

Summary
The conserved MAPKKK Dual-Leucine-zipper-bearing Kinases (DLKs) are key regulators of synaptic development and axon regeneration. Understanding the mechanism by which DLK kinases are activated is thus of central importance. Here, we show that C. elegans DLK-1 is activated by a novel Ca2+-dependent switch from inactive heteromeric to active homomeric protein complexes. A newly identified DLK-1 isoform, DLK-1S, shares identical kinase and leucine zipper domains with the previously described long isoform DLK-1L, but acts to inhibit DLK-1 function by binding to DLK-1L. The switch between homo- or hetero-meric DLK-1 complexes is influenced by Ca2+ concentration. A conserved hexapeptide in the DLK-1L C-terminus is essential for DLK-1 activity and is required for Ca2+ regulation. The mammalian DLK-1 homolog MAP3K13, also known as Leucine-Zipper-Kinase (LZK) contains an identical C-terminal hexapeptide and can functionally complement dlk-1 mutants, suggesting that the DLK activation mechanism is conserved. The DLK activation mechanism is ideally suited for rapid and spatially controlled signal transduction in response to axonal injury and synaptic activity.
Keywords: DLK-1, MAPKKK, MAP3K12, MAP3K13, synapses, axon regeneration, C. elegans, signal transduction
MAP kinase mediated signal transduction pathways have been implicated in many aspects of neuronal development and function (Huang and Reichardt, 2001; Ji et al., 2009; Mielke and Herdegen, 2000; Samuels et al., 2009; Subramaniam and Unsicker, 2010; Thomas and Huganir, 2004). As neurons are highly polarized cells receiving spatially segregated information, a critical aspect of MAP kinases is their ability to be locally regulated within cells and with tight temporal control. For example, in developing axons, local activation of p38 and Erk MAP kinases by the MAPKK MEK1/MEK2 is differentially required for BDNF and netrin-1 induced growth cone turning (Ming et al., 2002) and slit-2 induced growth cone collapse (Piper et al., 2006). Local activation of MAP kinases by neuronal excitation plays important roles in dendritic spine dynamics (Wu et al., 2001). Axonal injury can trigger activation of Erk at the injury site to regulate signal transduction via retrograde transport (Perlson et al., 2005). In a typical MAP kinase cascade, activation of the upstream MAPKKK is a critical control point for signal specificity and amplification (Chang and Karin, 2001). However, our knowledge of how MAPKKKs are activated in vivo by local neuronal signals remains limited.
The Dual-Leucine-zipper-bearing-Kinases (DLKs) are key regulators of synapse formation, axon regeneration, and axon degeneration from C. elegans to mammals (Collins et al., 2006; Hammarlund et al., 2009; Itoh et al., 2009; Miller et al., 2009; Nakata et al., 2005; Xiong and Collins, 2012; Xiong et al., 2010; Yan et al., 2009; Shin et al., 2012). The DLK kinases belong to the mixed-lineage family of MAPKKKs (Holzman et al., 1994). The hallmark of these kinases is a leucine-zipper domain, which can mediate protein dimerization or oligomerization and has been implicated in kinase activation (Nihalani et al., 2000). Although C. elegans and Drosophila each have only one gene encoding DLK kinase (Nakata et al., 2005; Collins et al., 2006), mammalian genomes encode two closely related DLK family kinases known as MAP3K12/DLK/MUK/ZPK (Blouin et al., 1996; Hirai et al., 1996; Holzman et al., 1994) and MAP3K13/LZK (Sakuma et al., 1997). Both kinases are widely expressed in the nervous system, and DLK/MAP3K12 was identified as a synapse-associated MAPKKK (Mata et al., 1996). The in vivo functions of these kinases were discovered through genetic studies of the synaptic E3 ubiquitin ligases known as PHR proteins, including C. elegans RPM-1, Drosophila Highwire, mouse Phr1, and human Pam (Collins et al., 2006; Lewcock et al., 2007; Nakata et al., 2005). Activated DLK kinases are targeted for degradation by these E3 ligases, resulting in a tight control of duration of signal transduction. In C. elegans, loss-of-function mutations in dlk-1 genetically suppress the neuronal defects of rpm-1 mutants, but dlk-1 mutants themselves are viable and grossly normal (Nakata et al., 2005). Constitutive activation of the DLK-1 pathway induces developmental defects mimicking rpm-1(lf) (Nakata et al., 2005). Moreover, expression of a constitutively active MAK-2, a downstream kinase of DLK-1, at synapses can disrupt synapse morphology and decrease synapse number (Yan et al., 2009), suggesting that local activation of the DLK-1 pathway plays important roles in synapse formation. In adult neurons, DLK-1 is essential for injured axons to regenerate, and its activity is required within a limited time window after injury (Hammarlund et al., 2009). These results indicate that the activation of DLK-1 must be precisely controlled in time and space by neuronal activity or injury.
