

Abstract
As parallel advances in cancer biology and drug development continue to elevate the role of targeted therapies in oncology, the need for imaging biomarkers that systematically measure the biology associated with therapeutic intervention has become more urgent. Although the molecular imaging community has a commitment to develop technologies to this end, few investigational radiotracers directly measure the biology of common oncogenic signaling pathways often addressed by targeted therapies. Visible progress has been achieved with a handful of radiotracers rationally designed to intercalate the patho-biology of prostate cancer, a molecularly heterogeneous disease nevertheless broadly defined by a fairly small repertoire of recurrent oncogenic lesions.
Introduction
On theoretical and practical grounds, a mandate for new imaging biomarkers that measure the output of oncogenic signaling pathways can be sensibly justified. As the oncology community now routinely advances targeted therapies alongside cytotoxic therapies into the clinic, it seems logical that cognate imaging biomarkers should complement this development by measuring those molecular events immediately impacted by targeted therapies, rather than reporting a symptom of overall tumor burden or health. Disappointingly (and for unclear reasons), in only a few settings have imaging biomarkers whose biology is distally related to the pharmacology of a targeted therapy impacted the approval process and patient care (vide infra). Nevertheless, regulatory agencies remain highly motivated to conditionally reshape drug approval criteria on the basis of any biomarker “reasonably likely to predict” clinical benefit (1, 2). With these considerations in mind, there appear to be ample opportunities for carefully designed imaging tools to streamline the clinical evaluation of experimental therapies.
One example of a malignancy that seems primed to benefit from the application of radiotracers measuring oncogenic signaling pathways is castration resistant prostate cancer (CRPC), the most advanced and fatal form of the disease. Indeed, many years of work have defined several oncogenic events that, in some permutation, drive the lethality of CRPC(3) (Figure 1). Two of the most frequently annotated phenomena—reactivation of androgen receptor (AR) signaling(4) and aberrant PI3K pathway signaling(5, 6)—are readily addressable by targeted therapies, some of which have or are undergoing clinical evaluation in CRPC(7, 8). Notably, two agents targeting the AR signaling axis—abiraterone acetate(9) and MDV3100(10)—extended survival in clinical trials, pointing to the substantial progress that has been made in comprehending the molecular determinants of CRPC. Moreover, that neither drug has been uniformly effective in the patient populations studied to date further supports a need to develop biomarkers that identify upfront patients most likely to respond to therapy, and/or more clearly indicate a molecular tumor response post therapy.
Figure 1. A brief synopsis of recurrent oncogenic lesions in CRPC.
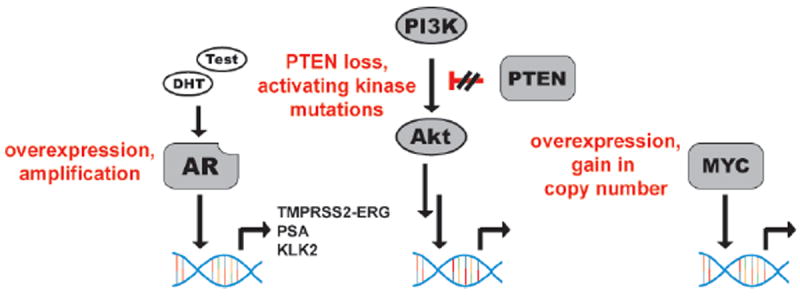
Pathological activation of AR signaling is one of the most clearly understood hallmarks of CRPC, occurring via gene amplification, receptor stabilization, or endocrine production of androgens. The transcriptional repertoire of AR is known to include several genes linked to the patho-biology of CRPC, including the TMPRSS2-ERG fusion protein. In addition, pathological activation of the PI3K signaling axis occurs very commonly in CRPC, principally via inactivation of the tumor suppressor PTEN. The downstream consequences of aberrant PI3K signaling are still being defined, but in some contexts, deregulation of transcriptional programs (e.g. HIF1α, Forkhead family transcription factors) seems to drive the pathology of this event. Finally, the transcription factor MYC is also a well-defined oncogenic driver of prostate cancer, with copy number alterations annotated in approximately 30% of patients.
Also (beyond invoking overall survival), defining responsive versus resistant sub-populations has been challenging for patients with CRPC. As distant prostate cancer metastases largely deposit in the bone, assessing objective response rates with radiological criteria (e.g. RECIST) is essentially ineffective(11). Appreciating this, oversight bodies like the Prostate Cancer Clinical Trials Consortium have promulgated more standardized criteria to interpret tumor progression in clinical trials with bone scans [i.e. 99mTc-MDP, and more recently, 18F-NaF (12)]. Though very accessible, this technology has several limitations (13). Most notably, it has been empirically demonstrated that radiographic responses can lag tumor responses to targeted therapies by months or even years. This phenomenon is rationalized on the basis of the mechanism of radionuclide localization to osteoblastic lesions. In both cases, the radiotracers target normal skeletal foci actively undergoing repair, rather than targeting the tumor itself, and normal bone healing can persist long after ablation of a nearby tumor. Consequently, even partial resolution of a bone scan is uncommon for therapies known to extend overall survival.
Into this vacuum, several groups have attempted to establish a role for positron emission tomography (PET) in the management of CRPC, owing to its sensitivity, non-invasiveness, and quantitative properties (14). Particular emphasis has been placed on small molecule radiotracers that might reveal a whole body assessment of a “metabolic tumor phenotype” (e.g. 18F-FDG, 18F-FACBC, 11C-methionine, 11C-acetate, 11C-choline). A thorough discussion of these approaches is beyond the scope of this review and several high quality treatments in the literature can be found elsewhere (15, 16). However, that none of these radiotracers have been adopted into widespread clinical use for treatment monitoring underscores their shortcomings. Principally, (although their respective mechanisms of tumor uptake are generally well characterized) that the deeper biological basis of their avidity for CRPC is not understood limits the hypotheses that one can reasonably advance to establish their clinical utility. Consequently, that many otherwise useful radiotracers cannot be cross-applied to CRPC for similar gains has elevated the criteria for radiotracer design.
The biomarker landscape and the emergence of molecular imaging in oncology
Prior to discussing the investigational radiotracers that are the topic of this review, the broader setting of biomarker development should be described to establish the context for their development. Generally, biomarkers can be defined according to the timing, and in turn, the purpose of their application(17). For instance, those assayed from treatment-naïve patients are intended to detect subclinical disease early, distinguish aggressive from indolent disease, or sort patient populations according to those most likely to respond to a given systemic therapy (screening, prognostic, or predictive biomarkers, respectively). Post therapy, biomarkers can be immediately applied to interpret the extent of target inhibition (pharmacodynamic biomarkers), or at early and intermediate intervals to assess tumor response parameters that could indicate an eventual increase in overall survival (clinical or surrogate endpoints).
