

Abstract
To understand the change of the dominant serogroup of Shigella spp., their antimicrobial resistance over more than two decades in Tianjin, their phylogenetic similarity and to determine their evolutionary biology by using REP-PCR and MLST in order to study their epidemiological character. Multi-locus Sequence Typing was performed to determine their lineage and phylogenetic similarity. REP-PCR typing was used to study the homology of their genomic DNA. The isolated rate of group D Shigella in 2009 and 2010 had obviously increased. Antimicrobial susceptibility test results showed that the resistant rates of the 1981-1983 Shigella flexneri to tetracycline, streptomycin and chloramphenicol varied from 76.47 to 100%, they were all sensitive to other antibiotics. During 2009-2010, the resistance rates of the isolated Shigella flexneri to gentamicin, amikacin, third and fourth Generation Cephalosporins and quinolones had increased. MLST results produced five sequence types and two sequence type complexes. REP-PCR showed DNA band similarities between the 1981-1983 and 2009-2010 strains. The dominant serogroup of Shigella in Tianjin has changed from Shigella flexneri to Shigella sonnei. Increased drug resistance of Shigella flexneri is higher than Shigella sonnei because a great variety of antibiotics has been used. The MLST results showed that the 1981-1983 strains had the same sequence type with some of the 2009-2010 strains. Combination of MLST and REP-PCR produced better discriminatory power than using either method alone.
Keywords: Shigella, multidrug resistance, REP-PCR, multi-locus sequence typing
Introduction
Shigella spp are Gram-negative bacteria belonging to the Enterobacteriaceae family. These bacteria are responsible for morbidity and mortality in high risk populations such as children under five years of age and the immunocompromised [1]. In China, Shigellosis epidemics are mainly caused by Shigella flexneri and shigella sonnei and there was a gradual increase in resistance to the commonly used antibiotics as first line treatment for dysentery in the last two decades. The ability of bacteria to acquire and disseminate exogenous genes via mobile genetic elements such as plasmids, transposons, insertion sequences and genomic islands has been the major factor in the development of multidrug-resistant strains [2,3]. In this study, we compare shigella serogroups by PCR based molecular typing and antimicrobial resistance of shigella spp. isolated from patients with diarrhea in 1981-1983 and recent shigella spp. isolated from patients with diarrhea at the out-patient section in three hospitals to provide the basis for scientific control in Tianjin.
Multi-locus Sequence Typing (MLST), a sequence-based genotyping technique first introduced by Maiden et al., provides reproducibility, comparability and transferability between laboratories [4,5]. MLST is based on sequencing the seven ‘housekeeping’ genes, which are under stabilizing selection. Like Multi-locus Enzyme Electrophoresis (MLEE), MLST uses variation that accumulates slowly, and which is expected to be selectively neutral, and achieves very high resolution by analyzing multiple loci [6]. The most important advantage of MLST over MLEE and the many typing methods that involve the comparisons of DNA fragments on gels are the unambiguity and electronic portability of nucleotide sequence data [7]. MLST has proved useful in characterizing and monitoring disease-causing and antibiotic resistance lineages of bacteria. Unlike other typing methods, MLST sequence data can be held through a central database and queried through a web server [8,9]. A surface antigen (serotyping or serovar) of organisms was the main source of differentiating bacteria. But, however, the enormous inventory of serovar, based mainly on an ever changing surface repertoire, throws an artificial and unreliable scenario of strain diversity [10,11]. It is therefore difficult to track strains whose molecular identity keeps changing according to the host and the environmental niches they inhabit and cross through [12,13]. The use of MLST in this study was to determine the clonal relationship within shigella spp. and to understand their evolutionary biology in Tianjin for the last two decades and compare their genetic similarity with different strains from different laboratories in the MLST database. MLST can be applied to almost all bacterial species and other haploid organisms, including those that are difficult to cultivate [8,14]. Although the results of MLST are highly reproducible, portable, and easy to interpret, MLST is complex and expensive to perform. Repetitive Extragenic Palindromic-PCR (REP-PCR) technique was used to characterize the strains, and to determine their genetic identity in the last two and half decades. REP-PCR is simple and cheap to perform; it can be applied to a bigger or smaller number of isolates [15]. REP-PCR has high resolution similar to that of Pulse Field Gel Electrophoresis (PFGE) but slightly lower discriminatory power than PFGE. The aims of this work were (i) To define the evolution and lineages of shigella strains isolated from consecutive dysentery during two and half decades in Tianjin (ii) To compare the phylogenetic similarity of strains isolated between 1981-1983 to those of 2009 and 2010. (iii) To understand their resistance characters with different antibiotics.
Materials and methods
Shigella spp susceptibility testing and serogroup distribution
Source of strains
66 strains of shigella were isolated and identified from clinical patients with diarrhea by stool cultures between May and October 2009 and 2010 at the Tianjin Children’s Hospital, The First General Hospital of Tianjin Medical University and the Second Hospital of Tianjin Medical University. 30 strains of shigella spp which were isolated between 1981-1983 and conserved by vacuum drying and stored at -70°C were collected from the Tianjin Institute of Infectious Diseases.
