

Abstract
Osteosarcoma (OS) is a rare malignant bone tumor with an overall incidence rate of 4.6 cases per million children aged 0-19 years in the United States. While the etiology of OS is largely unknown, its distinctive age-incidence pattern suggests that growth and development is crucial in genesis. Prior studies have suggested that variants in genes in the estrogen metabolism (ESTR) and insulin-like growth factor/growth hormone (IGF/GH) pathways are associated with OS. We examined 798 single nucleotide polymorphisms (SNPs) in 42 genes from these pathways in a case-parent study (229 complete triads and 56 dyads) using buccal cell samples. Relative risks (RR) and 95% confidence intervals (CI) associated with transmitting one or two copies of the variant were estimated using log-linear models. After Bonferroni correction, 1 SNP within the ESTR pathway (rs1415270: RR = 0.50 and 8.37 for 1 and 2 vs. 0 copies, respectively; p = 0.010), and two SNPs in the IGF/GH pathway (rs1003737: RR = 0.91 and 0.0001 for 1 and 2 vs. 0 copies, respectively; p <0.0001 and rs2575352: RR = 2.62 and 0.22 for 1 and 2 vs. 0 copies; p < 0.0001) were significantly associated with OS incidence. These results confirm previous findings that variation in the estrogen metabolism and bone growth pathways influence OS risk and further support a biologically and epidemiologically plausible role in OS development.
Keywords: Osteosarcoma, case-parent study, growth and development, insulin-like growth factor pathway, estrogen metabolism pathway
Introduction
Osteosarcoma (OS) is the most commonly occurring bone cancer in children <20 years of age, with about 440 new cases diagnosed annually in the United States and Canada [1,2]. Several lines of evidence link OS etiology to factors related to pubertal development and bone growth. First, incidence of OS is distributed bimodally, with the primary peak occurring during adolescence, corresponding to the pubertal growth spurt and its accompanying increase in endogenous sex and growth hormone levels [3-5]. OS incidence peaks earlier in females, which corresponds to the earlier onset of puberty in females [6]; however, overall incidence is higher in males, who are generally taller than females. The association of OS incidence with growth and puberty corroborates findings of associations among greater height [5,7-12], rate of growth [12], and birth weight [13,14] and increased risk of OS in both humans and canines, although not all studies have found such associations [15,16]. Furthermore, the most common sites of OS are areas of rapid bone growth during puberty [6,17,18].
There are few known causes of OS, and these account for only a small proportion of cases. The only widely accepted exogenous risk factors are high-dose ionizing radiation [19,20] and prior chemotherapy with alkylating agents [19,20]. In addition, there are several associated syndromes [21-23] including Li-Fraumeni Syndrome [24-26], hereditary retinoblastoma [27-29], Werner [30], Bloom [31], and Rothmund-Thomson Syndromes [32,33]. Because growth and puberty are heritable traits, it is possible that other forms of genetic variation may explain sporadic OS incidence.
In particular, evidence suggests that genes underlying pubertal growth might be key in OS etiology. Linear bone growth is under the influence of several genes, including those from the sex steroid hormone control pathway (e.g., estrogen metabolism (ESTR)) and insulin-like growth factor/growth hormone (IGF/GH) pathway, which regulate rates of bone growth [34-39] through release of estrogen, IGF1, and growth hormones. The timing and extent of adolescent bone growth is also under substantial genetic control, with heritability estimates ranging from 50 to 80% for the time of onset of puberty and peak bone mineral density [40,41]. Hence, variation in genes in the IGF/GH and ESTR pathways are reasonable candidates for exploration in OS etiology. A candidate gene study by Mirabello et al. identified 12 SNPs in the DNA repair gene and growth/hormone pathways as being associated with OS in a study of ~5000 tag SNPs in 255 candidate genes [3]. In the same population, Savage et al. identified two polymorphisms of the IGF2 receptor gene that were significantly more frequent in OS cases [42,43]. That study included adult and pediatric cases and a similar approach has not been taken in a study comprised solely of pediatric OS cases.
In order to assess the association of OS with genes putatively related to adolescent bone growth, we conducted a case-parent study consisting of nearly 300 cases of pediatric OS recruited through the Children’s Oncology Group and one or both of their biological parents. Using these triads/dyads, we employed log-linear regression to assess 798 SNPs across 42 candidate genes in biologically plausible pathways for an association with OS.