Beyond disease staging, an obvious clinical utility for functional imaging biomarkers pre therapy has not yet been realized, and more visible progress establishing a role for molecular imaging in oncology has been shown post systemic therapy. The first milestone (even predating the term “molecular imaging”) was the appreciation that a radiotracer measuring an aspect of tumor health could unambiguously demonstrate cancer cell death post therapy (and point to an eventual increase in overall survival), even when anatomical imaging did not necessarily depict a convincing regression in a mass once known to bear a cancerous lesion. Initially with 67Ga-citrate SPECT post cytoxic chemotherapy in lymphomas (18), and more recently with 18F-FDG PET post targeted therapies in lung cancer, gastrointestinal stromal tumors or melanoma(19-21), these precedents established and continue to support a unique virtue for monitoring tumor response with a functional imaging biomarker.
While it is quite encouraging that radiotracers reflective of clinical outcome can be empirically identified and validated without much prior appreciation of a functional relationship to the drug target, the phenomenological nature of these correlations makes it challenging to extract ubiquitous lessons that might instruct future radiotracer design. Indeed, of the many investigational radiotracers developed to monitor an aspect of tumor health to date, only a few have been shown in any context to clearly outperform the community’s current gold standard, 18F-FDG [one notable example is the proliferation marker 18F-FLT (22, 23)]. In this regard, a second milestone for the community was the appreciation that radiotracers can be rationally engineered to non-invasively assay the immediate biological properties of a given target protein.
Many groups have contributed to a general model showing that radiotracers imitating the pharmacology of an endogenous substrate, ligand, or a synthetic drug can serve as powerful pharmacodynamic biomarkers (24, 25). The model extends from the premise that a competition between radiotracer and a large excess of cold drug for binding to a mutual target protein can be exploited to interpret the extent of target blockade by drug. One prominent example is 16α-[18F]fluoro-17β-estradiol, a radiolabeled version of the endogenous female sex hormone, which has been effectively used to annotate estrogen receptor status in breast cancer patients, as well as to titrate the dose of antiestrogen therapies (26, 27).
As will be discussed in the following section, there are documented shortcomings to this approach, and a complementary model has advocated evaluating inhibition more directly by measuring the target’s downstream biology. A seminal example was the use of a radiolabeled F(ab’)2 derivative of Herceptin (a monoclonal antibody [mAb] to the receptor tyrosine kinase HER2) to interpret the impact of a dose of an inhibitor of the molecular chaperone HSP90 (28). Because HSP90 prevents HER2 degradation, the authors demonstrated that the downregulation of HER2 induced by HSP90 inhibition could be monitored by PET to quantify the extent of target inhibition in vivo. Beyond the immediate clinical significance of these findings, this study helped to establish a paradigm that has since been invoked repeatedly to develop pharmacodynamic imaging biomarkers for unrelated targets and malignancies (29, 30).
Because the potential for carefully designed radiotracers to broadly impact clinical practice is now very clear, I would submit that one of the next major frontiers for the molecular imaging community is to apply the aforementioned concepts to more systematically develop biomarkers matched to the patho-biology of validated oncogenic drivers in cancer. In the following sections, several investigational radiotracers engineered to address this end in CRPC will be described, with a particular emphasis on the biological rationale for their preparation, and the unmet need that warranted their development. The technologies will be outlined in chronological order of discovery, to emphasize how the lessons imparted by earlier technologies have empowered the development of newer radiotracers.
18F-16β-fluoro-5α-dihydrotestosterone (18F-FDHT), a radioligand designed to quantify AR occupancy by antiandrogens
The merit of inhibiting AR signaling in prostate cancer has been understood for decades(7), and the favorable results from two recent trials treating CRPC with highly potent AR inhibitors underscore the importance of AR in all stages of the disease. Although the clinical successes of the antiandrogen MDV3100 and the androgen biosynthesis inhibitor abiraterone acetate represent a milestone for the field, approximately 50% of patients fail to respond to either therapy. At least two hypotheses could account for this phenomenon. First, a patient’s disease may bear biological features that preclude response to even complete AR inhibition. Second, the dose of drug may be insufficient to impair AR signaling to the extent required to observe survival benefit.
The second hypothesis relates to the pharmacodynamic properties of the drug dose, and for antiandrogen therapies, understanding the extent to which drug binds AR could be greatly insightful. Expanding on previous observations conferred by researchers at Washington University in St. Louis (31-33), researchers at MSKCC showed that 18F-FDHT, a radiolabeled form of dihydrotestosterone that binds AR in prostate cancer lesions (34-36), can be used in-line with antiandrogen therapy to assess the extent of receptor blockade by drug (Figure 2A). A pilot study was conducted with 22 patients enrolled in the phase I/II portion of the MDV3100 trial to assess drug pharmacodynamics (10). Prior to beginning therapy, patients received an 18F-FDHT scan, and four weeks post initiation, a follow-up scan. Regardless of dose, post therapy 18F-FDHT SUVmax values almost uniformly declined (21 of 22 patients), strongly suggesting that MDV3100 effectively binds AR in vivo.
Figure 2. Monitoring the pharmacodynamics of antiandrogen therapies with the radioligand 18F-FDHT.
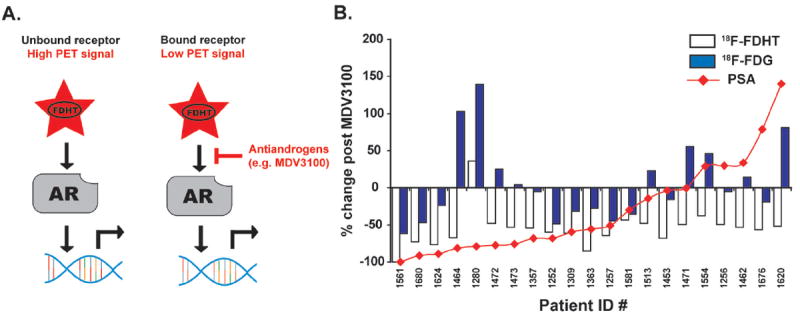
A. A schematic representation of how the radioligand 18F-FDHT is applied in man to assess AR expression levels and receptor occupancy by drug. In the context of CRPC, pathological activation of AR often occurs despite low circulating levels of androgens, allowing the radiotracer 18F-FDHT to bind AR in prostate cancer lesions. When applied post antiandrogen therapy, the absence of 18F-FDHT binding can indicate that AR is effectively engaged by drug. B. A pilot study showing that 18F-FDHT can be used to interpret dose selection of antiandrogens in man. Patients were scanned with 18F-FDHT prior to enrolling in the phase I/II trial, and after 4 weeks of therapy, scanned again to assess receptor blockade by MDV3100. Although SUVmax values almost uniformly declined in this cohort—pointing to effective engagement of AR by MDV3100—percent changes in serum PSA or 18F-FDG SUVmax values did not overlay in an interpretable fashion with these 18F-FDHT “responses”, further pointing to a need for imaging agents that measure AR pathway signaling output directly.
