

Abstract
The identities of toxic aggregate species in Huntington's disease pathogenesis remain ambiguous. While polyQ-expanded huntingtin (Htt) is known to accumulate in compact inclusion bodies inside neurons, this is widely thought to be a protective coping response that sequesters misfolded conformations or aggregated states of the mutated protein. To define the spatial distributions of fluorescently-labeled Htt-exon1 species in the cell model PC12m, we employed highly sensitive single-molecule super-resolution fluorescence imaging. In addition to inclusion bodies and the diffuse pool of monomers and oligomers, fibrillar aggregates ~100 nm in diameter and up to ~1–2 µm in length were observed for pathogenic polyQ tracts (46 and 97 repeats) after targeted photo-bleaching of the inclusion bodies. These short structures bear a striking resemblance to fibers described in vitro. Definition of the diverse Htt structures in cells will provide an avenue to link the impact of therapeutic agents to aggregate populations and morphologies.
The molecular details of protein misfolding and aggregation in neurodegenerative disorders may hold the key to therapeutic intervention1. In Huntington's disease (HD), neuronal degeneration is linked to an abnormal expansion of the polyglutamine (polyQ) tract in the huntingtin (Htt) protein to beyond 40 glutamines, caused by mutation of the HTT gene. While aggregate formation is the hallmark of this and several other disorders2,3, its significance in regard to toxicity remains controversial. It has been suggested that large contained aggregates may form in neurons as a protective response. These prominent inclusion bodies (IBs) have been shown to decrease mutant Htt levels elsewhere in the neuron, prolonging cell survival4. Recently, strong evidence for the toxicity of monomeric and small oligomeric species of mutant Htt that have adopted specific conformational states has also emerged5,6, but the great majority of cellular studies report on IBs alone.
Non-invasive, specific visualization of all of the Htt populations co-existing in cells, from low-molecular weight species (monomers, oligomers), to small aggregates (globules, fibers), and to IBs, would be an important addition to the arsenal of experimental techniques. By its nature, a single-molecule fluorescence approach is well-suited to the challenge of reporting on the locations of small numbers of proteins and the structures they assume. In vitro analysis of the aggregation pathway by various methods has identified a range of aggregate intermediates whose existence in neurons and role in pathology is under investigation7,8. As a recent promising methodological addition, super-resolution (SR) fluorescence microscopy of Htt fibers by single-molecule blinking was reported in vitro, highlighting the excellent targeting specificity and high signal-to-background ratio of this fluorescent approach9. SR microscopy typically achieves resolution of 20–50 nm full-width at half-maximum (FWHM) or 10–20 nm precision (σ), depending on the labels employed and the magnitude of non-specific background10. Recently, the method has been applied to protein aggregations in systems with relevance for Alzheimer's or Parkinson's diseases, however with limitations. For example, seeded aggregates of Aβ taken up by HeLa cells were imaged by the dSTORM method11, but it remained unclear if the aggregation occurred in the living cells or whether larger aggregates were, at least partially, endocytosed into the cell interior. The in vitro and in-cell Aβ aggregates had similar morphology, in contrast to results for α-synuclein, where aggregates were observed after microinjection of protein into cells12.
Results
Integrity of the cellular protein quality control machinery throughout the experiment can be regarded as a paramount requirement if protein fate is to be studied under conditions meaningful for pathology. We therefore employed direct cytosolic protein expression. A large body of evidence indicates that formation of N-terminal fragments of Htt containing exon 1 (ex1) by proteolytic cleavage plays an important role in pathogenesis and that mutant Htt-ex1 alone recapitulates the pathological features of HD in several mammalian cell culture models3, including PC12. We transiently transfected ex1 of Htt in NGF-treated sympathetic-neuron-like PC12m (subclone of PC12) cells (Fig. 1a). Htt-ex1 was visualized by C-terminal fusion to the enhanced yellow fluorescent protein, eYFP. The construct was overexpressed compared to endogeneous cytosolic huntingtin in order to produce a measurable aggregation response, where that propensity existed, within days after transfection.
Figure 1. Overview of fluorescence microscopy of sub-cellular huntingtin distributions.