Materials and methods
Study population
OS cases were identified through the Childhood Cancer Research Network (CCRN)--the registry component of the Children’s Oncology Group (COG) [44]. As part of registering all pediatric patients with the COG data center, the CCRN consent process allows for informed consent for the release of personal identifiers on patients and parents along with permission to contact in the future. Eligible cases were patients with a primary diagnosis of OS (ICCC 9180-9200; OMIM #259500) at age <20 years made at a North American COG institution between December 24, 2007 and March 31, 2010, who had at least one living biological parent willing to participate and understand English or Spanish. Initial contact was through an introductory letter followed by a telephone call during which study staff summarized the goals of the study and provided respondents with an opportunity to ask questions and assess eligibility. If consent over the telephone was given, assent forms (if appropriate), buccal cell collection kits (mouthwash or cytobrushes), a paper questionnaire, and return mailers were sent. The study sample consisted of the 290 cases who returned a buccal cell kit for themselves and at least one biological parent (mean age at diagnosis 12.9 years). Thus, this study population represents one of the largest and highly powered studies for assessing genetic variants associated with OS in a pediatric population.
SNP selection
Tag SNPs were selected as follows: first, transcripts and gene boundaries for gene symbols were obtained from Ensembl release 50. SNPs were then found by uploading the list of gene names to Ensembl’s BioMart, and then additional SNPs were obtained directly at hapmap. org [45] that were 20,000 bp up and downstream of gene boundaries using the data from the International HapMap Project Data Rel 23a/phase II Mar08 data file where tag SNP data was downloaded from the CEU population using the “tagger pairwise method” with an R-square cutoff of 0.8 and a minor allele frequency (MAF) cutoff of 0.0. The final set of SNPs were then chosen if they were annotated as verified in the dbSNP or SNP500 databases [46] or were coding SNPs predicted to be deleterious [47,48] or SNPs selected from the Phase I HapMap data using the Tagger algorithm with r2>0.8.
Genotyping assays
Buccal cell samples were stored at 4°C and batched for DNA extraction. Samples were centrifuged, cells pelleted and washed, and DNA was extracted using the Puregene kit (Gentra Systems, Minneapolis, MN). DNA was quantified, aliquoted and stored in hydration solution at -20°C. The Sequenom platform iPLEX Gold method was used for genotyping. Multiplexed PCR was performed in 5μl reactions on a 384-well plate. Reactions contain 0.5 U HotStar Taq polymerase (Qiagen), 100 nM primers, 1.25X HotStar Taq buffer, 1.625 mM MgC122, and 500 μM dNTPs. Enzyme activation was performed at 94°C for 15 minutes, followed by DNA amplification with 45 cycles at 94°C for 20 seconds, 56°C for 30 seconds, and 72°C for 1 minute, followed by a three minute extension at 72°C. Unincorporated dNTPs were removed using shrimp alkaline phosphatase (0.3 U, Sequenom, San Diego). SBE primers at concentrations from 0.625 μM to 1.25 μM and iPLEX enzyme and buffers in 9 μl reactions were used to carry out single base extension. Reactions were desalted and SBE products were measured using the MassARRAY system. TYPER software (Sequenom) was used to analyze mass spectra in order to generate genotype calls and allele frequencies.
DNA was quantified and normalized to a housekeeping gene. A total of 866 SNPs were selected and genotyped following our SNP selection procedure. SNPs were excluded if they had less than a 90% genotyping rate in our sample, were non-variable, or had a sample MAF of < 1%. Individuals were excluded if >10% genotypes were missing. If the individual excluded was a case, the entire triad/dyad was excluded. In addition, an algorithm was created to assess for discrepancy between the genotypes of the parents and their associated case child. Individuals were excluded whose genotypes demonstrated >5% inconsistency between the parents and the associated case child (e.g. paternal genotype contains two copies of the variant and the child’s genotype contains no copies). A total of 798 tag SNPs, 186 complete triads and 72 dyads met the above quality control criteria and were included for analysis. All original candidate genes had at least one SNP included in the final analysis except for IGFBP6.
