 10,000 × g for 10 min. The supernatant was centrifuged (15 min) at 10,000 × g. Proteins were separated using 4–15% and 15% Tris-HCl polyacrylamide gels (Bio-Rad) and blotted on a nitrocellulose membrane (Bio-Rad). Bands were detected using ECL Plus Western Blotting detection system (GE Healthcare Life Sciences). One-way ANOVA statistical analysis followed by Tukey's post hoc test (GraphPad InStat) was used to compare Atg7 levels (n = 3).
Ubiquitin, p62, and α-synuclein were homogenized in TNE buffer (50 mm Tris-HCl, pH 7.4, 100 mm NaCl, 5 mm EDTA) supplemented with 1% SDS, and protease inhibitor mixture (Roche) using a hand-held Polytron device and then sonicated for 20 s. Equivalent protein quantities were loaded on 4–15% polyacrylamide gels, and transferred onto a nitrocellulose membrane. To further ensure identical protein loading, 10% of the protein samples were run on separate gels and probed for actin, while equal blotting was verified by Ponceau S (Sigma) staining (data not shown). ImageJ (NIH) software was used to quantify the Western blots and the level of ubiquitination was normalized to the protein amount loaded. Paired two-tailed Student's t test was used to determine significance (n = 3).
Histology and immunohistochemistry.
Hematoxylin and eosin (H&E) staining of paraffin-embedded sections (6 μm) and Nissl staining were performed using standard methods as previously described (Savitt et al., 2005). For immunohistochemical analysis, animals were anesthetized with a lethal dose of pentobarbital (100 mg/kg) and perfused with 4% paraformaldehyde (Sigma). Brains were extracted, postfixed, and cryoprotected in 30% sucrose. Free-floating sections (40 μm) were harvested with a microtome and were processed for immunohistochemical analysis. Signal was generated using ABC vectastain (Vector Labs) and diaminobenzidine tablets (Sigma). For confocal microscopy, sections were mounted in Vectashield (Vector Laboratories) and visualized using a Zeiss LSM 510 microscope.
Antibodies.
The primary antibodies used include rabbit anti-tyrosine hydroxylase (TH) (1:1000), mouse anti-TH (1:1000), rabbit anti-LC3 (1:500) from Novus Biologicals, rabbit anti-Atg7 (1:1000, Sigma), rabbit anti-ubiquitin (1:1000, DAKO), human anti-K48-linked and anti-K63-linked polyubiquitin (1:500 and 1:1000, respectively; Genentech; Newton et al., 2008), and guinea pig anti-p62 (1:500; American Research Products). Goat biotin-conjugated anti-rabbit, anti-mouse, and anti-human were used for bright-field microscopy (all 1:500, Jackson ImmunoResearch). Goat Cy2-conjugated anti-rabbit, goat Cy3-conjugated anti-mouse, donkey Cy3-conjugated anti-human, and donkey Cy5-conjugated anti-guinea pig were used for double fluorescence labeling (all 1:500, Jackson ImmunoResearch). Horseradish peroxidase-conjugated goat anti-rabbit and anti-α-actin-peroxidase conjugate were used for Western blot analysis (both 1:10,000, Sigma). K48- and K63-linked polyubiquitin antibody specificity was verified using immunoblots loaded with purified polyubiquitin chains (Boston Biochem; data not shown). To illustrate the change in ubiquitin levels, the contrasts of the ubiquitin, K48, and K63 blots were uniformly modified (i.e., all ubiquitin blots were subject to the same contrast modifications).
Stereological analysis of dopaminergic neuronal cell number.
To quantify the dopamine neurons, every fourth section of the SNpc was TH and Nissl stained as previously described (Savitt et al., 2005). Cells were counted by nonbiased stereology using the optical fractionator method at 40× (StereoInvestigator 7, MBF Bioscience). A 40 × 40 μm counting frame and 100 × 100 μm grid were used, and counting was performed by a blinded investigator. One-way ANOVA statistical analysis followed by Tukey's post hoc test (GraphPad InStat) was used to compare TH cell counts (n = 5) and Nissl cell counts (n = 4–5 for all except 1-month-old Atg7flox/flox; En-Cre mice where n = 2). Ubiquitin-stained inclusions within SNpc cells were characterized and quantified using similar hardware and software at 100× magnification of random fields. The quick measure circle function was used to determine the average radius. An unpaired t test with Welch correction was used to determine significance.