While these data clearly support the continued use of 18F-FDHT to interpret dose selection for antiandrogens in patients, it is interesting to note that the 18F-FDHT “responses” did not overlay well with other conventional indicators of biological tumor response(37) (Figure 2B). Over the same time frame, ~40% of patients (9 of 22) profiled with 18F-FDHT showed rising 18F-FDG values, suggesting that tumor health was unaffected by MDV3100. Moreover, a non-overlapping group of patients comprising ~40% of the cohort (9 of 22) had stable or rising serum values of prostate specific antigen (PSA), an AR target gene, suggesting that AR function was not inhibited.
These observations raise the possibility that, in some contexts, engaging AR with an antiandrogen does not necessarily result in tumor response or AR inhibition. Indeed, symptoms of this phenomenon have been observed sporadically for many years. For instance, it is well known that in preclinical prostate cancer models bearing AR overexpression, the antiandrogen Casodex (bicalutamide) paradoxically acts as an agonist for AR [an effect that 18F-FDHT PET would not distinguish from authentic antagonism(4)]. More speculatively, PSA levels have been shown to occasionally decline after suspension of antiandrogen therapy in patients (i.e. “flutamide withdrawal syndrome”), perhaps recapitulating this preclinical observation in vivo (38).
While further research is required to establish the diagnostic value of post therapy declines in 18F-FDHT, the lessons from the proof-of-concept studies stress that there is a need to measure AR signaling output directly. Indeed, beyond the issues discussed above, it seems unlikely that 18F-FDHT PET would bear any meaningful information about the pharmacology of androgen biosynthesis inhibitors, since these drugs inhibit AR function without physically binding the receptor. As an aside, it is also well appreciated that 18F-FDHT is quite unstable in vivo—significant metabolism occurs within minutes post injection—further underscoring a need for second generation radiotracers targeting this oncogene(34). Consequently, the next two sections will describe two preclinical efforts to non-invasively measure AR signaling output itself.
89Zr-J591, a radiotracer designed to measure AR pathway signaling by quantifying relative changes in the expression of prostate specific membrane antigen
The rationale for developing an imaging biomarker reflective of AR pathway signaling also borrows heavily from insights conferred by studying serum levels of secreted AR target genes. AR regulated kallikreins—most notably, PSA—have been measured exhaustively in patient serum to screen for disease onset, develop prognoses, and monitor treatment response (39, 40). While useful, the shortcomings of this approach are well documented, and in principle, an imaging biomarker reflective of AR signaling could supplement these limitations. For example, circulating PSA levels are a depiction not only of changes in AR transcription, but also of secretion into pericellular space and leakage into the serum—two processes that are poorly understood and not necessarily AR regulated. It is also well documented that only a very small fraction of the total PSA produced in the prostate escapes into the serum(41), suggesting that a post-translational step (rather than AR-driven transcriptional changes per se) may be rate-limiting to PSA accumulation in the blood. Alternatively, measuring a gene with fewer degrees of freedom between AR transcription and expression (i.e. imaging a cell surface antigen) could confer more faithful measurements of AR signaling.
A further complication is that radiographic responses to androgen deprivation therapy in a patient can be mixed, with some lesions shrinking whereas others are stable or expanding, perhaps a reflection of the heterogeneity of metastatic lesions even within the same patient. Indeed, the recent work imaging patients with 18F-FDG and 18F-FDHT has revealed a highly diverse array of radiotracer uptake among CRPC lesions (37). By extension, the differential sensitivity of metastatic lesions to androgen deprivation therapy could be rationalized by different levels of AR inhibition. Because a serum biomarker reflects an average across all lesions, it would not inform as to whether AR inhibition varies at different sites, while an appropriately designed imaging biomarker could likely capture this behavior.
Among the genes shown to be regulated by AR, prostate specific membrane antigen (PSMA) emerged as an immediately attractive candidate biomarker owing to the large repertoire of tools available to image this protein, including reagents already cleared for clinical use (42-46) (Figure 3). Capitalizing on this information, Evans et al demonstrated in vitro and in vivo that androgens repress PSMA expression in multiple prostate cancer models, while antiandrogens upregulate expression (47). Genetic ablation of AR with siRNA in vitro confirmed that these phenomena are AR-mediated. The expression changes were also substantial enough to be quantitatively measured in vivo in human prostate cancer xenograft models through PET imaging with a radiolabeled version of a fully humanized antibody to PSMA, 64Cu-labeled J591(48).
Figure 3. Non-invasively measuring AR signaling pathway output with a radiotracer targeting PSMA.
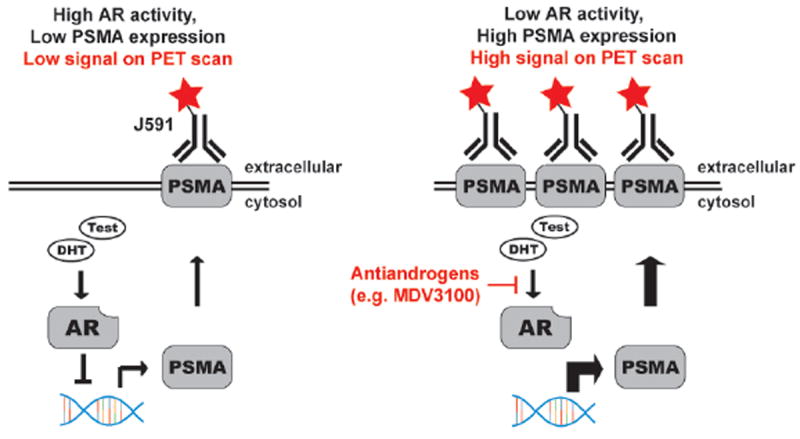
A schematic representation of the relationship between AR activity and PSMA expression, and the strategy to exploit this relationship for PET imaging. Several reports have shown that PSMA is an androgen repressed gene, and that AR inhibition elevates PSMA expression. ChIP-Seq data has shown AR to bind the PSMA gene, advancing the putative mechanism outlined in this figure.
In parallel, Holland et al. found that altering the radiolabeling strategy by appending zirconium-89 to J591 resulted in unprecedented tumor contrast ratios (>20:1 compared to muscle) in prostate cancer models(49). Moreover, because the half life of zirconium-89 is long (t1/2 ~78 h), images could be acquired out to 144 h post injection of 89Zr-J591. Both the tumor contrast and the magnitude of radiotracer uptake in the tumor improved continuously over time, likely due to antibody clearance from blood circulation and normal tissues. Collectively, these findings have motivated an ongoing clinical trial at MSKCC to evaluate 89Zr-J591 for prostate cancer diagnosis and treatment monitoring, and at time of press, first-in-man studies had commenced.