Despite extensive studies of DLK kinase function and their negative regulation by PHR proteins, the mechanisms by which DLK kinases are activated have remained elusive. Here we show that a newly identified short isoform, DLK-1S, shares identical kinase and leucine zipper domains with the previously reported long isoform DLK-1L, but binds to and inhibits the activity of the active DLK-1L. We identify a unique hexapeptide at the DLK-1L C-terminus that plays critical roles in DLK-1 isoform-specific interactions. We further show that mammalian MAP3K13 contains identical hexapeptide and can complement DLK-1 function. Our results reveal a novel mechanism for tight spatial and temporal control of MAPKKK activity in neurons.
Results
The dlk-1 locus encodes two isoforms with antagonistic functions
The previously reported C. elegans DLK-1 protein contains 928 amino acid residues, including a kinase domain (aa 133-382) and a leucine zipper (LZ, aa 459-480) (Figures 1A and 1B). By our analysis of new dlk-1 cDNA clones, and subsequently by RT-PCR and Northern blotting, we found that the dlk-1 locus generates a second shorter transcript by use of an alternative polyadenylation site in intron 7 (Figures 1A, S1A and Experimental Procedures). This transcript encodes a DLK-1 isoform of 577 residues. We here name the two isoforms DLK-1L (long) and DLK-1S (short). Both isoforms contain identical N-terminal kinase and LZ domains. The C-terminus of DLK-1S consists of 11 isoform-specific residues, whereas the DLK-1L-specific C-terminus contains 361 residues. Neither C-terminal domain contains known protein motifs. Analysis of ESTs (expressed sequence tags) for human and rat DLK family members indicates that these genes can also encode long and short isoforms (Figure 1B).
Figure 1. DLK-1 encodes two isoforms with different functions.
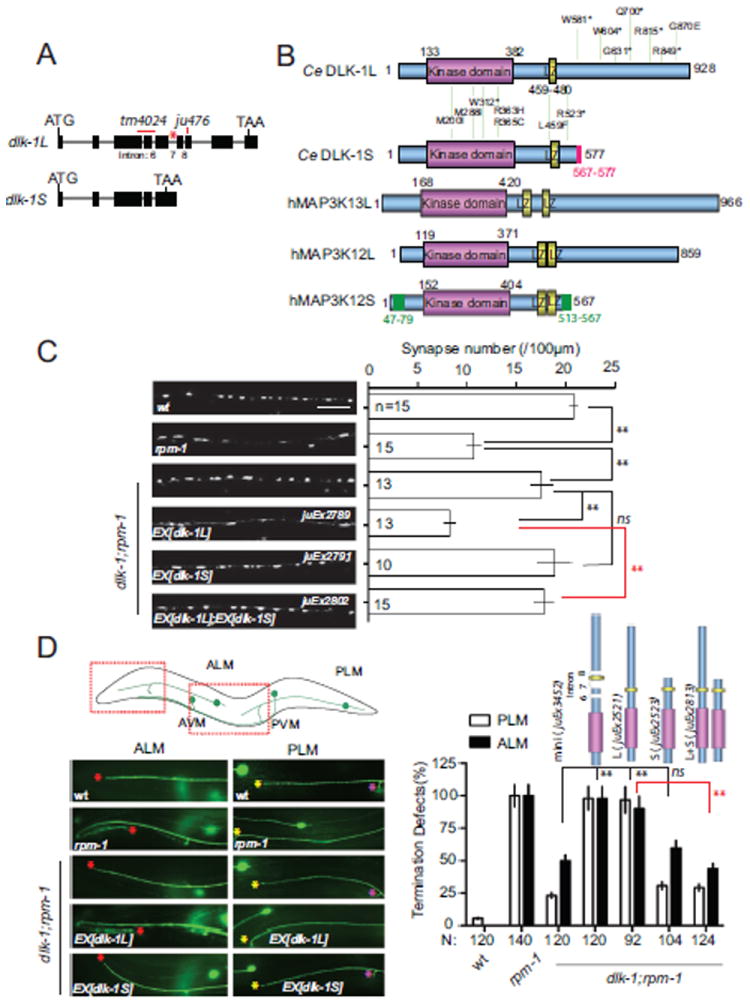
(A) Structure of the dlk-1 locus. Black boxes are exons; red star marks the alternative intron 7. The deletion allele tm4024 truncates kinase domain in both DLK-1L and DLK-1S followed by a premature stop codon. ju476 contains a 5bp insertion, inducing a premature stop codon only in DLK-1L.
(B) Schematic comparison of DLK-1 isoforms and their human homologs. Distribution of selected genetic loss of function mutations in DLK-1 is shown as amino acid change. The full list of dlk-1 mutations is in Supplemental Table 1. The accession# for human MAP3K13, MAP3K12L and S are EAW78225.1, NM_006301.3 and BC050050.1, respectively.