Statistical analysis
This study uses the case-parent design, which involved enrolling and genotyping OS cases and their parents [49,50] and testing whether the distribution of candidate susceptibility alleles in cases significantly deviates from what would be expected by Mendelian inheritance. We used a log-linear regression model to assess for significant over- or undertransmission of a variant allele among OS cases and their biological parents [51,52]. Briefly, the log-linear model estimates relative risks (RR) of possessing 0, 1, or 2 copies of the variant by assessing the joint likelihood of the case, maternal, and paternal genotypes where the statistical model is conditioned on the presence of disease in the offspring. A likelihood ratio test (LRT) determines the statistical significance for each SNP variant. The least common allele among cases was selected to serve as the variant for each SNP. Families were informative if at least one biological parent was available for genotyping. Estimation and inference were performed in the presence of missing parental data using a likelihood-based algorithm [51]. Bonferroni corrections were performed within each of two pathways--the ESTR pathway (29 genes, 519 SNPs after quality control) and the IGF/GH pathway (13 genes, 279 SNPs after quality control)--to correct for multiple statistical comparisons. Statistical analyses were performed using custom programs in R (http://www.r-project.org).
Results
A summary of case identification, enrollment, and DNA collection is provided in Figure 1. During the eligibility period, a total of 660 OS cases were identified at COG institutions. Of those, families of 602 (91%) cases consented to participation in the CCRN and potential future contact for study participation. Twenty of those who consented to CCRN were considered ineligible due to lack of an available biological parent (n=17) or lack of a parent who spoke either English or Spanish (n=3). Of those who were contacted and eligible (n=582), 445 families (76% contacted) gave consent over the phone and were sent a DNA kit, of which 290 (65% sent) were returned.
Figure 1.
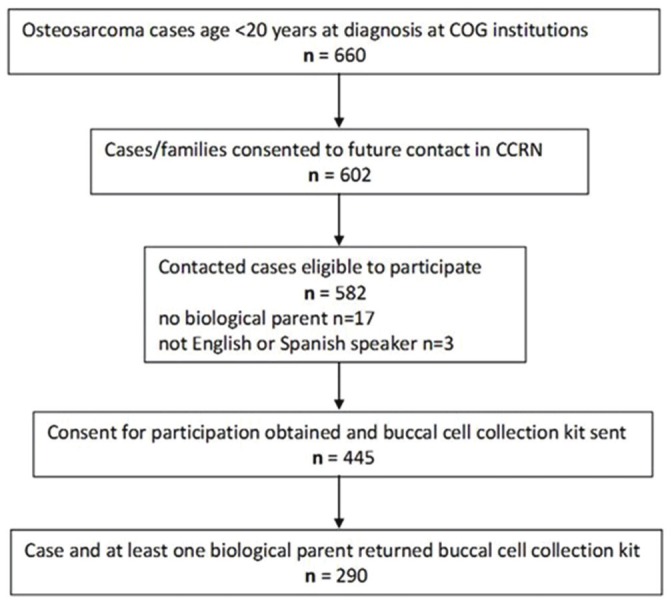
Flow chart describing the identification and enrollment of the cases that make up the study population from the initial population of childhood osteosarcoma cases treated at Children’s Oncology Group institutions during the eligibility period. Patient Identification, Contact, Consent, Enrollment, and Participation in a case-parent study of childhood osteosarcoma; April 1, 2007-March 31, 2010.