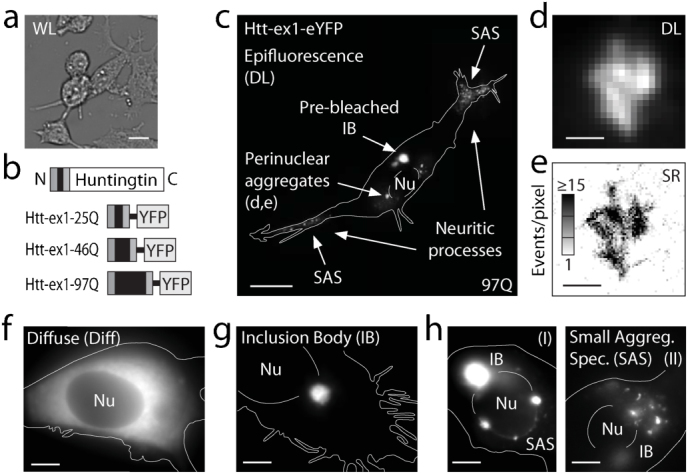
(a) Low-magnification white-light (WL) image of NGF-differentiated PC12m cell culture. (b) Schematic of full-length Htt (not to scale) and the Htt-ex1-xQ constructs fused at the C terminus to eYFP for expression experiments with x = 25, 46, 97. (c) Diffraction-limited (DL) fluorescence of a PC12m cell transfected with Htt-ex1-97Q-eYFP at high magnification (100x) with an evident inclusion body (IB, bright puncta). Before photo-bleaching, the same cell's image was dominated entirely by the brightness of the IB, reflecting its extremely high recruitment of Htt-ex1-97Q-eYFP. (CCD image taken without EM gain and at ~1% pump intensity for region of IB). Only after targeted partial photo-bleaching of the contents of the IB do other more intricate dimmer structures become appreciable (full image, composite of five regions). In general, perinuclear rings of compact aggregates and other small aggregate species can be discerned in many cells. Nu = Nucleus. (d) DL image and (e) super-resolution (SR) reconstruction (plotted as the number of localizations, or events, per pixel) of a perinuclear aggregate, showing the bundling of short fibers. DL examples of cells with (f) diffuse cytosolic distribution, (g) inclusion body, and (h) small aggregate species (round and elongated, fibrillar appearance, SAS) in a perinuclear (I) or dispersed cell-wide (II) arrangement, where the IB has been partially photo-bleached. Scale bars: 20 μm (a,c), 1 μm (d,e), 5 μm (f–h).
In this paper, we define the intracellular distribution of Htt-ex1 for three distinct glutamine repeat lengths, 25Q, 46Q and 97Q (Fig. 1b). From our observation of many cells by methods described below, three distinctive phenotype classes emerged. Most commonly, transfected cells featured a uniformly diffuse (Diff) Htt-ex1 distribution throughout the cell body and dendritic extensions, with evident exclusion from the nucleus (Fig. 1f and Supplementary Fig. S1). Among cells transfected with constructs containing Q-repeats beyond the disease threshold, a fraction exhibited strongly fluorescent perinuclear and cytoplasmic IBs, which were dense aggregates typically ~3–7 μm in diameter (Fig. 1g and Supplementary Fig. S1). A majority of cells with IBs contained additional small aggregates, herein referred to as small aggregate species (SAS, Fig. 1c,h and Supplementary Fig. S1), which became visible by a specialized protocol. The Htt-ex1 SAS were imaged far beyond the optical diffraction limit (Fig. 1d,e) in cells for the first time.
Making small aggregate species visible
When an IB is present, its fluorescence is extremely bright so that little else can be observed in the cell. To visualize the SAS, we therefore employed a specific procedure to reduce the extreme brightness of the IB. This involved targeted photo-bleaching of the IB, followed by the selective illumination of sub-cellular regions of interest (Fig. 2a). Transfected cells (Fig. 2b) were positioned such that the strongly fluorescent inclusion was centered in the excitation beam (Fig. 2c). The IB was then singled out for photo-bleaching by narrowing an iris diaphragm in the beam path. This solely exposed the targeted region to high-intensity light (~2.5 kW/cm2 at peak), strongly reducing the IB's fluorescence signal by many orders of magnitude (Fig. 2d), but preserving nearby fluorescent structures. Only then did the SAS become detectable (Fig. 2e). The disparate fluorescent signal level of the SAS compared to the IB represents an experimental obstacle that may have prevented earlier studies from detecting the diversity of aggregate populations in close spatial proximity.