89Zr-5A10, a radiotracer designed to measure AR pathway signaling by quantifying relative changes in the expression of free prostate specific antigen
Although 89Zr-J591 is an attractive lead agent, PSMA expression is not prostate-specific, the mechanism of AR repression of PSMA is not defined, and the clinical impact of AR-directed therapy on PSMA expression remains to be determined. In this regard, a radiotracer that targets a more thoroughly validated clinical biomarker of AR status could be a sensible alternative pending the outcome of the 89Zr-J591 trial. Appreciating this, Ulmert et al. recently disclosed a strategy to measure AR status by targeting PSA with an antibody-based radiotracer(50).
As a secreted protein, PSA is admittedly not an obvious imaging target, and one could reasonably argue that circulating antigens could confound tumor imaging by sequestering the radiotracer in serum. As serum PSA overwhelmingly exists in complexes with serpins (e.g. alpha-1 antichymotrypsin), the authors overcame this challenge by radiolabeling a monoclonal antibody (5A10) specifically reactive with uncomplexed, or “free” PSA (Figure 4A and B). Because of limited access to serpins, the tumor microenvironment is enriched in free PSA, and on this basis, it was hypothesized that the radiotracer would accumulate at or within a tumor cell. Consistent with this expectation, 89Zr-labeled 5A10 localized to multiple prostate cancer tumors in an AR and PSA-dependent manner, and effectively measured changes in PSA expression triggered by MDV3100 treatment. Moreover, 89Zr-5A10 readily detected osseous prostate cancer lesions, and was not cross-reactive with the non-malignant conditions that score on traditional bone scans. This data effectively established proof-of-concept, and pending the humanization of 5A10, the ultimate clinical utility of this strategy can be assessed.
Figure 4. Non-invasively measuring AR signaling pathway output with a radiotracer targeting free PSA.
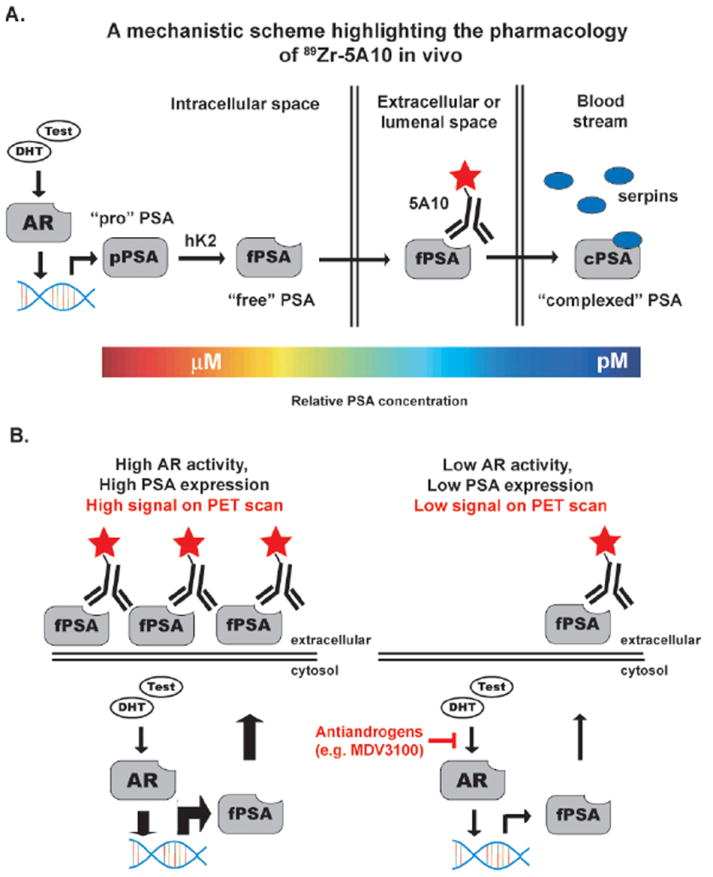
A. A schematic representation of the relationship between AR activity and PSA expression, and the strategy to exploit this relationship for PET imaging. The kallikrein PSA is an AR target gene, and has a complicated post transcriptional lifetime. The gene is transcribed as a catalytically inactive proenzyme (“pro” PSA), and is activated by the proteolytic activity of human kallikrein 2 (hK2). The pool of PSA with an unobscured catalytic cleft is referred to as “free” PSA, and can be secreted into extracellular space. Through an undefined mechanism, a small portion of free PSA will leak into the blood stream, whereupon it is rapidly and irreversibly complexed with serpins (e.g. alpha-1 antichymotrypsin). To target tumor-associated PSA, an antibody that specifically recognizes free PSA was radiolabeled (89Zr-5A10). The color bar at bottom highlights the gradient of PSA concentrations in the various tissue compartments, further underscoring the utility of targeting tumor associated PSA species for imaging. B. A schematic representation showing how 89Zr-5A10 PET can inform on the in vivo pharmacology of androgen deprivation therapies. The model was derived from that established with J591 PET.
One additional advantage to developing a radiotracer targeting a secreted protein is that any knowledge highlighting a shortcoming of the serum measurement provides an obvious clinical scenario to determine if the imaging tool can reveal any new and useful information. Indeed, the greatest hurdle to the approval of investigational radiotracers is demonstrating a clear clinical utility to regulatory agencies, and the attrition rate is currently high. As there are many partially flawed serum biomarkers in oncology (e.g. CA125 for ovarian cancer(51), carcinoembryonic antigen for colorectal cancer(52), and carbonic anhydrase 9 for renal cell carcinoma(53)), this paradigm may be useful to substantiate the approval of other radiotracers.
89Zr-transferrin, a radiotracer designed to measure aberrant MYC signaling by quantifying relative changes in the expression of the transferrin receptor
The previous sections have dealt with technologies designed to measure dimensions of the AR signaling axis. Several other oncogenic signaling events are known drive prostate cancer, suggesting a panel of biomarkers may be required to comprehensively manage the disease. One important oncogenic driver of prostate cancer is the transcription factor MYC. Through analysis of biopsies from early stages of disease, most estimates indicate that pathological activation of MYC occurs in ~30% of prostate cancers, and MYC copy number alterations are known to correlate with poor clinical outcome(54, 55). Moreover, in genetically engineered mouse models, prostate specific MYC overexpression leads to invasive adenocarcinoma(56). Nevertheless, very little is known about the role of MYC in clinical disease beyond what can be inferred from biopsy, and in particular, the impact of aberrant MYC activity on therapeutic intervention in CRPC is entirely unknown.
These observations argue strongly for a noninvasive biomarker of MYC status, and the model invoked for AR provided Holland et al with a clear avenue for radiotracer development(57). Among the target genes regulated by MYC, the transferrin receptor (TFRC) emerged as an obvious candidate(58), owing to several decades of work showing that transferrin (the serum ligand for TFRC) is a versatile scaffold for radionuclides [Figure 5 (59-61)]. Appreciating the quality of previously published work with 89Zr-labeled biomolecules(62, 63), the authors prepared 89Zr-transferrin (89Zr-Tf), and evaluated the properties of the radiotracer in preclinical models of MYC driven prostate cancer.