Demographics and clinico-pathologic characteristics of the cases are presented in Table 1. A majority (95%) of the families came from the United States. Cases were also primarily white (86%). Tumors were most often located in the long bones (92%). For the log-linear analysis, six families were not included because there was either a diagnosis of or a family history consistent with Li-Fraumeni Syndrome. An additional 30 triads were excluded due to excessive missing genotyping data from the case, and an additional 42 parents were excluded for the same reason. A further four fathers were excluded due to discordance above our 5% cutoff between paternal and progeny genotypes. A total of 68 SNPs were excluded, leaving a total of 798 SNPs for analysis, 519 on the ESTR pathway and 279 on the IGF/GH pathway. After Bonferroni correction (for 519 SNPs within ESTR and 279 within IGF/GH), two SNPs on the IGF/GH pathway remained significant (Table 2, Figure 2). The first SNP, rs1003737 is located in the intronic region of the IGF2R gene on chromosome 6, and was associated with a RR of 0.91 and 0.0001 for one and two copies versus no copies, respectively, of the minor allele (un-corr p<0.0001; corr-p <0.0001). The second SNP, rs2575352, is upstream of the IGFALS gene on chromosome 16 and was associated with a RR of 2.62 and 0.22 for one and two copies versus no copies of the minor allele, respectively (un-corr p<0.0001; corr-p <0.0001). An additional SNP from the ESTR pathway also maintained significance after Bonferroni correction: rs1415270 downstream of the androgen receptor (AR) gene of the X chromosome was associated with a RR of 0.53 and 8.26 for one and two copies, respectively, compared to no copies of the minor allele (un-corr p<0.0001; corr-p =0.02).
Table 1.
Demographic Characteristics from a Case-Parent Study of Childhood Osteosarcoma, Children’s Oncology Group, United States and Canada, 2007-2010 (n=290)
| Case Trait | Category | Count | % |
|---|---|---|---|
| Gender | Male | 170 | 58.6 |
| Female | 120 | 41.4 | |
| Age at diagnosis (years) | 0-4 | 2 | 0.7 |
| 5-9 | 51 | 17.6 | |
| 10-14 | 130 | 44.8 | |
| 15-19 | 107 | 36.9 | |
| Racea | White | 237 | 85.6 |
| Black | 21 | 7.6 | |
| Other | 19 | 6.9 | |
| Ethnicityb | Non-Hispanic | 239 | 86.0 |
| Hispanic | 39 | 14.0 | |
| Country of Residence | United States | 274 | 94.5 |
| Canada | 16 | 5.5 | |
| Tumor Locationc | Lower Limb | 221 | 77.5 |
| Upper Limb | 42 | 14.7 | |
| Chest | 6 | 2.1 | |
| Hip | 9 | 3.2 | |
| Spine | 2 | 0.1 | |
| Head | 5 | 1.8 | |
| Histology | Chondroblastic | 47 | 20.2 |
| Osteoblastic | 68 | 29.2 | |
| Fibroblastic | 16 | 6.9 | |
| Telangiectatic | 15 | 6.4 | |
| High grade | 74 | 31.8 | |
| Low grade | 5 | 2.1 | |
| Periosteal | 4 | 1.7 | |
| Mixed | 4 | 1.7 |
13 unknown;
12 unknown;
5 missing.
Table 2.
Significant SNPs from the IGF/GH and ESTR pathways and associated relative risks after correction for multiple testing from a study of childhood osteosarcoma
| Pathway | Gene | SNP id | Chromosome | Region | MAFa | RR1b | RR2c | Un-corr-p | Corr-pd |
|---|---|---|---|---|---|---|---|---|---|
| IGF/GH | IGF2R | rs1003737 | 6 | intronic | 0.30 | 0.91 | 0.0001 | <0.0001 | <0.0001 |
| IGFALS | rs2575352 | 16 | upstream | 0.35 | 2.62 | 0.22 | <0.0001 | <0.0001 | |
| ESTR | AR | rs1415270 | X | downstream | 0.22 | 0.53 | 8.26 | <0.0001 | 0.02 |
minor allele frequency;
relative risk for one vs. no copies;
relative risk for two vs. no copies;
Bonferroni corrected within pathway (519 SNPs for ESTR and 279 SNPs for IGF/GH).
Figure 2.
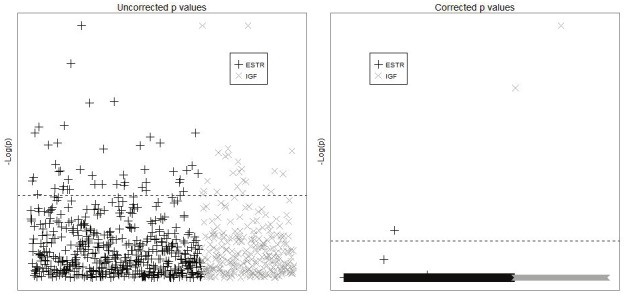
This figure depicts both the uncorrected p-values and the p-values after correction for multiple testing for each of the SNPs analyzed in the two pathways. We present the p-values (corrected and uncorrected) on the -log scale and provide a horizontal line so that the reader can easily see the threshold necessary for significance in this analysis. P-values for each SNP by pathway before and after Bonferroni correctiona α = 0.05. Inset box indicates pathway symbol, results for 798 SNPs in 42 candidate genes (29 ESTR and 13 IGF/GH). Dashed line represents significance threshold for α.