Figure 2. Targeted photo-bleaching of inclusion bodies and selective illumination of sub-cellular regions.

(a) Schematic of the procedure employed for bleaching a sample region containing a strongly fluorescent inclusion body (IB), to reduce its detrimental signal interference with nearby dimmer structures. The IB was first moved to the center of the excitation beam profile. This location was then singled out for photo-bleaching at high intensity (~2.5 kW/cm2 at peak) by narrowing an iris diaphragm in the excitation beam path. (b) White-light (WL) image of a transfected PC12m cell. (c) The diffraction-limited (DL) epi-fluorescence image of eYFP-labeled Htt-ex1 before bleachdown (boxed region in b) was entirely dominated by the IB signal (image recorded without EM gain). Nu = Nucleus. (d) After 12 minutes of bleach-down, the IB's signal had essentially disappeared. (e) Small aggregate species (SAS) were subsequently perceived (shown here with EM gain in a composite image of two regions). The residual fluorescence from the IB still had to be excluded by delimiting the regions of interest with the iris in each case, because the IB brightness slowly recovered over time as eYFP returned from very long-lived dark states. Scale bars: 10 µm (b–d), 5 µm (e).
Quantifying phenotype
With the capability to detect SAS as well as IB, we were able to quantify phenotype evolution of Htt-ex1 localization in cells for the three Q-repeat lengths. Counting experiments were performed using low-power laser excitation at 514 nm in a wide-field epi-fluorescence configuration. Cells were fixed at times t = 16, 24, 48, 96, 144, 192 hours (Fig. 3). The columns represent fractions of total numbers of transfected cells examined. Of these, the largest fraction at any given time-point exhibited diffuse cytosolic distribution (light gray column). A subset of cells exhibited one or more prominent inclusion bodies (white column). Of these cells with inclusion bodies, most contained small aggregate species as well, thus the dark gray column is always less or equally as tall as the white one. For the non-pathogenic Htt-ex1-25Q, the diffuse phenotype was observed in 100% of cells for all time points (Fig. 3a), in line with expectations for a Q-length below the disease threshold. In contrast, for 97Q, the fraction of cells containing IBs increased from ~5% at t = 16 hours to ~40% at the 192-hour time point (Fig. 3c). IBs developed significantly later for Htt-ex1 with 46 Q-repeats (above threshold, Fig. 3b).
Figure 3. Phenotype counting analysis of cellular huntingtin distributions for three repeat-lengths (25,46,97 Q-repeats) by high-magnification examination of NGF-differentiated PC12m cells.

(a,b,c) Count of Htt-ex1-transfected cells (fixed) classified as the three phenotypes defined in Fig. 1f–h for the three Q-repeat lengths, at time-points t = 16–192 hours post-transfection. Small aggregates were identified in a subset of the cells with bright inclusion bodies (indicated by brackets). (n = number of counting experiments of ~70–150 transfected cells each, N = total number of cells analyzed. Mean ± s.e.m.).
At low magnification, the bright IBs obscured any other, dimmer fluorescent features nearby in the sample. However, using high magnification, combined with targeted photo-bleaching of the region containing the IB (see Methods and Fig. 2), we were able to visualize the SAS as well. Some aggregates formed a perinuclear shell-like arrangement, similar to earlier descriptions13 (Fig. 1h (I) and Supplementary Video S3). In addition, SAS were frequently found to be dispersed throughout the entire cell (Fig. 1h (II), Fig. 4 and Supplementary Video S4). Thus, strikingly, aggregation is not exclusively defined by the intensely-fluorescent IBs and perinuclear aggregates alone, but also by a rich diversity of much smaller structures that can form within the same cell, as illustrated by the example in Fig. 1c.
Figure 4. Sub-cellular distributions of huntingtin aggregates.