Method
Bacterial isolation was done according to the Clinical Laboratory Procedures. Fresh stool specimens were inoculated into Salmonella Shigella (SS) and MacConkey agar plates, and were incubated at 37 °C for 24 hrs in an incubator. Suspected Shigella colonies were selected from pure culture for biochemical testing and serological identification with standard antiserum typing kits. The 1981-1983 strains were resuscitated in Luria Bertani (LB) nutrient broth and inoculated on blood agar for identification and serological typing. The Shigella antiserum was purchased from Lanzhou Institute of Biological Products. Serological test was done by slide agglutination, using normal saline as a control.
Bacteria susceptibility testing
Antimicrobial susceptibility testing was carried out according to the Clinical Laboratory Standards Institute (CLSI) recommended criteria for disk diffusion method. The antimicrobial disks used include: ampicillin 10μg, piperacillin 100μg, cefazolin 30μg, ceftazidime 30μg, cefotaxime 30μg, ceftriaxone 30μg, cefepime 30μg, imipenem 30μg, tetracycline 30μg, chloramphenicol 30μg, streptomycin 10μg, amikacin 30μg, gentamicin 10μg, cotrimoxazole 125μg, norfloxacin 30μg, and levofloxacin 5μg. The antimicrobial disks were purchased from Beijing Biological products Inspection. Strain ATCC 25922 was used as a Susceptibility quality control. Data was analyzed using SPSS 19 statistical software and quantity one 4.5 software for statistical and band pattern analysis respectively.
REP-PCR and MLST analyses
DNA extraction
Bacteria DNA were extracted using the boiling method. The crude DNA was then refined by extraction with equal volumes of Chloroform. The refined DNA was used for REP-PCR and MLST analyses.
Polymerase chain reaction and REP-PCR
17 of the 30 strains of S. flexneri (isolated in 1981-1983) that showed varying degree of drug resistance and the 29 strains of S. flexneri and 20 strains of S. sonnei isolated in 2009-2010 were typed by REP-PCR to produce optimal conditions of repeatedly legible fingerprint, but it was only the results of S. flexneri were analyzed using quantity one 4.5 software to obtain a dendrogram showing band similarity between strains, (Figure 2). REP-PCR was carried out in MygeneTM Series Peltier Thermal Cycler machine. Briefly, the primer 5’GCGCCGTCATGCGGCATT 3’ was used under the following conditions: An initial temperature of 94°C for 2 minutes with 30 cycles of 1 min. at 94°C, 1 min. at 40°C, and 8 min. at 65°C, with a final extension of 16 min. at 65°C. The PCR reaction was done in a total volume of 25μl, with 5μl of DNA template, 2μl of primer, 12.5μl of premix and 5.5μl of sterilized water. The reaction was analyzed on a 1% agarose gel for 6 hours at 50V. Photos of electrophoresis results were taken by UV scanning machine for further analysis, (Figure 1).
Figure 2.

Dendrogram showing band similarity between the 1981-1983 and 2009-2010 shigella flexneri by REP-PCR.
Figure 1.

REP-PCR. Lanes M, molecular size marker (DL 2000); lanes A and B: S. flexneri isolated in 1981-1983; lanes C, D and E: S. sonnei isolated in 2009-2010; lanes F, G, H, I, J, K and L: S. flexneri isolated in 2009-2010.
Multi-Locus Sequence Typing (MLST)
Multi-locus Sequence typing was performed using a standardized protocol for E. coli maintained at the MLST database at the ERI website (http://mlst.ucc.ie/). Five strains in all were selected to perform MLST to get a better comparison and to understand the evolution of shigella spp. in Tianjin. The selected strains were: one strain in the 1981-1983 isolates (187), one shigella flexneri isolated in 2010 (3402), two S.sonnei isolated each in 2009 (N6) and 2010 (AB101) respectively and one shigella boydii isolated in 2009 (N2B5) to determine the lineage of shigella spp. in this study, (Figure 3).
Figure 3.

the electrophoretic results showing band sizes of the seven housekeeping genes. Lane M, molecular size marker (DL 2000). Lanes A recA; Lanes B, purA; Lanes C, mdh; Lanes D, icd; Lanes E, gyrB; Lanes F, fumC; Lanes G, adk.
Results
Serogroup distribution
In 1981-1983, 30 strains were Shigella flexneri, while in 2009-2010, 66 strains of shigella were collected, in which 29 strains were Shigella flexneri, accounted for 43.94%, 36 strains were Shigella Sonnei, accounted for 54.55%; and one strain was S. boydii accounted for 1.52%. S. dysentery was not detected. Because only one S. boydii was isolated, it was included only in the MLST analysis.
Shigella clinical isolates for drug sensitivity tests analysis
The results of the resistance rate of the groups of Shigella bacteria to the commonly used antibiotics are shown in (Table 1).