Figure 5. Non-invasively measuring MYC signaling with a radiotracer targeting TFRC.
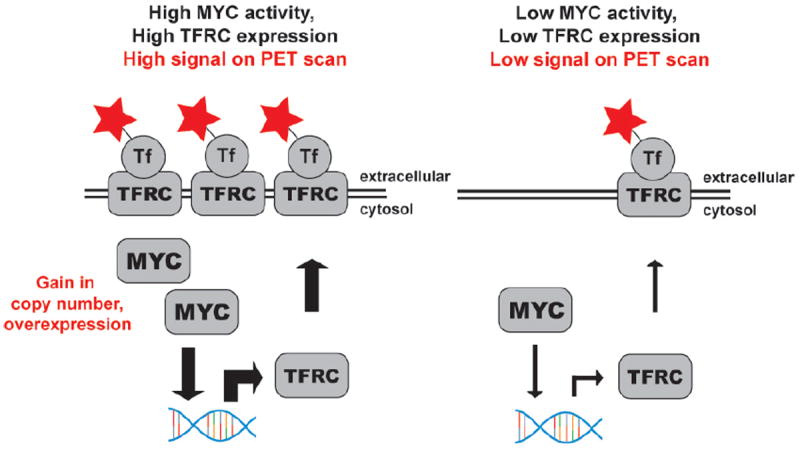
A schematic representation of the relationship between MYC activity and TFRC expression, and the strategy to exploit this relationship for PET imaging. TFRC is a validated MYC target gene in many cancers, including prostate cancer. The soluble ligand of TFRC, the serum protein transferrin (Tf), was radiolabeled to target the relative changes in TFRC expression that occur with fluctuations in MYC signaling.
Unlike previous strategies (60, 64) to image TFRC expression levels (e.g. 67Ga-citrate), 89Zr-Tf produced high contrast PET images with exceptional resolution and low uptake in normal tissues. The radiotracer sensitively measured treatment-induced changes in MYC and TFRC expression in MycCaP tumors, a murine prostate cancer model bearing an androgen regulated, inducible MYC allele (65). The superior pharmacokinetic properties of 89Zr-Tf also allowed the detection of spontaneously developing prostate cancer in a transgenic MYC prostate cancer model (56). Notably, 89Zr-Tf detected regions of aberrant MYC signaling independent of the histopathological stage of disease, imaging foci that appeared well prior to histological or anatomic evidence of adenocarcinoma.
While this work is of immediate relevance to the mouse modeling community (PET imaging of prostate cancer with common radiotracers like 18F-FDG is not useful because the prostate is obscured by bladder accumulation of radiotracer), 89Zr-Tf could also become a powerful imaging biomarker in man for cancer detection and for assessing response to therapy. Particularly in light of recent reports demonstrating that JQ1—an inhibitor of the epigenetic protein BRD4—exerts its anti-tumor effects by downregulating MYC, this radiotracer may be suitable for monitoring response to this promising new therapy(66-68). Finally, TFRC is also regulated by MYC in other cancers, including lymphoma(69) and breast(70), which may further broaden the significance of this technology.
Emerging themes and concluding remarks
As the oncology community continues to substantiate the hypothesis that many cancers can be managed with cleaner pharmacological agents targeting disease-specific molecular aberrations, the molecular imaging community is positioned to serve as a powerful foil to accelerate discovery and streamline the clinical evaluation of experimental therapeutics. At the forefront, many years of work deconstructing the molecular determinants of CRPC have facilitated the rational design of several novel radiotracers to address the patho-biology of two highly visible oncogenes. The radiotracers described herein are most obviously applied as pharmacodynamic biomarkers or early and intermediate response indicators, although one could reasonably imagine extending this paradigm to other classes of biomarkers. The promise of this proof-of-concept work can be summarized into a handful of themes that will hopefully empower future radiotracer development programs for CRPC and other malignancies:
Favoring biological events directly impacted by oncogenic signaling pathways may enhance the informational content of PET scans. The sporadic success of radiotracers targeting gross tumor properties, the complications associated with interpreting the multiple equilibria that govern ambient serum biomarker concentrations, and the inspiring example set with pharmacodynamic imaging biomarkers, argues for developing imaging biomarkers derived from molecular events more immediately affected by oncogene activity to evaluate targeted therapies.
Mining transcriptional profiling data is a highly effective approach to triage candidate imaging biomarkers. The examples presented herein primarily focus on exploiting as imaging biomarkers target genes whose expression is driven by oncogenic transcription factors. This model draws on previous insights garnered principally from studying secreted biomarkers in oncology, and should scale well to other oncogenic signaling pathways. One particularly attractive aspect of this approach is that it capitalizes on the wealth of expression data publicly available from preclinical models and clinical specimens, providing an extensive list of genes that can be cross-referenced over many independently derived data sets to improve confidence in target selection.
Deliberately targeting an “imageable” form of a serum biomarker could more clearly define a unique clinical utility for investigational imaging agents. For several serum biomarkers used in the management of cancer, a vast body of epidemiological data has clearly highlighted virtues and shortcomings. Given the challenges associated with demonstrating a clear clinical utility for investigational radiotracers, attention from the imaging community should be paid, where appropriate, to targets bearing a strong epidemiological history to more readily highlight the unique virtues of an imaging tool.
While the data summarized in this review make a case for a philosophy of de novo radiotracer design from carefully curated imaging targets, it is worth noting that this approach is not the only manner in which to discover imaging biomarkers reflective of aberrant oncogenic signaling. For instance, in a highly instructive study, Palaskas et al. analyzed the molecular basis of 18F-FDG avidity in breast cancer biopsies with an annotated PET history (70). An unbiased survey of transcriptional changes identified a genetic signature indicative of elevated MYC activity in basal-like breast cancer tissues that were avid for 18F-FDG, and immunohistochemical analysis of patient tissue showed a strong correlation between MYC overexpression and high 18F-FDG SUVmax values. These findings raise the possibility that other radiotracers engineered to measure a general property of tumor health may in some contexts be ascribed to readily interpretable oncogenic events. In this regard, retrospectively defining the patho-biological events that drive the upregulation of the machinery responsible for retaining a radiotracer at or within a tumor cell could be a highly attractive and expedient approach to expand the clinical utility of an investigational radiotracer, particularly among those for which successful feasibility studies in man have already been conducted.