Discussion
We employed a candidate gene based approach in a case-parent study to assess the involvement of genes in biologically plausible pathways for a possible association with pediatric OS. The IGF/GH and ESTR pathways are reasonable candidates for analysis due to previously established associations of growth and puberty with OS. The heritability of growth and hormone regulation provides additional rationale for examining genes in these pathways [3-7,10,11,40,41].
After Bonferroni correction for multiple statistical tests, we identified one SNP in each of three genes of interest as significant. In the IGF/GH pathway, the IGF2R gene (on chromosome 6) plays a role in activating growth factor beta and degrading IGF2, while IGFALS (on chromosome 16) increases the half life of IGF. SNPs in the IGF2R gene were previously reported to be associated with increased risk of OS. However, the SNP found in our study, rs1003737, was in low LD with the previously reported SNPs (r-squared 0.10) [42]. The third SNP is located on the X chromosome about 20 kb downstream of the AR gene in a region that plays a role in the N-terminal, the DNA-binding, and the androgen-binding domains [42,43,45]. To our knowledge, neither the particular function of these specific SNPs nor their involvement in other malignancies has been reported.
Previous studies used a case-control approach, which is limited by the need to identify an appropriate control group and the possibility of population stratification in genetic analyses. In contrast, for the case-parent approach, no controls are needed and population stratification is inherently corrected for. The statistical model employed in a case-parent design also allows for the estimation of RRs rather than odds ratios-- the standard measure of association in case-control studies. Further, our focus on pediatric malignancies is a strength given the availability and willingness of biological parents to participate in research, particularly when compared to the underwhelming participation rates for population based controls. Including solely pediatric OS cases could also improve our understanding of etiology should it differ from that of older patients; this is especially the case in light of our large sample size, given the rarity of the tumor. We appear to have taken the first comprehensive look at genetic variation in the ESTR pathway as an OS risk factor. Our identification of a significant SNP associated with the AR gene suggest that this pathway might also be of biological relevance. It should also be noted that while Bonferroni corrections were used in both our analyses and that of previous studies, we elected to correct at the pathway level and thus our criteria for statistical significance was considerably more stringent. This could partially explain both the reduction in number of significant SNPs compared to the findings by Mirabello et al. as well as our failure to confirm significant SNPs from that study.
While we did identify statistically significant SNPs in our proposed pathways, we recognize that interpretation of our results must be considered in the context of the caveats associated with a candidate gene based approach, primarily the reliance on current knowledge regarding the biology of OS and the limitation in the number of SNPs examined compared to a genome-wide approach. Despite these shortcomings, we selected a biologically plausible set of genes based on previous research indicating the primacy of the IGF/GH and ESTR pathways in promoting linear bone growth [34] and on preliminary data implicating the IGF/GH axis in adult and pediatric OS [42], which made candidate genes in the IGF/GH and ESTR pathways obvious choices for a study in children. Confirmation of these results in future studies would lend additional support to the association of bone growth pathways in pediatric OS.
Acknowledgements
This research was supported by National Institutes of Health grants U01CA122371, T32 CA099936-S1, K05 CA157439, U01CA98543, U10CA13539, the Children’s Cancer Research Fund, Minneapolis, MN, grant 97834 from the Canadian Institutes for Heath Research (CIHR), and the intramural research program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute.