Locomotor activity.
Novelty-induced locomotor activity was assessed in the open field over a 6 h period during the wake cycle using activity chambers with infrared photo beams (San Diego Instruments). Horizontal and vertical activities were automatically recorded. Atg7flox/flox control, Atg7flox/flox; DAT-Cre, and DAT-Cre mice at 7 and 12 months of age (n = 4–6 for each cohort) were subjected to open-field analysis. The total number of beam breaks including central and peripheral areas over 6 h was recorded and analyzed. The results were analyzed using two-way repeated-measures ANOVA (SigmaStat).
HPLC-electrochemical analysis of neurotransmitters.
The monoamine levels were quantified by HPLC as follows. Mice were killed by decapitation, the brains were extracted immediately and the striatum was dissected and weighed. Tissue was homogenized using 50 volumes (v/w) of ice-cold 10 mm perchloric acid and sonication for 20 s on ice. After centrifugation at 20,000 × g for 30 min, the supernatants were filtered through polytetrafluoroethylene acrodiscs (0.22 mm average pore diameter; Millipore S.A.) and aliquots of these were directly analyzed. Samples (10 ml) were analyzed using HPLC with electrochemical detection (Antec Leyden) following standard protocols. The mobile phase to detect dopamine and 5-HT (5-hydroxytryptamine) consisted of 50 mm perchloric acid, 8 mm sodium phosphate, 0.1 mm EDTA, and 500 mg/L octane sulfonic acid, 12.5% methanol, and pH adjusted to 6.0. For norepinephrine and metabolites the mobile phase consisted of the same buffer but with 10% methanol and 50 mm citric acid. Flow rate was maintained at 0.5 ml/min. Relative content was calculated. The results were analyzed by one-way ANOVA and Dunn's method (SigmaStat).
Statistical analysis.
All data are presented as mean + SEM.
Results
We generated tissue-specific Atg7 conditional knock-out mice using the Cre/lox technique. Atg7flox/flox mice were bred with mice expressing Cre recombinase downstream of either the dopamine transporter (DAT) or Engrailed-1 (En) promoters. Engrailed-1 expresses Cre widely in the hindbrain, cerebellum, and midbrain, including the dopaminergic cells of the SNpc and ventral tegmental area, whereas the DAT promoter drives Cre exclusively in dopaminergic cells (Fig. 1A). A reduction in the levels of Atg7 protein in Atg7flox/flox; En-Cre mice verified appropriate gene targeting and inactivation in the model system (Fig. 1B,C). Although Engrailed-1 is expressed widely, there are cells and cell processes in those regions where the En promoter is not active. As a result, there is residual Atg7 immunoreactivity in those protein lysates.
Figure 1.

Generation of mice with the conditional deletion of Atg7. A, Cre recombinase activity (blue coloration) in the SNpc, ventral tegmental area (VTA), hindbrain (HB), and cerebellum (CB) of mice expressing Cre downstream of either the DAT or the En promoters. Scale bar, 500 μm. B, Western blot analysis of Atg7 (∼75 kDa) expression in the midbrain/hindbrain (MHB), cerebellum (CB), and cortex (CTX) of juvenile (1-month-old) Atg7 flox/flox control and Atg7 flox/flox; En-Cre mice. C, Quantitation of Atg7 levels in the indicated brain regions of Atg7 flox/flox control mice (black) and Atg7 flox/flox; En-Cre mice (gray). *p < 0.05, **p < 0.01, difference in Atg7 flox/flox; En-Cre mice compared with age-matched Atg7 flox/flox controls. D, Survival rate of Atg7 flox/flox; DAT-Cre mice (black) and Atg7 flox/flox; En-Cre mice (red) compared with Atg7 flox/flox control mice (gray). E, Atg7 flox/flox; DAT-Cre mice (black) are hyperactive at 7 months compared with both controls: Atg7 flox/flox mice (light gray) and DAT-Cre mice (dark gray). *p < 0.05, difference in Atg7 flox/flox; DAT-Cre mice compared with age-matched Atg7 flox/flox controls.
Mice with the conditional Atg7 deletion are born healthy and indistinguishable from control littermates. Although the survival rate of Atg7flox/flox; DAT-Cre mice is comparable to that of the Atg7flox/flox controls, Atg7flox/flox; En-Cre mice displayed a 76% reduction in survival by 1 year of age (Fig. 1D). The more widespread loss of autophagic function in the Atg7flox/flox; En-Cre mice compared with the isolated effect on dopamine neurons in the Atg7flox/flox; DAT-Cre mice is the likely cause of increased mortality.