Moving forward, one outstanding concern for the oncology and molecular imaging communities should be the logic of continuing to argue that a single radiotracer can emerge as an instructive endpoint in clinical trials. Indeed, the documented molecular heterogeneity of some cancers (71) as well as the fact that oncogenes like AR or MYC can regulate hundreds of genes (72, 73), should continue to raise doubts as to whether, even for a single oncogene, one cognate imaging biomarker may be broadly applicable to patient populations. Speaking to this point, many promising predictive biomarkers constitute molecular signatures derived from a panel of genetic events, rather than a single “smoking gun” event per se (74). In this regard (and despite the remarkable precedent established with 67Ga-citrate and 18F-FDG), the clinical merit of documenting tumor response to therapy with an imaging biomarker must be continuously reevaluated. Ultimately, molecular imaging tools will almost certainly be best invoked as one of many endpoints in clinical trials, combining their considerable virtues with other emerging noninvasive technologies [e.g. profiling circulating tumor cells in patient serum(75)] that might supplement their shortcomings.
Significance.
That variable treatment responses or emergent resistance phenotypes are often documented in man argues strongly for diagnostic technologies that can be realistically applied post therapy to capture the dynamic patterns of disease response. The purpose of this review is to describe a collection of radiotracers developed to measure the patho-biology of prostate cancer for improved treatment monitoring, placing particular emphasis on the biological rationale for their preparation. A chronological description of radiotracer development programs is outlined, primarily to stress how an ongoing dialectic between earlier and more contemporary imaging technologies has accelerated discovery.
Acknowledgments
I would like to thank Drs. Charles L. Sawyers, Jason S. Lewis, Ingo K. Mellinghoff, Howard I. Scher, Steven M. Larson, Hedvig Hricak, Naga Vara Kishore Pillarsetty, Igor Vivanco, and Brett S. Carver for instructive discussions, and Dr. Josef J. Fox for assistance in compiling clinical data.
Financial Support: M.J.E. was supported by the Geoffrey Beene Cancer Research Center of MSKCC, the Brain Tumor Center at MSKCC, and a training grant in molecular imaging from the National Institutes of Health (R25-CA096945).
Footnotes
Conflicts of Interest: The author declares no conflicts of interest.
References
- 1.Prentice RL. Surrogate and mediating endpoints: current status and future directions. J Natl Cancer Inst. 2009;101(4):216–7. doi: 10.1093/jnci/djn515. [DOI] [PubMed] [Google Scholar]
- 2.Wagner JA, Williams SA, Webster CJ. Biomarkers and surrogate end points for fit-for-purpose development and regulatory evaluation of new drugs. Clin Pharmacol Ther. 2007;81(1):104–7. doi: 10.1038/sj.clpt.6100017. [DOI] [PubMed] [Google Scholar]
- 3.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23(32):8253–61. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 4.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10(1):33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 5.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 18(1):11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Majumder PK, Sellers WR. Akt-regulated pathways in prostate cancer. Oncogene. 2005;24(50):7465–74. doi: 10.1038/sj.onc.1209096. [DOI] [PubMed] [Google Scholar]
- 7.Chen Y, Clegg NJ, Scher HI. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol. 2009;10(10):981–91. doi: 10.1016/S1470-2045(09)70229-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sarker D, Reid AH, Yap TA, de Bono JS. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin Cancer Res. 2009;15(15):4799–805. doi: 10.1158/1078-0432.CCR-08-0125. [DOI] [PubMed] [Google Scholar]
- 9.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 364(21):1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet. 375(9724):1437–46. doi: 10.1016/S0140-6736(10)60172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scher HI, Morris MJ, Kelly WK, Schwartz LH, Heller G. Prostate cancer clinical trial end points: “RECIST”ing a step backwards. Clin Cancer Res. 2005;11(14):5223–32. doi: 10.1158/1078-0432.CCR-05-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scher HI, Halabi S, Tannock I, Morris M, Sternberg CN, Carducci MA, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26(7):1148–59. doi: 10.1200/JCO.2007.12.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Even-Sapir E, Metser U, Mishani E, Lievshitz G, Lerman H, Leibovitch I. The detection of bone metastases in patients with high-risk prostate cancer: 99mTc-MDP Planar bone scintigraphy, single- and multi-field-of-view SPECT, 18F-fluoride PET, and 18F-fluoride PET/CT. J Nucl Med. 2006;47(2):287–97. [PubMed] [Google Scholar]
- 14.Fox JJ, Morris MJ, Larson SM, Schoder H, Scher HI. Developing imaging strategies for castration resistant prostate cancer. Acta Oncol. 50(Suppl 1):39–48. doi: 10.3109/0284186X.2011.572914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hricak H, Choyke PL, Eberhardt SC, Leibel SA, Scardino PT. Imaging prostate cancer: a multidisciplinary perspective. Radiology. 2007;243(1):28–53. doi: 10.1148/radiol.2431030580. [DOI] [PubMed] [Google Scholar]
- 16.Fox JJ, Schoder H, Larson SM. Molecular imaging of prostate cancer. Curr Opin Urol. doi: 10.1097/MOU.0b013e32835483d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.www.biomarkersconsortium.org
- 18.Gasparini M, Bombardieri E, Castellani M, Tondini C, Maffioli L, Devizzi L, et al. Gallium-67 scintigraphy evaluation of therapy in non-Hodgkin’s lymphoma. J Nucl Med. 1998;39(9):1586–90. [PubMed] [Google Scholar]
- 19.McArthur GA, Puzanov I, Amaravadi R, Ribas A, Chapman P, Kim KB, et al. Marked, Homogeneous, and Early [18F]Fluorodeoxyglucose-Positron Emission Tomography Responses to Vemurafenib in BRAF-Mutant Advanced Melanoma. J Clin Oncol. 30(14):1628–34. doi: 10.1200/JCO.2011.39.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Contractor KB, Aboagye EO. Monitoring predominantly cytostatic treatment response with 18F-FDG PET. J Nucl Med. 2009;50(Suppl 1):97S–105S. doi: 10.2967/jnumed.108.057273. [DOI] [PubMed] [Google Scholar]
- 21.Weber WA, Figlin R. Monitoring cancer treatment with PET/CT: does it make a difference? J Nucl Med. 2007;48(Suppl 1):36S–44S. [PubMed] [Google Scholar]