References
- 1.McLaughlin. Canadian Cancer Statistics. Toronto: 2005. [Google Scholar]
- 2.Ries L. SEER Cancer Statistic Review. 1975-2002. Bethesda MD: 2004. [Google Scholar]
- 3.Mirabello L, Yu K, Berndt SI, Burdett L, Wang Z, Chowdhury S, Teshome K, Uzoka A, Hutchinson A, Grotmol T, Douglass C, Hayes RB, Hoover RN, Savage SA. A comprehensive candidate gene approach identifies genetic variation associated with osteosarcoma. BMC Cancer. 2011;11:209. doi: 10.1186/1471-2407-11-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mirabello L, Troisi RJ, Savage SA. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. Int J Cancer. 2009;125:229–234. doi: 10.1002/ijc.24320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fraumeni JF Jr. Stature and malignant tumors of bone in childhood and adolescence. Cancer. 1967;20:967–973. doi: 10.1002/1097-0142(196706)20:6<967::aid-cncr2820200606>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 6.Staheli LT. Normative data in pediatric orthopedics. J Pediatr Orthop. 1996;16:561–562. doi: 10.1097/00004694-199609000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Gelberg KH, Fitzgerald EF, Hwang S, Dubrow R. Growth and development and other risk factors for osteosarcoma in children and young adults. Int J Epidemiol. 1997;26:272–278. doi: 10.1093/ije/26.2.272. [DOI] [PubMed] [Google Scholar]
- 8.Cotterill SJ, Wright CM, Pearce MS, Craft AW. Stature of young people with malignant bone tumors. Pediatr Blood Cancer. 2004;42:59–63. doi: 10.1002/pbc.10437. [DOI] [PubMed] [Google Scholar]
- 9.Longhi A, Pasini A, Cicognani A, Baronio F, Pellacani A, Baldini N, Bacci G. Height as a risk factor for osteosarcoma. J Pediatr Hematol Oncol. 2005;27:314–318. doi: 10.1097/01.mph.0000169251.57611.8e. [DOI] [PubMed] [Google Scholar]
- 10.Mirabello L, Pfeiffer R, Murphy G, Daw NC, Patino-Garcia A, Troisi RJ, Hoover RN, Douglass C, Schuz J, Craft AW, Savage SA. Height at diagnosis and birth-weight as risk factors for osteosarcoma. Cancer Causes Control. 2011;22:899–908. doi: 10.1007/s10552-011-9763-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Withrow SJ, Powers BE, Straw RC, Wilkins RM. Comparative aspects of osteosarcoma. Dog versus man. Clin Orthop Relat Res. 1991:159–168. [PubMed] [Google Scholar]
- 12.Tjalma RA. Canine bone sarcoma: estimation of relative risk as a function of body size. J Natl Cancer Inst. 1966;36:1137–1150. [PubMed] [Google Scholar]
- 13.Gelberg KH, Fitzgerald EF, Hwang SA, Dubrow R. Fluoride exposure and childhood osteosarcoma: a case-control study. Am J Public Health. 1995;85:1678–1683. doi: 10.2105/ajph.85.12.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartley AL, Birch JM, McKinney PA, Teare MD, Blair V, Carrette J, Mann JR, Draper GJ, Stiller CA, Johnston HE, et al. The Inter-Regional Epidemiological Study of Childhood Cancer (IRESCC): case control study of children with bone and soft tissue sarcomas. Br J Cancer. 1988;58:838–842. doi: 10.1038/bjc.1988.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Operskalski EA, Preston-Martin S, Henderson BE, Visscher BR. A case-control study of osteosarcoma in young persons. Am J Epidemiol. 1987;126:118–126. doi: 10.1093/oxfordjournals.aje.a114643. [DOI] [PubMed] [Google Scholar]
- 16.Buckley JD, Pendergrass TW, Buckley CM, Pritchard DJ, Nesbit ME, Provisor AJ, Robison LL. Epidemiology of osteosarcoma and Ewing’s sarcoma in childhood: a study of 305 cases by the Children’s Cancer Group. Cancer. 1998;83:1440–1448. doi: 10.1002/(sici)1097-0142(19981001)83:7<1440::aid-cncr23>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 17.Price CH. Primary bone-forming tumours and their relationship to skeletal growth. J Bone Joint Surg Br. 1958;40-B:574–593. doi: 10.1302/0301-620X.40B3.574. [DOI] [PubMed] [Google Scholar]