In addition, we performed a locomotor activity analysis of these mice as a measure of bradykinesia. Paradoxically, the Atg7flox/flox; DAT-Cre mice were found to be more active than control mice during the wake cycle (Fig. 1E). This trend continued in older mice as well, but was no longer statistically significant when compared with controls. Interestingly, the Atg7flox/flox; En-Cre mice displayed decreased locomotor activity, tremulousness, weight loss, and a wide-based and ataxic gait presumably due to the additional brainstem, midbrain, spinal cord, and cerebellar pathology (data not shown).
The effect of Atg7 deficiency on dopaminergic neuron survival was investigated using immunohistochemical techniques and stereological analysis. We observed an age-dependent loss of TH-immunoreactive cells in the SNpc of mice with the conditional deletion of Atg7 (Fig. 2A). Adult Atg7flox/flox; DAT-Cre and Atg7flox/flox; En-Cre mice displayed a 40% and 60% loss of SNpc dopamine neurons, respectively, compared with adult Atg7 control mice (Fig. 2B). The corresponding decrease in Nissl-positive neuron count and lack of significant difference in age- and genotype-matched Nissl and TH counts further support the loss of neurons rather than loss of cellular TH immunoreactivity (Fig. 2C). Despite a trend toward fewer cells, Atg7-deficient juvenile mice did not show a significant loss of TH neurons compared with controls (Fig. 2B). Consistent with these data, HPLC analysis confirmed an age-dependent decrease in striatal dopamine content in the affected adult brain (Fig. 2D). We detected a 55.5% and a 64.5% reduction in striatal dopamine in adult Atg7flox/flox; DAT-Cre and Atg7flox/flox; En-Cre mice, respectively, whereas a significant decrease in striatal dopamine was not observed in juvenile mice. These results suggest that dopamine neurons are sensitive to impaired autophagy in vivo and are associated with a progressive age-dependent cell loss. Autophagy impairment in the SNpc also resulted in a modest increase in reactive astrocytes as indicated by GFAP staining (Fig. 2E). Additionally, as expected from previous work, we observed Purkinje cell loss in the Atg7flox/flox; En-Cre mice (Komatsu et al., 2006) (Fig. 2F).
Figure 2.
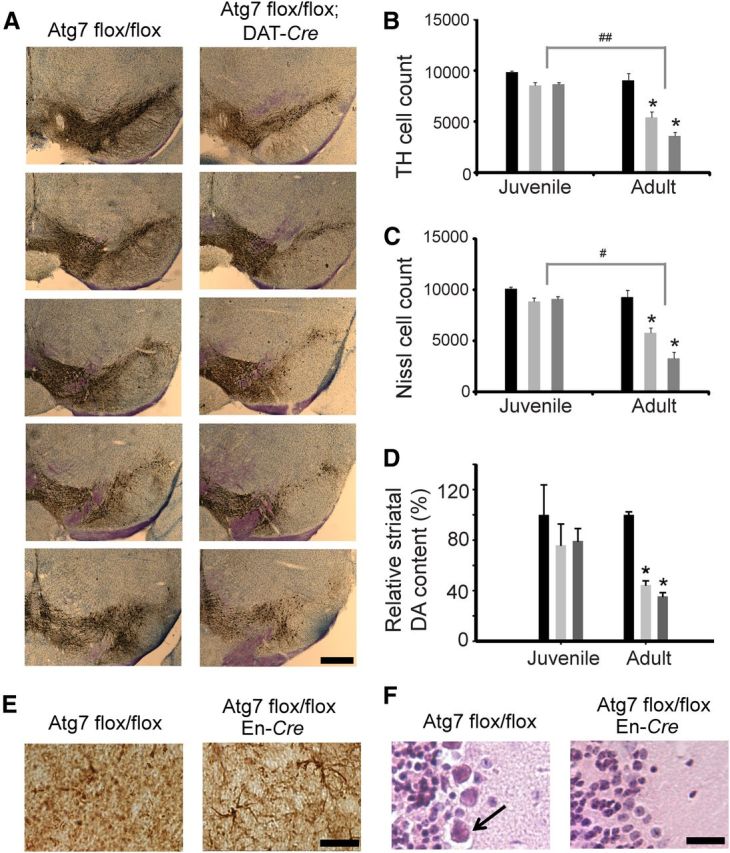