- 22.Solit DB, Santos E, Pratilas CA, Lobo J, Moroz M, Cai S, et al. 3’-deoxy-3’-[18F]fluorothymidine positron emission tomography is a sensitive method for imaging the response of BRAF-dependent tumors to MEK inhibition. Cancer Res. 2007;67(23):11463–9. doi: 10.1158/0008-5472.CAN-07-2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen W, Cloughesy T, Kamdar N, Satyamurthy N, Bergsneider M, Liau L, et al. Imaging proliferation in brain tumors with 18F-FLT PET: comparison with 18F-FDG. J Nucl Med. 2005;46(6):945–52. [PubMed] [Google Scholar]
- 24.Hicks JW, VanBrocklin HF, Wilson AA, Houle S, Vasdev N. Radiolabeled small molecule protein kinase inhibitors for imaging with PET or SPECT. Molecules. 15(11):8260–78. doi: 10.3390/molecules15118260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mankoff DA, Link JM, Linden HM, Sundararajan L, Krohn KA. Tumor receptor imaging. J Nucl Med. 2008;49(Suppl 2):149S–63S. doi: 10.2967/jnumed.107.045963. [DOI] [PubMed] [Google Scholar]
- 26.Mortimer JE, Dehdashti F, Siegel BA, Katzenellenbogen JA, Fracasso P, Welch MJ. Positron emission tomography with 2-[18F]Fluoro-2-deoxy-D-glucose and 16alpha-[18F]fluoro-17beta-estradiol in breast cancer: correlation with estrogen receptor status and response to systemic therapy. Clin Cancer Res. 1996;2(6):933–9. [PubMed] [Google Scholar]
- 27.Linden HM, Kurland BF, Peterson LM, Schubert EK, Gralow JR, Specht JM, et al. Fluoroestradiol positron emission tomography reveals differences in pharmacodynamics of aromatase inhibitors, tamoxifen, and fulvestrant in patients with metastatic breast cancer. Clin Cancer Res. 17(14):4799–805. doi: 10.1158/1078-0432.CCR-10-3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith-Jones PM, Solit DB, Akhurst T, Afroze F, Rosen N, Larson SM. Imaging the pharmacodynamics of HER2 degradation in response to Hsp90 inhibitors. Nat Biotechnol. 2004;22(6):701–6. doi: 10.1038/nbt968. [DOI] [PubMed] [Google Scholar]
- 29.Dumont RA, Hildebrandt I, Su H, Haubner R, Reischl G, Czernin JG, et al. Noninvasive imaging of alphaVbeta3 function as a predictor of the antimigratory and antiproliferative effects of dasatinib. Cancer Res. 2009;69(7):3173–9. doi: 10.1158/0008-5472.CAN-08-3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dumont RA, Deininger F, Haubner R, Maecke HR, Weber WA, Fani M. Novel (64)Cu- and (68)Ga-labeled RGD conjugates show improved PET imaging of alpha(nu)beta(3) integrin expression and facile radiosynthesis. J Nucl Med. 52(8):1276–84. doi: 10.2967/jnumed.111.087700. [DOI] [PubMed] [Google Scholar]
- 31.Dehdashti F, Picus J, Michalski JM, Dence CS, Siegel BA, Katzenellenbogen JA, et al. Positron tomographic assessment of androgen receptors in prostatic carcinoma. Eur J Nucl Med Mol Imaging. 2005;32(3):344–50. doi: 10.1007/s00259-005-1764-5. [DOI] [PubMed] [Google Scholar]
- 32.Choe YS, Lidstrom PJ, Chi DY, Bonasera TA, Welch MJ, Katzenellenbogen JA. Synthesis of 11 beta-[18F]fluoro-5 alpha-dihydrotestosterone and 11 beta-[18F]fluoro-19-nor-5 alpha-dihydrotestosterone: preparation via halofluorination-reduction, receptor binding, and tissue distribution. J Med Chem. 1995;38(5):816–25. doi: 10.1021/jm00005a009. [DOI] [PubMed] [Google Scholar]
- 33.Bonasera TA, O’Neil JP, Xu M, Dobkin JA, Cutler PD, Lich LL, et al. Preclinical evaluation of fluorine-18-labeled androgen receptor ligands in baboons. J Nucl Med. 1996;37(6):1009–15. [PubMed] [Google Scholar]
- 34.Beattie BJ, Smith-Jones PM, Jhanwar YS, Schoder H, Schmidtlein CR, Morris MJ, et al. Pharmacokinetic assessment of the uptake of 16beta-18F-fluoro-5alpha-dihydrotestosterone (FDHT) in prostate tumors as measured by PET. J Nucl Med. 51(2):183–92. doi: 10.2967/jnumed.109.066159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larson SM, Morris M, Gunther I, Beattie B, Humm JL, Akhurst TA, et al. Tumor localization of 16beta-18F-fluoro-5alpha-dihydrotestosterone versus 18F-FDG in patients with progressive, metastatic prostate cancer. J Nucl Med. 2004;45(3):366–73. [PubMed] [Google Scholar]
- 36.Zanzonico PB, Finn R, Pentlow KS, Erdi Y, Beattie B, Akhurst T, et al. PET-based radiation dosimetry in man of 18F-fluorodihydrotestosterone, a new radiotracer for imaging prostate cancer. J Nucl Med. 2004;45(11):1966–71. [PubMed] [Google Scholar]
- 37.Fox JJ, Autran-Blanc E, Morris MJ, Gavane S, Nehmeh S, Van Nuffel A, et al. Practical approach for comparative analysis of multilesion molecular imaging using a semiautomated program for PET/CT. J Nucl Med. 52(11):1727–32. doi: 10.2967/jnumed.111.089326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scher HI, Kelly WK. Flutamide withdrawal syndrome: its impact on clinical trials in hormone-refractory prostate cancer. J Clin Oncol. 1993;11(8):1566–72. doi: 10.1200/JCO.1993.11.8.1566. [DOI] [PubMed] [Google Scholar]
- 39.Lilja H, Ulmert D, Vickers AJ. Prostate-specific antigen and prostate cancer: prediction, detection and monitoring. Nat Rev Cancer. 2008;8(4):268–78. doi: 10.1038/nrc2351. [DOI] [PubMed] [Google Scholar]
- 40.Ulmert D, O’Brien MF, Bjartell AS, Lilja H. Prostate kallikrein markers in diagnosis, risk stratification and prognosis. Nat Rev Urol. 2009;6(7):384–91. doi: 10.1038/nrurol.2009.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stege RH, Tribukait B, Carlstrom KA, Grande M, Pousette AH. Tissue PSA from fine-needle biopsies of prostatic carcinoma as related to serum PSA, clinical stage, cytological grade, and DNA ploidy. Prostate. 1999;38(3):183–8. doi: 10.1002/(sici)1097-0045(19990215)38:3<183::aid-pros2>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 42.Wright GL, Jr, Grob BM, Haley C, Grossman K, Newhall K, Petrylak D, et al. Upregulation of prostate-specific membrane antigen after androgen-deprivation therapy. Urology. 1996;48(2):326–34. doi: 10.1016/s0090-4295(96)00184-7. [DOI] [PubMed] [Google Scholar]
- 43.Noss KR, Wolfe SA, Grimes SR. Upregulation of prostate specific membrane antigen/folate hydrolase transcription by an enhancer. Gene. 2002;285(1-2):247–56. doi: 10.1016/s0378-1119(02)00397-9. [DOI] [PubMed] [Google Scholar]