Table 1.
Comparison of antimicrobial resistance rates in different years and different serogroups
| Drug | 1981-1983 | 2009-2010 | X21 | P1 | X22 | P2 | ||
|---|---|---|---|---|---|---|---|---|
| S.flexneri | S.flexneri | S.sonnei | Total | |||||
| (n=30 st) | (n=29 st) | (n=36 st) | (n=65 st) | |||||
| Ampicillin | 13.3 | 96.6 | 91.7 | 93.9 | ----- | 0.000* | ----- | 0.622 |
| Piperacillin | 13.3 | 51.7 | 86.1 | 70.8 | 10.0 | 0.002* | 9.18 | 0.002* |
| Cefazolin | 6.7 | 48.3 | 11.1 | 27.7 | 12.9 | 0.002* | 11.07 | 0.001* |
| Ceftazidime | 0.0 | 10.3 | 0.0 | 4.6 | ----- | 0.112 | ----- | 0.084 |
| Cefotaxime | 0.0 | 37.9 | 5.6 | 20.0 | 14.0 | 0.000* | 10.52 | 0.001* |
| Ceftriaxone | 0.0 | 41.4 | 5.6 | 21.5 | 15.58 | 0.000* | 12.20 | 0.000* |
| Cefepime | 0.0 | 20.7 | 0.0 | 9.2 | 4.83 | 0.028θ | 5.92 | 0.015θ |
| Imipenem | 0.0 | 0.0 | 0.0 | 0.0 | ---- | ---- | ---- | ---- |
| Tetracycline | 100.0 | 86.2 | 88.9 | 887.7 | ---- | 0.052 | 0.00 | 1.000 |
| Chloramphenicol | 83.3 | 62.1 | 2.8 | 29.2 | 3.37 | 0.006* | 27.3 | 0.000* |
| Streptomycin | 76.7 | 75.9 | 72.2 | 73.9 | 0.01 | 0.940 | 0.11 | 0.740 |
| Gentamicin | 0.0 | 3.4 | 77.8 | 44.6 | ----- | 0.492 | 35.92 | 0.000* |
| Amikacin | 0.0 | 24.1 | 0.0 | 10.8 | 6.07 | 0.014θ | 7.39 | 0.007θ |
| Norfloxacin | 0.0 | 13.8 | 0.0 | 6.2 | ----- | 0.052 | ----- | 0.035α |
| Levofloxacin | 0.0 | 10.3 | 0.0 | 4.6 | ----- | 0.112 | ----- | 0.084 |
| Cotrimoxazole | 46.7 | 100.0 | 91.7 | 95.4 | 21.22 | 0.000* | ----- | 0.247 |
Note: P1 represents S.flexneri of 1983 and 2009 from the chi-square test P value; P2 represents S.flexneri and S.sonnei of 2009 from the chi-square test P value; X21 represents the chi-square for P1, X22 represents the chi-square for P2;
= Pearson chi-square,
= Yates’ correction,
= Fisher’s Exact test.
From Table 1: The resistance rate of all Shigella flexneri isolated between 1981-1983 to the traditional antibiotics ( tetracycline, streptomycin ,chloramphenicol) other than gentamicin was higher and varied from 76.47 to 100%, while the 2009-2010 Shigella strains showed susceptibility to tetracycline, streptomycin, chloramphenicol, their resistance rate was slightly lower, but the difference was not statistically significant (P> 0.05). Cotrimoxazole resistance in 26 years increased significantly from 46.70% to 100.00%. While The 1981-1983 Shigella bacteria were 100% sensitive to amikacin, gentamicin, the third and fourth generation cephalosporins, quinolones and imipenem, the 2009-2010 Shigella strains showed different levels of increasing drug resistance to the third and fourth generation cephalosporins and quinolones. According to Table 1, the resistance of Shigella flexneri increased significantly than Shigella sonnei. There was a rapid change of resistance rate especially for the third and fourth generation cephalosporins, (P <0.05). The resistance rate of the 2009-2010 Shigella flexneri to amikacin and gentamicin was higher than that of the 1981-1983 strains to varying degrees, but there was no significant difference (P> 0.05). But, however, the differences between the resistance rate of the 2009-2010 Shigella flexneri and Shigella sonnei to amikacin and gentamicin was statistically significant (P <0.05).
To enhance better explanation for the multidrug resistance difference in this study, serogroups isolated in different years were classified into three groups. The 1981-1983 S. flexneri as group I , 2009-2010 S. flexneri as group II and the 2009-2010 S. sonnei as group III. Multidrug resistance (MDR) is defined as the resistance of a microbe to more than 3 antibiotics. The results showed that groups II and III have higher resistance rate than Group I (Table 2).
Table 2.