Progressive loss of TH neurons. A, Impaired autophagy induced TH-positive neuronal cell loss in adult Atg7 flox/flox; DAT-Cre mice compared with an age-matched Atg7 flox/flox control. Scale bar, 500 μm. B, Stereological analysis of TH-positive cell counts in the SNpc. C, Stereological analysis of Nissl-positive cell counts in the SNpc. The progressive loss of Nissl-positive neurons corresponds to the TH-positive neuronal loss. For B and C, *p < 0.001, statistically significant differences versus control animals at the same age. #p < 0.01, ##p < 0.001, statistically significant differences between animals of the same Cre line but different ages. D, HPLC was used to measure the relative content of dopamine in juvenile (1 month) and adult (7–9 month) mice. *p < 0.05, statistically significant differences versus age-matched control Atg7 flox/flox. For B–D, Atg7 flox/flox control mice (black), Atg7 flox/flox; DAT-Cre mice (light gray), and Atg7 flox/flox; En-Cre mice (dark gray). E, GFAP staining in the SNpc of Atg7 flox/flox control and Atg7 flox/flox; En-Cre mice. Scale bar, 25 μm. F, Purkinje cells (arrow) are present in the cerebellum of control mice as indicated by H&E stain. Additional pathology in Atg7 flox/flox; En-Cre mice includes the loss of Purkinje cells. Scale bar, 25 μm.
Given the PD-like neuronal loss in Atg7-deficient mice, we examined the tissue for the presence of inclusions using H&E staining of paraffin-embedded SNpc sections. Neuronal inclusions are present in the SNpc of Atg7-deficient mice, but not Atg7flox/flox controls (Fig. 3A). The inclusions were often perinuclear, but also appeared in the neuropil. They stained intensely for ubiquitin and for the ubiquitin-binding protein and autophagy substrate p62 (Komatsu et al., 2007). Interestingly, although we did not observe significant cell loss in the SNpc of juvenile Atg7flox/flox; DAT-Cre mice, inclusions were present and were significantly smaller (1.1 ± 0.23 vs 1.7 ± 0.43 μm average diameter, p = 0.0013) than those seen at older ages (Fig. 3B). Similar inclusions were detected in Atg7flox/flox; En-Cre mice (Fig. 3B). We verified the presence of ubiquitin using immunoblots and demonstrated an increase in polyubiquitinated proteins in the SDS-soluble fractions of midbrain/hindbrain lysates of Atg7flox/flox; En-Cre mice compared with control mice while ubiquitin levels remained unchanged in the cortex of the Atg7 conditional knock-out mice (Fig. 3C,D).