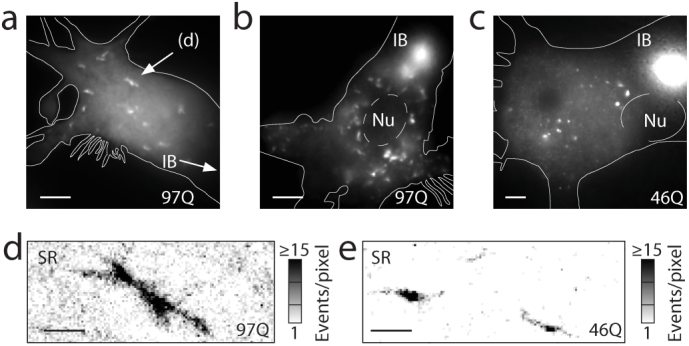
(a,b,c) Small aggregates dispersed throughout the cell bodies form for both 46Q (example e, from a different image plane in c) and 97Q (a,b,d) at time points beyond 16 hrs post-transfection. Their observation became possible after significantly reducing the brightness of the inclusion body (IB) through our targeted photo-bleaching and imaging protocol described in Fig. 2. Only remnants of the bright IB remain. Nu = nucleus. (d,e) Super-resolution (SR) reconstructions of fibrillar aggregates. Scale bars: 5 μm (a–c), 1 μm (d,e).
Super-resolution imaging of small aggregate species
In conventional epifluorescence imaging, SAS appeared as poorly-defined diffraction-limited (DL) objects with round or oblong shapes. To extract further structural detail, we subjected the small aggregate species to examination by SR fluorescence microscopy. The light-induced blinking of fluorescent proteins exploited here is one example of a suite of fluorescence-based single-molecule techniques that can deliver sub-diffraction-limit SR images10. Crucially, genetically expressed probes can be utilized to study protein super-structures in cells at high resolution14. Illuminated with 514 nm light (~2 kW/cm2), eYFP molecules were forced into long-lived dark states. Sparse subsets of eYFP then returned to emit bursts of ~2000 photons per molecule (on average). These bright “blinks” sampled the underlying spatial arrangements (structure) of Htt-ex1 proteins. In a continuous fluorescence recording of these blinking events, single molecules were imaged in each frame and localized with high spatial precision (σ ≈ 15–20 nm) by Gaussian fitting (Supplementary Fig. S2), and SR images of the aggregates were reconstructed (example in Fig. 1e). The increase in information over the diffraction-limited image (Fig. 1d) is substantial, revealing bundled shorter fibers as observed before in vitro9, suggestive of a three-dimensional structure. Fig. 4d shows the SR reconstruction of a small aggregate (from the field of view in Fig. 4a) with fiber-like linear segments. The diffuse localizations outside of the fiber in the SR reconstruction result from monomeric and possibly small oligomeric Htt-ex1 that was not cleared from the cytosol by addition to the IB or other aggregates. This residual monomeric/oligomeric background, which varied among cells, did not compromise our ability to image the SAS co-existing with this pool of very small species. SAS also formed in the axonal and dendritic processes extending from the PC12m cell bodies (Fig. 5a,b and Fig. 1c, top right). Blockage of neuronal processes (by larger aggregates) may have toxic effects, if anterograde and retrograde transport is impaired. Aggregates both in the cell body (Figs. 1 and 4) and the processes (Fig. 5c–e) strongly resembled the fibers examined by SR fluorescence and AFM in vitro9.
Figure 5. Aggregate species in neuritic processes imaged by super-resolution microscopy.

(a,b) Diffraction-limited and super-resolution image of a cellular process (large field). The cell was transfected with fluorescently labeled Htt-ex1-97Q for 24 hours before fixation. Scale bar: 5 μm. (c,d,e) Super-resolution reconstructions of individual small aggregate species, with comparisons to diffraction-limited widefield epifluorescence. Histogram bin width (pixel size) is 50 nm. Scale bars: 500 nm. (f) Histograms in the transverse direction across the fibers indicated in (d,e) show apparent widths of ~80 and ~90 nm (FWHM) respectively.