Comparison of multiple drug resistance of Shigella spp. in different years
| time | Total no. of Strains | No. of strains of multiple drug resistant | percentage of multiple drug resistant (%) |
|---|---|---|---|
| Group I | 30 | 16 | 53.3 |
| Group II | 29 | 28 | 96.60 |
| Group III | 36 | 29 | 80.56 |
| Total | 95 | 73 | 76.84 |
To compare the rates of multiple drug resistant of the three groups I, II and III strains of Shigella species. I Vs II, Fisher’s exact test. P value < 0.05. There were significant differences. II Vs III, P value = 0.148, No significant difference.
REP-PCR analysis
The strains were divided into 41 genotypes (Figure 2). In the dendrogram, it could be seen that strains 166 and F2 are identical with a similarity coefficient of approximately 97% to strain 181. Similarly, strains 184, AB105, AB4, N1 are identical, with a partial similarity of approximately 96% to strain 1. Even though the 1981-1983 S. flexneri strains have taken over 26 years, they still had genetic similarity to that of S. flexneri isolated in 2009-2010 that caused frequent infection in Tianjin but with different resistance characters. Because most of the cases studied here were caused by very similar or identical strains, it is possible that these strains could survive in the environment either in contaminated water and food or in carriers such as humans and other animals [16].
MLST analysis
From Tables 3 and 4, Six new allele types (adk, 45; fumC, 240; icd, 99; purA, 159, recA, 142) were found in one of my strains, AB101. In addition to that, one new sequence type (ST 2208) was found in this study. Three ST complexes (ST 152, ST243, ST245) have been reported in the MLST database. Two of my strains (N6 and 3402) which were isolated in 2009 and 2010 respectively fall in ST 152 complex (allele types 11, 63, 7, 1, 14, 7, 7 in the order of adk, fumC, gyrB, icd, mdh, purA and recA) and they were predicted as the ancestors or founding members in this complex by BURST. In these two strains, one is shigella flexneri (3402) and the other is shigella sonnei (N6). Another two of my strains (187 and N2B5) which were isolated in 1983 and 2009 respectively fall in ST 245 complex (allele types 6, 61, 6, 11, 13, 3, 50 in the order of adk, fumC, gyrB, icd, mdh. purA and recA) and they were predicted as ancestors or founding members in this ST complex by BURST. The 1983 strain was shigella flexneri while the 2009 strain was shigella boydii. According to the MLST database for E.coli, they have a very close genetic similarity (the same allelic profile) with other shigella spp. isolated in Canada and Germany in 1982 and 1997 respectively. However, they differ in one allele from strains isolated from other parts of China, Japan and Philippine (Table 4). The one new ST (ST2208) which was found in this study did not fall in to any ST complex. This strain differ from the rest of the isolates in this study in all the seven loci, instead, it bears relation with Enteropathogenic E.coli (EPEC) and Enterhemorragic E.coli (EHEC) in one of its locus (mdh91) (Table 4).
Table 3.
MLST results of 5 clinical isolates of shigella species analyzed in this study
| Strain name | MLST name | Group name | ST | ST | Year isolated | adk | fumC | gyrB | icd | mdh | purA | recA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 187 | 187/83 | S.flexneri | 245 | 245 | 1983 | 6 | 61 | 6 | 11 | 13 | 3 | 50 |
| N6 | N6/09 | S.sonnei | 152 | 152 | 2009 | 11 | 63 | 7 | 1 | 14 | 7 | 7 |
| N2B5 | N2B5/09 | S.boydii | 245 | 245 | 2009 | 6 | 61 | 6 | 11 | 13 | 3 | 50 |
| 3402 | 3402/10 | S.flexneri | 152 | 152 | 2010 | 11 | 63 | 7 | 1 | 14 | 7 | 7 |
| AB101 | AB101/10 | S.sonnei | 2208 | none | 2010 | 45 | 240 | 192 | 99 | 91 | 159 | 142 |
ST= sequence types of the respective strains.
Table 4.