Figure 3.
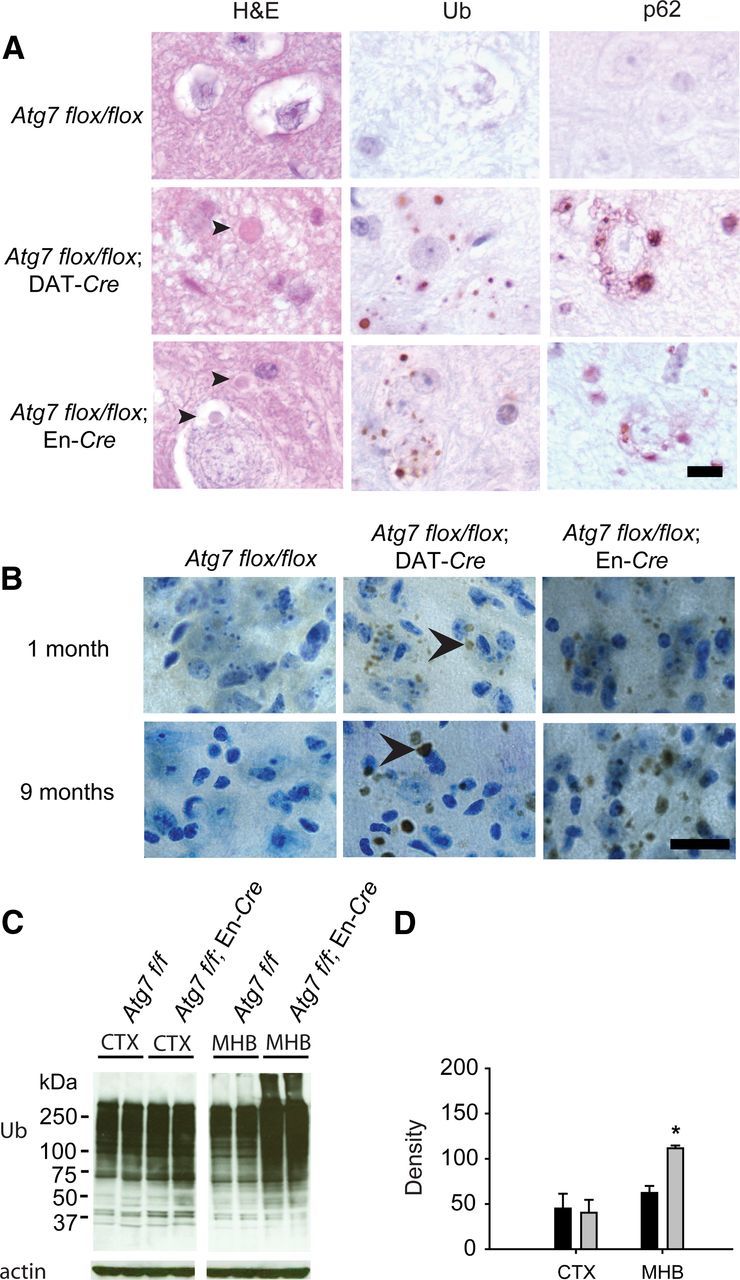
Detection of inclusions in Atg7 conditional knock-out mice. A, Inclusions (arrowheads) are present in the Atg7 conditional knock-out sections of 10-month-old mice. Ubiquitin (Ub)- and p62-positive inclusions are present in the cytoplasm of neuronal cells. Scale bar, 10 μm. B, Ubiquitin-containing inclusions (arrowheads) are present in juvenile (1-month-old) mice but are larger in size in adult (9-month-old) Atg7 flox/flox; DAT-Cre mice and Atg7 flox/flox; En-Cre mice. Scale bar, 20 μm. Cells were counterstained with Nissl. C, Western blot analysis of ubiquitin (Ub) in cortex (CTX) and midbrain/hindbrain (MHB) lysates obtained from Atg7 flox/flox control and Atg7 flox/flox; En-Cre mice. D, Quantitation of ubiquitin content (*p < 0.05). Atg7 flox/flox control mice (black) and Atg7 flox/flox; En-Cre mice (light gray).
As polyubiquitin is known to exist in various forms depending on the lysine residue involved in the linkage, we sought to determine whether a particular ubiquitin linkage serves as a specific autophagy signal using linkage-specific antibodies (Newton et al., 2008). The anti-K48-linked polyubiquitin antibody stains inclusions within dopamine neurons in the SNpc of Atg7flox/flox; DAT-Cre mice and in Atg7flox/flox; En-Cre mice (Fig. 4A). The accumulation of this form of ubiquitin was confirmed in midbrain/hindbrain lysates of Atg7flox/flox; En-Cre mice using immunoblots (Fig. 4B). Based on the assessment of two animals, there was a 54% increase in total K48 ubiquitin and a 45% increase in high molecular weight K48 ubiquitin in Atg7flox/flox; En-Cre mice compared with Atg7flox/flox controls. Staining for K63-linked ubiquitin in autophagy-deficient mice suggests little colocalization of p62 and K63-linked polyubiquitin in aggregates of dopamine neurons in the SNpc of the conditional knock-outs (data not shown). Western blot analysis of K63-linked polyubiquitin in Atg7flox/flox and Atg7flox/flox; En-Cre midbrain/hindbrain lysates indicates the K63 protein is not increased in Atg7-deficient animals compared with control.
Figure 4.