Discussion
The linearity of the SAS structures was in agreement with original reports by electron microscopy15, but our fluorescence method allowed measurements in many cells showing apparent widths of the linear fibers of ~80–100 nm (FWHM, Fig. 5f) and up to 150 nm in some segments where additional Htt-ex1 seemed to have been recruited (Fig. 6). The single-molecule analysis differentiated individual linear fibers (Fig. 5d,e) from more compact localization patterns, such as the close bundling of such fibers (Fig. 5c), the two cases being indistinguishable in the blurred diffraction-limited image (additional examples in Fig. 6).
Figure 6. Small aggregates, imaged with super-resolution microscopy, based on the light-induced blinking of single eYFP fused to Htt-ex1.

(a) – (h) (Left) Diffraction-limited (DL) images, (middle) super-resolution (SR) reconstructions of structures as histograms of localizations. The thin linear segments in the boxed regions were analyzed to extract the thickness of fibers. This was done by constructing transverse histograms (right), integrating along the fiber length. The thinnest cases represent individual fibers, which had an apparent width of ~80–100 nm (full-width at half-maximum). A number of examples, e.g. (b), (e), (g), see also Fig. 1d,e, Fig. 4d and Fig. 5c, suggest the crossing, contact or bundling of shorter (≤ 2 µm) such fibers, in agreement with observations of aggregate formation in in vitro assays9. For these, cross-sections can range up to 150 nm (e.g. in (e)). Note that the orientation of aggregates in the cell is putatively random, allowing for better or worse structural insight from 2D representations, even at SR, depending on the aggregate's orientation and its projection onto the 2D image plane. All cytosolic aggregates shown are from cells expressing labeled Htt-ex1-97Q (a–f) and Htt-ex1-46Q (g,h). All scale bars are 500 nm.
SAS were identified in most, but not all, IB-containing cells (Fig. 3b,c). If SAS are precursors to the IB, one would expect SAS to be found in some cells independently of IBs, but this is not observed. Our data suggest that any initially formed species (oligomeric and SAS) are quickly sequestered into the IBs, and finer time resolution is needed to observe the initial stages.
As the SAS defined here are only observed for polyQ-expanded Htt-ex1 variants, they may be linked to development of toxicity. By comparison to the unperturbed aggregate properties reported here, it will be possible to assess the role of added therapeutic agents in direct cause-and-effect experiments. The creation of stably-transfected cell lines expressing mutant huntingtin with suitable fluorescent labels for SR microscopy will enable more specialized investigations. Combining our method with toxicity assays will allow detailed studies of aggregate populations in the context of cell death.
In summary, solving the problem of excessive in-plane fluorescence from the bright inclusions by targeted photo-bleaching enabled visualization of a range of structures at high magnification and resolution. We expect that protein aggregation processes and aggregate morphologies related to other neurodegenerative disorders will be examined by similar super-resolution approaches that utilize single-molecule sensitivity and active control of fluorescence emitters. Other optical super-resolution approaches including stimulated emission depletion (STED) microscopy or saturated structured illumination microscopy (SSIM), which can feature comparable resolutions, may prove equally useful for such structural investigations in the future. Optical methods share the potential for minimally-invasive, live-cell, time-lapse investigations of aggresome-type inclusion body formation and fibrillar aggregate growth. The ultimate goal is to visualize the addition of small numbers of protein molecules to the aggregate structures as they are forming and evolving over time. Extending experiments to multi-color fluorescence, the interactions between protein quality control agents such as chaperonins and aggregating Htt can be elucidated in cellulo.
Methods
PC12m cell model
We used PC12m cells, a sub-clone of the original PC12 neuronal model cell line developed by L. Greene (Columbia University). PC12m was selected for its enhanced attachment to plastics, facilitating culture work, in M. Montminy's laboratory (Salk Institute, La Jolla). PC12m cells are well suited for imaging experiments in culture due to their moderate thickness of 10–20 μm when adhered to the microscope slide. PC12 and its sub-clones, differentiated into sympathetic-neuron-like cells, have served as models for early pathogenic events in Huntington's Disease with fluorescent-protein fusions to huntingtin (e.g.16,17).