- 18.Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer. 2009;115:1531–1543. doi: 10.1002/cncr.24121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hawkins MM, Wilson LM, Burton HS, Potok MH, Winter DL, Marsden HB, Stovall MA. Radiotherapy, alkylating agents, and risk of bone cancer after childhood cancer. J Natl Cancer Inst. 1996;88:270–278. doi: 10.1093/jnci/88.5.270. [DOI] [PubMed] [Google Scholar]
- 20.Tucker MA, D’Angio GJ, Boice JD Jr, Strong LC, Li FP, Stovall M, Stone BJ, Green DM, Lombardi F, Newton W, et al. Bone sarcomas linked to radiotherapy and chemotherapy in children. N Engl J Med. 1987;317:588–593. doi: 10.1056/NEJM198709033171002. [DOI] [PubMed] [Google Scholar]
- 21.Malkin D, Li FP, Strong LC, Fraumeni JF Jr, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 22.Hickson ID. RecQ helicases: caretakers of the genome. Nat Rev Cancer. 2003;3:169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 23.Benedict WF, Fung YK, Murphree AL. The gene responsible for the development of retinoblastoma and osteosarcoma. Cancer. 1988;62:1691–1694. doi: 10.1002/1097-0142(19881015)62:1+<1691::aid-cncr2820621306>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 24.Li FP, Fraumeni JF Jr, Mulvihill JJ, Blattner WA, Dreyfus MG, Tucker MA, Miller RW. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48:5358–5362. [PubMed] [Google Scholar]
- 25.Varley JM. Germline TP53 mutations and Li-Fraumeni syndrome. Hum Mutat. 2003;21:313–320. doi: 10.1002/humu.10185. [DOI] [PubMed] [Google Scholar]
- 26.Porter DE, Holden ST, Steel CM, Cohen BB, Wallace MR, Reid R. A significant proportion of patients with osteosarcoma may belong to Li-Fraumeni cancer families. J Bone Joint Surg Br. 1992;74:883–886. doi: 10.1302/0301-620X.74B6.1447251. [DOI] [PubMed] [Google Scholar]
- 27.Wong FL, Boice JD Jr, Abramson DH, Tarone RE, Kleinerman RA, Stovall M, Goldman MB, Seddon JM, Tarbell N, Fraumeni JF Jr, Li FP. Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA. 1997;278:1262–1267. doi: 10.1001/jama.278.15.1262. [DOI] [PubMed] [Google Scholar]
- 28.Hansen MF, Koufos A, Gallie BL, Phillips RA, Fodstad O, Brogger A, Gedde-Dahl T, Cavenee WK. Osteosarcoma and retinoblastoma: a shared chromosomal mechanism revealing recessive predisposition. Proc Natl Acad Sci USA. 1985;82:6216–6220. doi: 10.1073/pnas.82.18.6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chauveinc L, Mosseri V, Quintana E, Desjardins L, Schlienger P, Doz F, Dutrillaux B. Osteosarcoma following retinoblastoma: age at onset and latency period. Ophthalmic Genet. 2001;22:77–88. doi: 10.1076/opge.22.2.77.2228. [DOI] [PubMed] [Google Scholar]
- 30.Goto M, Miller RW, Ishikawa Y, Sugano H. Excess of rare cancers in Werner syndrome (adult progeria) Cancer Epidemiol Biomarkers Prev. 1996;5:239–246. [PubMed] [Google Scholar]
- 31.German J, Ellis NA, Proytcheva M. Bloom’s syndrome XIX. Cytogenetic and population evidence for genetic heterogeneity. Clin Genet. 1996;49:223–231. doi: 10.1111/j.1399-0004.1996.tb03778.x. [DOI] [PubMed] [Google Scholar]
- 32.Leonard A, Craft AW, Moss C, Malcolm AJ. Osteogenic sarcoma in the Rothmund-Thomson syndrome. Med Pediatr Oncol. 1996;26:249–253. doi: 10.1002/(SICI)1096-911X(199604)26:4<249::AID-MPO5>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 33.Kansara M, Thomas DM. Molecular pathogenesis of osteosarcoma. DNA Cell Biol. 2007;26:1–18. doi: 10.1089/dna.2006.0505. [DOI] [PubMed] [Google Scholar]
- 34.Juul A. The effects of oestrogens on linear bone growth. Hum Reprod Update. 2001;7:303–313. doi: 10.1093/humupd/7.3.303. [DOI] [PubMed] [Google Scholar]