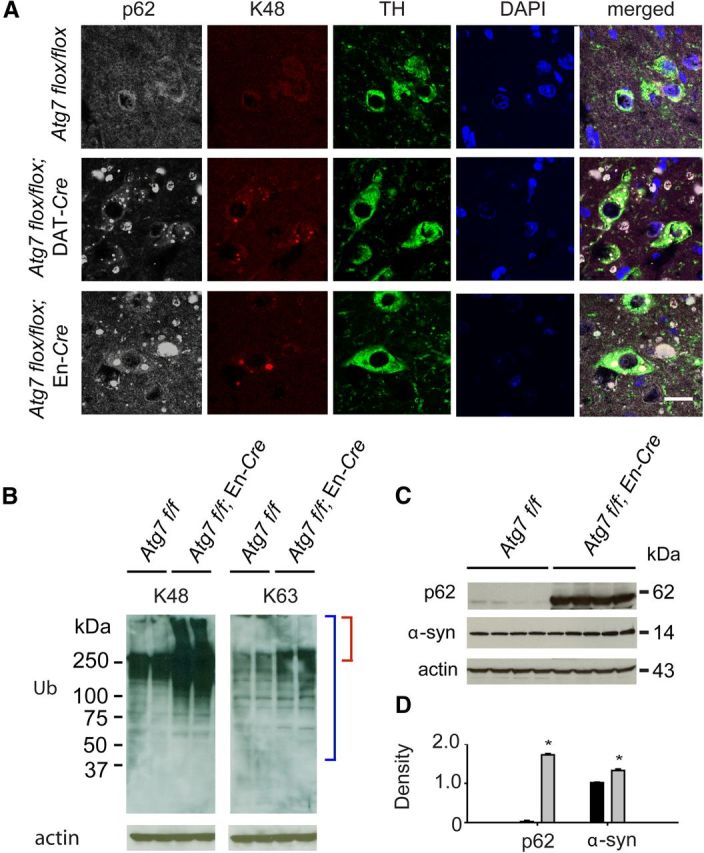
Ubiquitin-linked protein changes and α-synuclein accumulation resulting from autophagy impairment. A, Colocalization of p62- and K48-linked polyubiquitin in aggregates within dopamine neurons of the SNpc in both Atg7 flox/flox; DAT-Cre and Atg7 flox/flox; En-Cre mice. Scale bar, 10 μm. B, K48 ubiquitin chains are significantly increased in midbrain/hindbrain lysates of Atg7flox/flox; En-Cre mice compared with Atg7flox/flox mice, whereas K63 protein levels did not increase in the conditional knock-out. Similar results were obtained in repeat experiments. Blue brackets indicate total ubiquitin while red brackets indicate high-molecular-weight ubiquitin. C, Western blot of α-synuclein (α-syn) and p62 levels in SDS-soluble lysates of midbrain/hindbrain regions. D, Quantitation of p62 and α-syn content (*p < 0.05). Atg7 flox/flox control mice (black) and Atg7 flox/flox; En-Cre mice (light gray).
Inclusions containing p62 were observed in the dopaminergic cells of Atg7-deficient mice and in addition were found to colocalize with K48-linked polyubiquitin (Fig. 4A). The accumulation of this protein was verified using immunoblots showing a dramatic increase in p62 levels in midbrain/hindbrain lysates taken from autophagy-deficient mice (Fig. 4C,D). Although α-synuclein inclusions were not present (data not shown), using immunoblot analysis, we demonstrated elevated levels of low-molecular-weight (14 kDa) α-synuclein in the brains of mice with impaired autophagy (Fig. 4C,D). This provides in vivo evidence that α-synuclein levels are influenced by the process of autophagy.
Discussion
Selective impairment of autophagy in the substantia nigra of Atg7flox/flox; DAT-Cre and Atg7flox/flox; En-Cre mice results in a progressive loss of dopamine neurons, reduction in dopamine level in the striatum, and an accumulation of protein aggregates. The autophagy-based animal model described here is suitable to study the progressive nature of dopaminergic cell loss and supports a role for autophagy in neurodegeneration. Protein aggregation is a hallmark of PD pathogenesis (Savitt et al., 2006). In the autophagy-based mouse model, these inclusions contain both ubiquitin and p62. Interestingly, the protein p62 binds both ubiquitinated proteins and LC3, thus providing a functional link between proteins targeted for degradation by ubiquitin and the autophagy pathway (Bjørkøy et al., 2005; Pankiv et al., 2007). In addition, p62 also accumulates in various neurodegenerative conditions (Kuusisto et al., 2001). Further analysis of the Atg7flox/flox; DAT-Cre and Atg7flox/flox; En-Cre mice demonstrates that the polyubiquitin consists mainly of K48 linkages. Thus, K48-linked polyubiquitin, which is known to target proteins to the ubiquitin-proteasome degradation pathway (Komatsu et al., 2006), may also be involved in autophagy-mediated degradation. Although K63 polyubiquitin levels were not significantly elevated, we cannot exclude the possibility that K63 levels are increased due to limitations of the reagents used.