Cell culture
PC12m cells were grown to confluency at 37°C in a 5% CO2, 96% relative humidity incubator in tissue-culture treated flasks (75 cm2, BD Biosciences) in cell culture medium (10% fetal bovine serum (FBS), 90% phenol red free DMEM, both Gibco). Cells were passaged with 1:5 dilution at least 3 times prior to experiments. Each passage consisted of a 5 min incubation with 2 ml of trypsin replacement (TrypLE Express, Gibco), addition of 8 ml cell media to deactivate the enzyme, a brief centrifugation (<2 min, <400 rcf), followed by media replacement to remove any residual trypsin replacement. Cell suspensions were then added to 6-well tissue culture plates (BD Biosciences). Cells grew to a monolayer (>90% confluency in 1–2 days), whereupon differentiation into sympathetic-neuron-like cells was achieved by treatment with neural growth factor (NGF 2.5S, ~100ng/ml, Invitrogen) for 48 hrs. At this time, cells were transfected with the plasmid DNA for fluorescently labeled Htt-ex1 utilizing a modified protocol for Lipofectamine 2000 (Invitrogen).
Transfection and sample preparation
Complexes were prepared using 2 µg of DNA and 6 µl of Lipofectamine 2000 in 100 µl of Opti-MEM 1 media (Gibco). DNA-transfection reagent complexes were then added to cells and incubated for 5 hrs, at which point the transfection-complex-containing medium was aspirated and each well was treated with 0.5 ml TrypLE Express for 5 min. Cells were suspended in 4–8 ml of NGF-supplemented media and were divided between 2–4 wells (to achieve appropriate cell densities for imaging) in 6-well plates containing microscope slides. These slides (standard no. 1 coverslips, Fisher) were pre-cleaned, ozone-treated and coated with 500 μl of either 8.4 μg/ml fibronectin (EMD Biosciences) in 1x PBS (pH 7.4, Gibco) or 0.460 mg/ml collagen type-I (EMD Biosciences) in 0.02N acetic acid for 1 hr, and rinsed with 1x PBS before use. At defined time points post-transfection (addition of transfection complexes is t = 0), cells were imaged alive, or fixed in 4% paraformaldehyde (PFA, Electron Microscopy Sciences) and washed for the extended, detailed cell-by-cell examination and super-resolution imaging. For the 144- and 192-hr time points, cells were transferred to microscope slides at least 48 hrs prior to experiments but kept on 6-well tissue culture wells in the interim. This adjustment ensured that the surface treatment of the glass slides was relatively fresh and prevented the cell layer from peeling off during fixation washing steps.
Plasmid for transient transfection of fluorescently labeled huntingtin
The first 94 amino acids of huntingtin, encoding the full exon1 (Htt-ex1: MATLEKLMKAFESLKSF[25Q]PPPPPPPPPPPQLPQPPPQAQPLLPQPQPPPPPPPPPPGPAVAEEPLHRPGS) with a 25Q polyglutamine tract and mutant forms (46Q and 97Q respectively) were cloned into a mammalian expression vector (pcDNA3.1(-), Invitrogen) using XhoI and BamHI restriction enzymes (New England Biolabs), to result in CMV promoter-controlled C-terminal fusion constructs to enhanced yellow fluorescent protein (eYFP).
Fluorescence imaging
All imaging was performed on a custom optical setup built around an inverted microscope stage (Olympus IX-71). The pumping light (514 nm, Coherent Sapphire, 50 mW) was focused into the back aperture of a 1.4 NA oil-immersion objective (Olympus UPLSAPO, 100x), achieving standard Köhler illumination for wide-field laser excitation (~30 μm spot size, FWHM) in epi-fluorescence mode. Fluorescence was selected by a dichroic mirror (Semrock, Di01-R532), 525 nm long pass filter (Semrock, BLP01-514R-25) and a band pass filter for eYFP emission (Semrock, FF01-578/105-25). Neutral density filters were employed in the excitation beam path to switch between a low-intensity (~20 W/cm2) cell-scanning mode for assessing cellular fluorescence and characterizing phenotypes, and the high-intensity mode (~1–2 kW/cm2) required for efficient eYFP blinking14,18,19 and fast image acquisition. The image was formed on a highly sensitive electron-multiplying charge coupled device (EMCCD) camera (Andor iXon DU897) that allowed high frame rates. All data shown was extracted from 35-ms-duration frames. Appropriate averaging of 100 frames was applied to epi-fluorescence images (low intensity), to provide a fair comparison of the diffraction-limited epi-fluorescence mode and the single-molecule-localization-based super-resolution reconstructions.