- 35.Bland R. Steroid hormone receptor expression and action in bone. Clin Sci (Lond) 2000;98:217–240. [PubMed] [Google Scholar]
- 36.Le Roith D, Bondy C, Yakar S, Liu JL, Butler A. The somatomedin hypothesis: 2001. Endocr Rev. 2001;22:53–74. doi: 10.1210/edrv.22.1.0419. [DOI] [PubMed] [Google Scholar]
- 37.Gill MS, Tillmann V, Veldhuis JD, Clayton PE. Patterns of GH output and their synchrony with short-term height increments influence stature and growth performance in normal children. J Clin Endocrinol Metab. 2001;86:5860–5863. doi: 10.1210/jcem.86.12.8116. [DOI] [PubMed] [Google Scholar]
- 38.Styne DM. Physiology of Puberty. Adolesc Med. 1994;5:171–188. [PubMed] [Google Scholar]
- 39.Wang J, Zhou J, Cheng CM, Kopchick JJ, Bondy CA. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J Endocrinol. 2004;180:247–255. doi: 10.1677/joe.0.1800247. [DOI] [PubMed] [Google Scholar]
- 40.Palmert MR, Boepple PA. Variation in the timing of puberty: clinical spectrum and genetic investigation. J Clin Endocrinol Metab. 2001;86:2364–2368. doi: 10.1210/jcem.86.6.7603. [DOI] [PubMed] [Google Scholar]
- 41.Brown LB, Streeten EA, Shuldiner AR, Almasy LA, Peyser PA, Mitchell BD. Assessment of sex-specific genetic and environmental effects on bone mineral density. Genet Epidemiol. 2004;27:153–161. doi: 10.1002/gepi.20009. [DOI] [PubMed] [Google Scholar]
- 42.Savage SA, Woodson K, Walk E, Modi W, Liao J, Douglass C, Hoover RN, Chanock SJ. Analysis of genes critical for growth regulation identifies Insulin-like Growth Factor 2 Receptor variations with possible functional significance as risk factors for osteosarcoma. Cancer Epidemiol Biomarkers Prev. 2007;16:1667–1674. doi: 10.1158/1055-9965.EPI-07-0214. [DOI] [PubMed] [Google Scholar]
- 43.Savage SA, Mirabello L. Using epidemiology and genomics to understand osteosarcoma etiology. Sarcoma. 2011;2011:548151. doi: 10.1155/2011/548151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steele JR, Wellemeyer AS, Hansen MJ, Reaman GH, Ross JA. Childhood cancer research network: a North American Pediatric Cancer Registry. Cancer Epidemiol Biomarkers Prev. 2006;15:1241–1242. doi: 10.1158/1055-9965.EPI-06-0447. [DOI] [PubMed] [Google Scholar]
- 45.The International HapMap Consortium. The International HapMap Project. Nature. 2003 Dec 18;426:789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 46.Chanock S. Genetic variation and hematology: single-nucleotide polymorphisms, haplotypes, and complex disease. Semin Hematol. 2003;40:321–328. doi: 10.1016/s0037-1963(03)00198-7. [DOI] [PubMed] [Google Scholar]
- 47.Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12:436–446. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahsan H, Hodge SE, Heiman GA, Begg MD, Susser ES. Relative risk for genetic associations: the case-parent triad as a variant of case-cohort design. Int J Epidemiol. 2002;31:669–678. doi: 10.1093/ije/31.3.669. [DOI] [PubMed] [Google Scholar]
- 50.Weinberg CR, Umbach DM. Choosing a retrospective design to assess joint genetic and environmental contributions to risk. Am J Epidemiol. 2000;152:197–203. doi: 10.1093/aje/152.3.197. [DOI] [PubMed] [Google Scholar]
- 51.Weinberg CR, Wilcox AJ, Lie RT. A log-linear approach to case-parent-triad data: assessing effects of disease genes that act either directly or through maternal effects and that may be subject to parental imprinting. Am J Hum Genet. 1998;62:969–978. doi: 10.1086/301802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bergemann TL, Huang Z. A new method to account for missing data in case-parent triad studies. Hum Hered. 2009;68:268–277. doi: 10.1159/000228924. [DOI] [PMC free article] [PubMed] [Google Scholar]