Portability and comparative analysis of strains in this study and their related STs in the MLST database
| Strain | ST | adk | fumC | gyrB | icd | mdh | purA | recA | Country | City | Year | G.Name |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 187 | 245 | 6 | 61 | 6 | 11 | 13 | 3 | 50 | China | Tianjin | 1983 | S.flexneri |
| N2B5 | 245 | 6 | 61 | 6 | 11 | 13 | 3 | 50 | China | Tianjin | 2009 | S.bovdii |
| 245 | 6 | 61 | 6 | 11 | 13 | 3 | 50 | Germany | 1997 | S.flexneri | ||
| 245 | 6 | 61 | 6 | 11 | 13 | 3 | 50 | Canada | 1982 | S.flexneri | ||
| 651 | 6 | 149 | 6 | 11 | 13 | 3 | 50 | China | S.flexneri | |||
| 634 | 6 | 61 | 6 | 123 | 13 | 3 | 50 | China | S.flexneri | |||
| 633 | 6 | 61 | 6 | 11 | 13 | 98 | 50 | Japan | S.flexneri | |||
| 628 | 6 | 61 | 6 | 11 | 13 | 3 | 55 | Philippine | S.flexneri | |||
| 629 | 6 | 145 | 6 | 11 | 13 | 3 | 55 | Korea | S.flexneri | |||
| 3402 | 152 | 11 | 63 | 7 | 1 | 14 | 7 | 7 | China | Tianjin | 2010 | S.flexneri |
| N6 | 152 | 11 | 63 | 7 | 1 | 14 | 7 | 7 | China | Tianjin | 2009 | S.sonnei |
| 152 | 11 | 63 | 7 | 1 | 14 | 7 | 7 | Germany | Gotting | 1997 | S.sonnei | |
| 151 | 11 | 62 | 7 | 1 | 14 | 7 | 7 | Germany | Passau | 1997 | S.sonnei | |
| 241 | 11 | 63 | 6 | 1 | 14 | 7 | 7 | 1997 | S.sonnei | |||
| 1757 | 11 | 63 | 9 | 1 | 14 | 7 | 7 | |||||
| 1756 | 11 | 63 | 7 | 1 | 14 | 186 | 7 | |||||
| 1755 | 11 | 63 | 7 | 1 | 14 | 11 | 7 | |||||
| AB101 | 2208 | 45 | 240 | 192 | 99 | 91 | 159 | 142 | China | Tianjin | 2010 | S.sonnei |
| 788 | 142 | 67 | 13 | 150 | 91 | 115 | 11 | Germany | 2007 | EPEC | ||
| 725 | 13 | 18 | 10 | 13. | 91 | 14 | 79 | Germany | 1987 | EPEC | ||
| 653 | 76 | 39 | 60 | 13 | 91 | 8 | 29 | Spain | 2004 | none | ||
| 593 | 13 | 13 | 10 | 13 | 91 | 14 | 79 | Germany | 1990 | EHEC | ||
| 412 | 95 | 111 | 91 | 99 | 68 | 70 | 76 | Bangladesh | S.bovdii |
Comparison of REP-PCR and MLST result
The isolates which were genotyped with REP-PCR, and MLST-based approach was employed to investigate their population structure and phylogenetic relationships to closely related species. Because of the small number of isolates typed by MLST as compared to those typed by REP-PCR there was not enough data to make a complete comparison between the banding patterns of REP-PCR results and MLST sequence types. However, with the few strains analyzed by MLST, it was found that two isolates (187 and N2B5) which had the same REP-PCR banding patterns also have the same allelic profile, thus the same ST (ST245). Similarly, two other isolates (N6 and 3402) which have similar REP-PCR patterns possessed the same ST. However, AB101 which have different REP-PCR banding patterns from the above four strains had different sequence type (ST2208). According to the two molecular typing methods (REP-PCR and MLST) used, the 1983 isolate (187) has the same genetic constitution with some of the shigella spp. isolated in 2009 and 2010.
BURST analysis of STs in this study and their related STs in the MLST database
Based upon related sequence type (BURST) divided the strains into three groups and two clonal complexes with two ST that were predicted as founding members. ST152 was the predicted ancestral genotype for group one and an asterisk was marked on it. This ST also defines the largest number of single locus variants (SLVs) and it corresponds to the highest frequency (FREQ). The same applies to group two with ST245 as the ancestral genotype because it defines the largest number of SLVs and corresponding to the highest frequency (FREQ). The ancestral genotype is within the central ring. Bold circles indicate SLVs and faint circles indicate double locus variants (DLVs). The larger bold circles enclose the SLVs of the ancestral genotype and the faint circles enclose DLVs of the ancestral genotype. However, strain AB101 was described as a singleton because its allelic numbers differ from all the strains analyzed by MLST in this study, (Figure 4).
Figure 4.

BURST analysis of ST in this study and their related ST in the MLST database.
MLST phylogenetic and minimal spanning tree analyses
The allelic profile data of the strains in this study and their related STs in the E.coli MLST database was used to construct a rooted phylogenetic tree by a neighbor joining method using the Phylogeny Inference Package (PHYLIP) suite of programs in the pubmlst website (http://pubmlst.org/analysis/.). The program divided the allelic profile data into three clusters (A, B, C). It could be seen from the tree that ST152 occupied the root of the tree indicating that it is the predicted ancestor in this analysis. The minimal spanning tree (MST, Figure 5) and the MLST phylogenetic tree have very similar evolutionary arrangement of sequence type (ST). ST152 being the predicted ancestor in the phylogenetic tree, it also served as the predicted founder in the minimal spanning tree. A combination of the two trees can give a stronger prediction that ST152 was the ancestor of all the STs analyzed in this study.
Figure 5.

Phylogenetic tree of the allelic profiles data of strains in this study and their related strains in E.coli MLST database. The blue fonts are the strains analyzed in this study and the black fonts are their related strains in the MLST database.