The microscope sample slide and the micrometer translation stage (Semprex, KMI53) were allowed to thermally equilibrate before a super-resolution recording began. In some cases, the microscope slide was adhered to the aluminum sample plate with tape to further enhance stability. Avoiding unnecessary temperature fluctuations in the laboratory and allowing the coarse focus knob to rest before recording helped to keep sample drift to a minimum.
No substantial lateral (xy) drift was present during the ~4–10 minute total acquisition. Continuous sample drift during the short acquisition window of several minutes would manifest itself as a tendency of points to be systematically offset in an ordered way at later times in the acquisition compared to earlier localizations. This effect, which would lead to additional broadening of the observed structures (i.e. fiber widths), was not observed (Supplementary Fig. S2). Consistency in the feature sizes of thin linear segments, ~80–120 nm thickness at several orientations within the same fields of view, further supports this conclusion (Fig. 6). Localizations contributing to the reconstructed image were obtained after a short (5–15 s) initial bleachdown (30–300 s for denser aggregates), but before all eYFP molecules had been bleached. The useful frame range of single-molecule images typically comprised the first five minutes of a recording.
Selective imaging of small aggregates in close proximity to strongly fluorescent inclusions
The highly fluorescent inclusion bodies hindered visualization of the diverse collection of smaller, dimmer aggregate structures which co-exist in many cells in close proximity to the inclusions. Such sizeable brightness disparities of neighboring objects across the sample represent a general problem in wide-field microscopy with an area detector operating at a common gain/pixel. The single, immensely bright IB fully exploits the dynamic range of the detector, while the adjacent dimmer features are not detected, either because the pump light has to be reduced accordingly or due to bright signal spill-over onto the adjacent camera pixels. Consequently, while the inclusions are clearly visible (e.g.16,20), they usually remain the predominant fluorescent feature detected and reported in the cells, especially in work at low magnification and with limited fluorescence dynamic range. We used an iris placed centrally in the excitation path before the focusing Köhler lens to limit excitation to a much smaller region of the sample (Fig. 2). In this way, we selectively photo-bleached IBs, by illumination over minutes at ~2.5 kW/cm2 intensity, and reduced their signal magnitude substantially. We subsequently visualized many dimmer fluorescent species (even in close proximity to the inclusion) by translating the sample, moving the inclusion outside the iris-patterned excitation beam profile and imaging aggregates with minimal signal interference.
Analysis
The extraction of Htt-ex1 single-molecule coordinates from recordings was performed in a manner similar to previous reports14,21,22,23,24. In brief, single molecules were identified by selecting bright pixels which exceeded the average background by at least ~5 times its standard deviation. Positions were determined by fitting pixelated single-molecule images to symmetric two-dimensional Gaussian functions, which describe the microscope point spread function (PSF), and extracting the center coordinates, (x,y). Beside requiring fit convergence, filters were applied to exclude spurious localizations arising from background noise or overlapping molecules. Reconstructed SR images were computed as histograms, i.e. counts of localization events falling within xy domains (50×50 nm bins). Fiber widths were calculated from Gaussian fits to similar histograms in the direction transverse to the fiber's long axis. The statistical localization precision of single molecules was estimated from repetitive localizations of a subset of molecules appearing as multiple isolated clusters25 (Supplementary Fig. S2).
Author Contributions
S.J.S. and L.E.W. designed and performed the experiments, and analyzed the data. W.E.M. supervised the project. S.J.S., L.E.W., W.C.D. and W.E.M. wrote the paper. All authors jointly conceived the study and discussed the results.