The accumulation and aggregation of α-synuclein is another characteristic feature of PD (Polymeropoulos et al., 1997; Krüger et al., 1998; Webb et al., 2003). We show that selective Atg7 deficiency leads to the accumulation of low-molecular-weight α-synuclein. Overexpression of α-synuclein is also thought to impair autophagy (Winslow et al., 2010), suggesting the presence of a cycle of impairment and accumulation. Prior studies have shown that α-synuclein is degraded by both macroautophagy and chaperone-mediated autophagy (CMA; Webb et al., 2003; Cuervo et al., 2004; Vogiatzi et al., 2008). Interestingly, aberrant A53T α-synuclein is thought to be associated specifically with CMA (Cuervo et al., 2004; Winslow et al., 2010). Thus, future studies using the autophagy-based mouse model in conjunction with the A53T α-synuclein transgenic mice may provide further insight into autophagy-dependent processing of abnormal α-synuclein.
In summary, the age-related pathology observed in mice with Atg7 deficiency in SNpc dopaminergic neurons suggests that autophagy impairment is a valuable model for elucidating the molecular mechanisms involved in the pathology of Parkinson's disease. Furthermore, these mice provide a powerful tool to assess the additional effect of PD-associated mutations, such as those in α-synuclein, Parkin, or LRRK2, on neurodegeneration and inclusion formation. The results will lend insight into the role of autophagy in the pathological processes of these disease-causing mutations. Such new models of PD will be crucial for the development of novel therapeutic targets and agents to combat this progressive neurodegenerative disease.
Footnotes
This study was supported by the Parkinson's Disease Foundation and NIH/NINDS Grants K08 NS052624 and P50 NS 38377, and the JPB Foundation. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. We acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with The Johns Hopkins Hospital and The Johns Hopkins University School of Medicine and the Foundation's Parkinson's Disease Program No. M-1. We thank Dr. Masaaki Komatsu, Dr. Xiaoxi Zhuang, and Dr. Alexandra Joyner for providing floxed Atg7 mice, DAT-Cre mice, and En-Cre mice, respectively.
The authors declare no competing financial interests.
References
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung ZH, Ip NY. The emerging role of autophagy in Parkinson's disease. Mol Brain. 2009;2:29. doi: 10.1186/1756-6606-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- Jaeger PA, Wyss-Coray T. All-you-can-eat: autophagy in neurodegeneration and neuroprotection. Mol Neurodegener. 2009;4:16. doi: 10.1186/1750-1326-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel RA, Turnbull DH, Blanquet V, Wurst W, Loomis CA, Joyner AL. Two lineage boundaries coordinate vertebrate apical ectodermal ridge formation. Genes Dev. 2000;14:1377–1389. [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O. AlaSOPro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Kuusisto E, Salminen A, Alafuzoff I. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport. 2001;12:2085–2090. doi: 10.1097/00001756-200107200-00009. [DOI] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Matsumoto ML, Wertz IE, Kirkpatrick DS, Lill JR, Tan J, Dugger D, Gordon N, Sidhu SS, Fellouse FA, Komuves L, French DM, Ferrando RE, Lam C, Compaan D, Yu C, Bosanac I, Hymowitz SG, Kelley RF, Dixit VM. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell. 2008;134:668–678. doi: 10.1016/j.cell.2008.07.039. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Plowey ED, Cherra SJ, 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008;105:1048–1056. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Savitt JM, Jang SS, Mu W, Dawson VL, Dawson TM. Bcl-x is required for proper development of the mouse substantia nigra. J Neurosci. 2005;25:6721–6728. doi: 10.1523/JNEUROSCI.0760-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitt JM, Dawson VL, Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest. 2006;116:1744–1754. doi: 10.1172/JCI29178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. 2008;283:23542–23556. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O'Kane CJ, Rubinsztein DC. alpha-Synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol. 2010;190:1023–1037. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X, Masson J, Gingrich JA, Rayport S, Hen R. Targeted gene expression in dopamine and serotonin neurons of the mouse brain. J Neurosci Methods. 2005;143:27–32. doi: 10.1016/j.jneumeth.2004.09.020. [DOI] [PubMed] [Google Scholar]