Discussion
Shigella spp. is one of the most common causes of shigellosis which can cause epidemics worldwide. Currently, bacillary dysentery caused by shigella is still one of the most frequently-occurring diseases in children in China. The disease can occur all year round, especially in summer and autumn. In developed countries S. sonnei is the dominant specie, while in developing countries including China, S. flexneri has been the main pathogen causing dysentery in children and the immunocompromised [17]. In recent years, S. sonnei infection rate has increased [18]. The Statistical results showed that the dominant serogroup of shigella in Tianjin has changed from S. flexneri to S. sonnei, contrary to Wang Xiaojuan’s report. The reason for the rapid increase infection rate of shigella sonnei in Tianjin may be related to the misuse of antibiotics on one hand, and the broad use of disinfectants on the other hand. The changes in the antibiotics used in treating dysentery and the improvement in health conditions might not be unrelated in the change of resistance and the dominating serogroup. In the 20th century, there was a gradual increasing resistance of shigella spp. against the cheap and traditional antibiotics (cotrimoxazole, tetracycline ampicillin etc.) frequently used in the late 80s for the treatment of shigella infection. These drugs are no longer suitable choices for patients with suspected shigella infections, which is in the same view with Agasan [17]. In the last couple of years, with the application of the third and fourth generation cephalosporin, the resistant rate of shigella against these drugs was increasing. The resistant rate against some kinds of the third generation cephalosporin was up to 40%. Because of the low rate use of chloramphenicol [19], the resistant rate against it was decreased. Does this suggest some resistance can be avoided by rotation of antibiotics to resolve the problem, still remain to be a large and long-term observation?
In addition to that, between each group of Shigella, the resistance rate was somewhat different. The resistance rate of shigella flexneri collected in 2009 was higher than that of Shigella sonnei. Despite the large, extensive, and long-term use of Quinolones, they still maintain a higher sensitivity to Shigella, suggesting that quinolones are better choice for diarrhea caused by Shigella. When we compare the distribution of multi-drug resistant strains of Shigella isolated in different years, the 2009-2010 strains presented significantly increased multi- drug resistant than the 1981-1983 strains (P <0.05), this prompted us that the resistance may be resulted from the wide use of antibiotics. Physicians should request antimicrobial susceptibility testing and choose drugs according to the results of sensitive antibiotics. The change of dominant serogroup was a significant epidemiological study.
Multi-locus sequence typing (MLST) was developed as an attractive sequence based typing technique because it provides reproducibility, comparability, and transferability between laboratories [20]. The use of multi loci is essential to achieve the resolution required to provide meaningful relationships among strains and is particularly important because of clones diversify with age, as a result of mutational or recombination events, and might be typed incorrectly if only a single locus was examined [8,14]. ST complexes (ST245 AND ST243) have been reported previously [14]. In this study, three STs (ST152, ST245, and ST2208) were found. Two of my strains belonged to ST245 complex, two belong to ST152 complex and one (ST2208) did not belonged to any ST complex. Comparing the band similarities of strains in the REP-PCR and the allelic profiling of the MLST sequence results, we found that a combination of the two methods to type bacteria is more discriminatory than typing based on MLST alone. Taking into consideration the relatedness of the strains in this study with E.coli in the MLST database, it was only ST2208 that was closely related to E. coli (EPEC and EHEC) by possessing the same allele, mdh91 with ST593, ST653, ST725 and ST788 respectively. This is in concordant with Seon Young Choi et al that shigella spp. belong to the Escherichia coli superfamily, consisting of three main clusters and a number of outliers [21-23]. As determined by using BURST and the minimum spanning tree (MST) analysis (Figures 4 and 6), the STs analyzed were grouped into two clonal complexes (CC), i.e. groups of closely related STs differing by no more than two allele from another member of the group. MLST is a sequence based typing technique that has overcome other molecular typing methods for tracking disease outbreaks.
Figure 6.
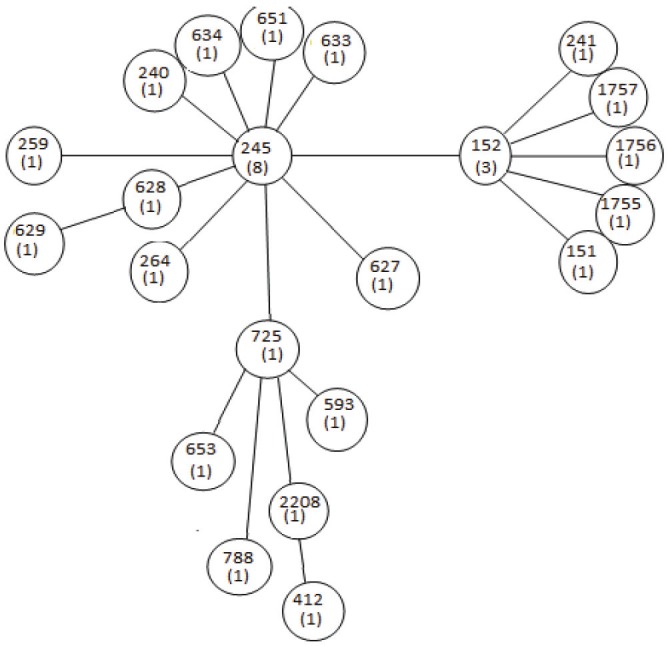
Minimal Spanning tree of the allelic profile data of the strains analyzed in this study and their related strains in the E.coli MLST database showing distances of evolution.