- 44.Hillier SM, Maresca KP, Femia FJ, Marquis JC, Foss CA, Nguyen N, et al. Preclinical evaluation of novel glutamate-urea-lysine analogues that target prostate-specific membrane antigen as molecular imaging pharmaceuticals for prostate cancer. Cancer Res. 2009;69(17):6932–40. doi: 10.1158/0008-5472.CAN-09-1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holmes EH. PSMA specific antibodies and their diagnostic and therapeutic use. Expert Opin Investig Drugs. 2001;10(3):511–9. doi: 10.1517/13543784.10.3.511. [DOI] [PubMed] [Google Scholar]
- 46.Smith-Jones PM, Vallabahajosula S, Goldsmith SJ, Navarro V, Hunter CJ, Bastidas D, et al. In vitro characterization of radiolabeled monoclonal antibodies specific for the extracellular domain of prostate-specific membrane antigen. Cancer Res. 2000;60(18):5237–43. [PubMed] [Google Scholar]
- 47.Evans MJ, Smith-Jones PM, Wongvipat J, Navarro V, Kim S, Bander NH, et al. Noninvasive measurement of androgen receptor signaling with a positron-emitting radiopharmaceutical that targets prostate-specific membrane antigen. Proc Natl Acad Sci U S A. 108(23):9578–82. doi: 10.1073/pnas.1106383108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bander NH, Trabulsi EJ, Kostakoglu L, Yao D, Vallabhajosula S, Smith-Jones P, et al. Targeting metastatic prostate cancer with radiolabeled monoclonal antibody J591 to the extracellular domain of prostate specific membrane antigen. J Urol. 2003;170(5):1717–21. doi: 10.1097/01.ju.0000091655.77601.0c. [DOI] [PubMed] [Google Scholar]
- 49.Holland JP, Divilov V, Bander NH, Smith-Jones PM, Larson SM, Lewis JS. 89Zr-DFO-J591 for immunoPET of prostate-specific membrane antigen expression in vivo. J Nucl Med. 51(8):1293–300. doi: 10.2967/jnumed.110.076174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ulmert D, Evans MJ, Holland JP, Rice SL, Wongvipat J, Pettersson K, Abrahamsson PA, Scardino PT, Larson SM, Lilja H, Lewis JS, Sawyers CL. Imaging androgen receptor signaling with a radiotracer targeting free prostate specific antigen. Cancer Discovery. 2012;2(4):320–7. doi: 10.1158/2159-8290.CD-11-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Markman M. Limitations to the use of the CA-125 antigen level in ovarian cancer. Curr Oncol Rep. 2003;5(4):263–4. doi: 10.1007/s11912-003-0063-1. [DOI] [PubMed] [Google Scholar]
- 52.Duffy MJ. Carcinoembryonic antigen as a marker for colorectal cancer: is it clinically useful? Clin Chem. 2001;47(4):624–30. [PubMed] [Google Scholar]
- 53.Tostain J, Li G, Gentil-Perret A, Gigante M. Carbonic anhydrase 9 in clear cell renal cell carcinoma: a marker for diagnosis, prognosis and treatment. Eur J Cancer. 46(18):3141–8. doi: 10.1016/j.ejca.2010.07.020. [DOI] [PubMed] [Google Scholar]
- 54.Jenkins RB, Qian J, Lieber MM, Bostwick DG. Detection of c-myc oncogene amplification and chromosomal anomalies in metastatic prostatic carcinoma by fluorescence in situ hybridization. Cancer Res. 1997;57(3):524–31. [PubMed] [Google Scholar]
- 55.Gurel B, Iwata T, Koh CM, Jenkins RB, Lan F, Van Dang C, et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod Pathol. 2008;21(9):1156–67. doi: 10.1038/modpathol.2008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4(3):223–38. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 57.Holland JP, Evans MJ, Rice SL, Wongvipat J, Sawyers CL, Lewis JS. Annotating MYC oncogene status with 89Zr-transferrin. Nature Medicine. 2012 doi: 10.1038/nm.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O’Donnell KA, Yu D, Zeller KI, Kim JW, Racke F, Thomas-Tikhonenko A, et al. Activation of transferrin receptor 1 by c-Myc enhances cellular proliferation and tumorigenesis. Mol Cell Biol. 2006;26(6):2373–86. doi: 10.1128/MCB.26.6.2373-2386.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aloj L, Jogoda E, Lang L, Caraco C, Neumann RD, Sung C, et al. Targeting of transferrin receptors in nude mice bearing A431 and LS174T xenografts with [18F]holo-transferrin: permeability and receptor dependence. J Nucl Med. 1999;40(9):1547–55. [PubMed] [Google Scholar]
- 60.Vavere AL, Welch MJ. Preparation, biodistribution, and small animal PET of 45Ti-transferrin. J Nucl Med. 2005;46(4):683–90. [PubMed] [Google Scholar]
- 61.Som P, Oster ZH, Matsui K, Guglielmi G, Persson BR, Pellettieri ML, et al. 97Ru-transferrin uptake in tumor and abscess. Eur J Nucl Med. 1983;8(11):491–4. doi: 10.1007/BF00598908. [DOI] [PubMed] [Google Scholar]
- 62.Ruggiero A, Holland JP, Hudolin T, Shenker L, Koulova A, Bander NH, et al. Targeting the internal epitope of prostate-specific membrane antigen with 89Zr-7E11 immuno-PET. J Nucl Med. 52(10):1608–15. doi: 10.2967/jnumed.111.092098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Holland JP, Caldas-Lopes E, Divilov V, Longo VA, Taldone T, Zatorska D, et al. Measuring the pharmacodynamic effects of a novel Hsp90 inhibitor on HER2/neu expression in mice using Zr-DFO-trastuzumab. PLoS One. 5(1):e8859. doi: 10.1371/journal.pone.0008859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Macapinlac HA, Scott AM, Larson SM, Divgi CR, Yeh SD, Goldsmith SJ. Gallium-67-citrate imaging in nuclear oncology. Nucl Med Biol. 1994;21(5):731–8. doi: 10.1016/0969-8051(94)90044-2. [DOI] [PubMed] [Google Scholar]
- 65.Watson PA, Ellwood-Yen K, King JC, Wongvipat J, Lebeau MM, Sawyers CL. Context-dependent hormone-refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res. 2005;65(24):11565–71. doi: 10.1158/0008-5472.CAN-05-3441. [DOI] [PubMed] [Google Scholar]
- 66.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 468(7327):1067–73. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 146(6):904–17. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 478(7370):524–8. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006;16(4):253–64. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 70.Palaskas N, Larson SM, Schultz N, Komisopoulou E, Wong J, Rohle D, et al. 18F-fluorodeoxy-glucose positron emission tomography marks MYC-overexpressing human basal-like breast cancers. Cancer Res. 71(15):5164–74. doi: 10.1158/0008-5472.CAN-10-4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 366(10):883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138(2):245–56. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19(1):1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sawyers CL. The cancer biomarker problem. Nature. 2008;452(7187):548–52. doi: 10.1038/nature06913. [DOI] [PubMed] [Google Scholar]
- 75.Danila DC, Fleisher M, Scher HI. Circulating tumor cells as biomarkers in prostate cancer. Clin Cancer Res. 17(12):3903–12. doi: 10.1158/1078-0432.CCR-10-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]