Supplementary Material
Supplementary Information
Supplementary Video S3
Supplementary Video S4
Acknowledgments
We thank M. Retzlaff for the gift of the plasmid DNA of Htt-ex1 fused to eYFP and C. M. Livingston for help with DNA amplification and sequencing as well as insightful discussions. This work was funded in part by Award Number PN2EY016525 from the National Eye Institute of the U.S. National Institutes of Health, a U.S. National Science Foundation Graduate Research Fellowship (W.C.D.), and the Gabilan Stanford Graduate Fellowship (W.C.D.).
References
- Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 49–60 (2003). [DOI] [PubMed] [Google Scholar]
- Scherzinger E. et al. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: Implications for Huntington's disease pathology. Proc. Natl. Acad. Sci. USA 96, 4604–4609 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imarisio S. et al. Huntington's disease: from pathology and genetics to potential therapies. Biochem. J. 412, 191–209 (2008). [DOI] [PubMed] [Google Scholar]
- Arrasate M., Mitra S., Schweitzer E. S., Segal M. R. & Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–810 (2004). [DOI] [PubMed] [Google Scholar]
- Nagai Y. et al. A toxic monomeric conformer of the polyglutamine protein. Nat. Struct. Mol. Biol 14, 332–340 (2007). [DOI] [PubMed] [Google Scholar]
- Miller J. et al. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat. Chem. Biol. 7, 925–934 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur A. K. et al. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 16, 380–389 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legleiter J. et al. Mutant Huntingtin fragments form oligomers in a polyglutamine length-dependent manner in vitro and in vivo. J. Biol. Chem. 285, 14777–14790 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duim W. C., Chen B., Frydman J. & Moerner W. E. Sub-diffraction imaging of Huntingtin protein aggregates by fluorescence blink-microscopy and atomic force microscopy. ChemPhysChem 12, 2387–2390 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerner W. E. Microscopy beyond the diffraction limit using actively controlled single molecules. J. Microsc. 246, 213–220 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski Schierle G. S. et al. In situ measurements of the formation and morphology of intracellular β-amyloid fibrils by super-resolution fluorescence imaging. J. Am. Chem. Soc. 133, 12902–12905 (2011). [DOI] [PubMed] [Google Scholar]
- Roberti M. J. et al. Imaging nanometer-sized α-synuclein aggregates by superresolution fluorescence localization microscopy. Biophys. J. 102, 1598–1607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martindale D. et al. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat. Genetics 18, 150–154 (1998). [DOI] [PubMed] [Google Scholar]
- Biteen J. S. et al. Superresolution imaging in live Caulobacter crescentus cells using photoswitchable EYFP. Nat. Methods 5, 947–949 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M. et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993 (1997). [DOI] [PubMed] [Google Scholar]
- van Roon-Mom W. M. C. et al. Mutant huntingtin activates Nrf2-responsive genes and impairs dopamine synthesis in a PC12 model of Huntington's disease. BMC Molecular Biology 8, 84–96 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostol B. L. et al. Mutant huntingtin alters MAPK signaling pathways in PC12 and striatal cells: ERK1/2 protects against mutant huntingtin-associated toxicity. Hum. Mol. Gen. 15, 273–285 (2006). [DOI] [PubMed] [Google Scholar]
- Dickson R. M., Cubitt A. B., Tsien R. Y. & Moerner W. E. On/off blinking and switching behavior of single green fluorescent protein molecules. Nature 388, 355–358 (1997). [DOI] [PubMed] [Google Scholar]
- Fölling J. et al. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat. Methods 5, 943–945 (2008). [DOI] [PubMed] [Google Scholar]
- Legleiter J. et al. Monoclonal antibodies recognize distinct conformational epitopes formed by polyglutamine in a mutant huntingtin fragment. J. Biol. Chem. 284, 21647–21658 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E. et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science 313, 1642–1645 (2006). [DOI] [PubMed] [Google Scholar]
- Rust M. J., Bates M. & Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–796 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess S. T., Girirajan T. P. K. & Mason M. D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 91, 4258–4272 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff H., Galbraith C. G., Galbraith J. A. & Betzig E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat. Methods 5, 417–423 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. D., Sahl S. J., Lew M. D. & Moerner W. E. The double-helix microscope super-resolves extended biological structures by localizing single blinking molecules in three dimensions with nanoscale precision. Appl. Phys. Lett. 100, 153701 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Video S3
Supplementary Video S4