Even though serological typing method in this study designated the strains into different serogroups, REP-PCR and MLST analysis showed that isolates from different serogroup can be genetically identical. This is in concordant with Sara et al that virulence factors and antigens are frequently under selective pressure driven by host innate and acquired immunity and this may contribute to antigenic protein diversity directly exposed to host defense systems. Therefore, genes encoding such factors are more likely to undergo mutations over a short period of time compared with genes necessary for basic metabolic processes, such as the ‘housekeeping’ genes used in MLST [12].
Compared with other DNA typing methods, REP-PCR typing offers a simple, rapid, and highly discriminatory means of identifying and comparing shigella strains. The method can also be applied to other species responsible for nosocomial infection [24], making it a versatile and cost-effective tool for use in hospital diagnostic laboratories. It should be noted that the relationships shown in the dendrogram represent statistical relationships between banding patterns. REP-PCR is fast becoming the most widely used method of DNA typing. The technique is easy to perform, cheaper and can be applied to larger or smaller numbers of isolates. Rep-PCR shows broader species applicability and better discriminatory power than either plasmid profiling or genomic fingerprinting [25]. Strains that had similar or the same band patterns in REP-PCR belonged to the same ST in MLST analysis. A combination of REP-PCR and MLST typing techniques may provide stronger discriminatory ability to investigate the population structure, phylogenetic relationship and the evolution of closely related spp. if the numbers of strains analyzed in both techniques are the same. Finally, several studies have shown Rep-PCR to have good correlation with PFGE results but, in general, with slightly less discriminatory power.
Acknowledgments
I thank my colleagues, more especially Wang Lei and Men Kun for their enormous help during my laboratory work. I would like to send my thanks and appreciation to my supervisor, (Professor Wei Dian Jun) for providing funds for this research. I also send my gratitude to the Infectious Disease Institute for providing me with the 1981-1983 S. flexneri stains.
References
- 1.Thong KL, Hoe SL, Puthucheary SD, Yasin RM. Detection of virulence genes in Malaysian Shigella species by multiplex PCR assay. BMC Infect Dis. 2005;14:5–8. doi: 10.1186/1471-2334-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rowe-Magnus DA, Guerout AM, Mazel D. Bacterial resistance evolution by recruitment of super-integron gene cassettes. Mol Microbiol. 2002;43:1657–1669. doi: 10.1046/j.1365-2958.2002.02861.x. [DOI] [PubMed] [Google Scholar]
- 3.Sire JM, Macondo EA, Perrier-Gros-Claude JD, Siby T, Bahsoun I, Seck A, Garin B. Antimicrobial resistance in Shigella species isolated in Dakar, Senegal (2004-2006) Senegal Jpn J Infect Dis. 2008;61:307–309. [PubMed] [Google Scholar]
- 4.Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant DA, Feavers IM, Achtman M, Spratt BG. Multi-locus Sequence Typing: A Portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci USA. 1998;95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Urwin R, Maiden MC. Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol. 2003;10:479–487. doi: 10.1016/j.tim.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 6.Tartof SY, Solberg OD, Manges AR, Riley LW. Analysis of a uropathogenic Escherichia coli clonal group by multilocus sequence typing. J Clin Microbiol. 2005;43:5860–5864. doi: 10.1128/JCM.43.12.5860-5864.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nemoy LL, Kotetishvili M, Tigno J, Keefer-Norris A, Harris AD, Perencevich EN, Johnson JA, Torpey D, Sulakvelidze A, Morris JG Jr, Stine OC. Multilocus sequence typing versus pulsed-field gel electrophoresis for characterization of extended-spectrum beta-lactamase-producing Escherichia coli isolates. J Clin Microbiol. 2005;43:1776–1781. doi: 10.1128/JCM.43.4.1776-1781.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan MS, Maiden MC, Spratt BG. Database-driven Multi locus Sequence Typing ( MLST) of Bacteria Pathogens. Bioinformatics. 2001;17:1077–1083. doi: 10.1093/bioinformatics/17.11.1077. [DOI] [PubMed] [Google Scholar]
- 9.Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004;186:1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tiruneh M. Serodiversity and Antimicrobial Resistance Pattern of Shigella Isolates at Gondar University of Teaching Hospital, Norwest Ethiopia. Jpn J Infect Dis. 2009;62:93–97. [PubMed] [Google Scholar]
- 11.Escobar-paramo P, Giudicelli C, Parsot C, Denamur E. The evolutionary history of shigella and enteroinvasive Escherischia coli revised. J Mol Evol. 2003;57:140–148. doi: 10.1007/s00239-003-2460-3. [DOI] [PubMed] [Google Scholar]
- 12.Dias RC, Moreira BM, Riley LW. Use of fimH single-nucleotide polymorphisms for strain typing of clinical isolates of Escherichia coli for epidemiologic investigation. J Clin Microbiol. 2010;48:483–488. doi: 10.1128/JCM.01858-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woodward DL, Clark CG, Caldeira RA, Ahmed R, Soule G, Bryden L, Tabor H, Melito P, Foster R, Walsh J, Ng LK, Malcolm GB, Strockbine N, Rodgers FG Canadian Public Health Laboratory Network. Identification and characterization of Shigella boydii 20 serovar nov. a new and emerging Shigella serotype. J Med Microbiol. 2005;54:741–748. doi: 10.1099/jmm.0.46095-0. [DOI] [PubMed] [Google Scholar]
- 14.Choi SY, Jeon YS, Lee JH, Choi B, Moon SH, von Seidlein L, Clemens JD, Dougan G, Wain J, Yu J, Lee JC, Seol SY, Lee BK, Song JH, Song M, Czerkinsky C, Chun J, Kim DW. Multilocus sequence typing analysis of Shigella flexneri isolates collected in Asian countries. J Med Microbiol. 2007;56:1460–1466. doi: 10.1099/jmm.0.47322-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Penatti MP, Hollanda LM, Nakazato G, Campos TA, Lancellotti M, Angellini M, Brocchi M, Rocha MM, Dias da Silveira W. Epidemiological characterization of resistance and PCR typing of Shigella flexneri and Shigella sonnei strains isolated from bacillary dysentery cases in Southeast Brazil. Brazil Journal of Medical and Biological research. 2007;40:249–258. doi: 10.1590/s0100-879x2007000200012. [DOI] [PubMed] [Google Scholar]
- 16.Rotz LD, Khan AS, Lillibridge SR, Ostroff SM, Hughes JM. Public health assessment of potential biological terrorism agents. Emerg Infect Dis. 2002;8:225–230. doi: 10.3201/eid0802.010164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agasan A, Reddy S, Wiliams G, Perry W, Backer M, Ramon A, et al. High Prevalence of Antimicrobial Resistance among Shigella Isolates to Agents Commonly Used for Treatment , NARMS 1999 and International Conference on Emerging Infectious Disease. Atlanta, GA: 2000. Jul, [Google Scholar]
- 18.Kuo CY, Su LH, Perera J, Carlos C, Chiu CH. Antimicrobial susceptibility of Shigella isolates in eight Asian countries, 2001-2004. J Microbiol Immunol Infect. 2008;41:107–111. [PubMed] [Google Scholar]
- 19.Kotloff KL, Winickoff JP, Ivanoff B, Clemens JD, Swerdlow DL, Sansonetti PJ, Adak GK, Levine MM. Global burden of Shigella infectious: implications for vaccine development and implementation of control strategies. J Bull World Health Organ. 1999;77:651–666. [PMC free article] [PubMed] [Google Scholar]
- 20.Ahmed AM, Futruta K, Shimomura K, Kasama Y, Shimamoto T. Genetic characterization of multidrug resistance in Shigella spp. from Japan. J Med Microbiol. 2006;55:1685–1691. doi: 10.1099/jmm.0.46725-0. [DOI] [PubMed] [Google Scholar]
- 21.Johnson JR, Owens KL, Clabots CR, Weissman SJ, Cannon SB. Phylogenetic relationships among clonal groups of extraintestinal pathogenic Escherichia coli as assessed by multi-locus sequence analysis. Microbes Infect. 2006;8:1702–1713. doi: 10.1016/j.micinf.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 22.Lacher DW, Steinsland H, Blank TE, Donnenberg MS, Whittam TS. Molecular evolution of typical enteropathogenic Escherichia coli: clonal analysis by multilocus sequence typing and virulence gene allelic profiling. J Bacteriol. 2007;189:342–350. doi: 10.1128/JB.01472-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sturenburg E, Mack D. Extended-spectrum beta-lactamases: implications for the clinical microbiology laboratory, therapy, and infection control. J Infect. 2003;47:273–295. doi: 10.1016/s0163-4453(03)00096-3. [DOI] [PubMed] [Google Scholar]
- 24.Drews SJ, Lau C, Andersen M, Ferrato C, Simmonds K, Stafford L. Laboratory based surveillance of travel-related Shigella sonnei and Shigella flexneri in Alberta from 2002 to 2007. Global Health. 2010;1:6–20. doi: 10.1186/1744-8603-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Georghiou PR, Hamill RJ, Wright CE, Versalovic J, Koeuth T, Watson DA, Lupski JR. Molecular epidemiology of infections due to Enterobacter aerogenes: identification of hospital associated strains by molecular techniques. Clin Infect Dis. 1995;20:84–94. doi: 10.1093/clinids/20.1.84. [DOI] [PubMed] [Google Scholar]