(C-D) The DLK-1S isoform does not provide normal function of DLK-1, and antagonizes DLK-1L. (C) Images of SNB-1-GFP puncta expressed in the GABAergic motor neurons (juIs1 [Punc-25-SNB-1-GFP]) in young adults of genotypes indicated. The graph shows quantification of SNB-1-GFP puncta number of corresponding genotypes. n = number of animals; scale bar, 10 μm. Similar changes are observed for the presynaptic active zone protein UNC-10/RIM (see Figure S2A). (D) Images show touch neuron axons labeled by muIs32 [Pmec-7-GFP] in young adults in regions illustrated above. In wild type ALM axons end before the tip of the nose (red stars) and PLM axons end before ALM cell bodies (yellow stars). In rpm-1 both ALM and PLM axons overextend. The PLM neuron also has one synaptic branch near the vulva (purple stars), which is absent in half of rpm-1 animals. Quantification of axon termination defects of PLM and ALM is shown as % of total # of animals. Quantification of PLM synaptic branch shows a similar pattern (not shown). Data is shown as mean ± SEP in D. Statistics in this and subsequent figures: Student’s t test for synaptic puncta and Fisher exact test for axonal morphology. *, P < 0.05; **, P < 0.01; ns, not significant.
To gain clues about the functions of the two isoforms of DLK-1, we took advantage of our extensive collection of genetic loss of function mutations in dlk-1, all of which were isolated as suppressors of rpm-1(lf) (Nakata et al., 2005). A large number of missense mutations affect conserved residues in the kinase domain (Figures 1B, S1B, S1C, and Table S1); one mutation (ju591) changes the conserved Leu at residue 459 in the LZ domain (Figure 1B). The strong loss-of-function phenotypes induced by these mutations are consistent with the essential roles of the kinase and LZ domains (Figure S1C). Unexpectedly, another set of strong loss-of-function mutations affect the C-terminus specific to DLK-1L, and are not predicted to affect DLK-1S (Figures 1B and S1C, Table S1). RT-PCR analysis showed that DLK-1S transcripts were produced at normal levels in the C-terminal mutants (Figure S1D). These observations raised the possibility that DLK-1S does not have the same activity as DLK-1L.
To more directly address the role of DLK-1S, we assayed its function in synaptogenesis and developmental axon outgrowth, using transgenic rescue of the phenotypes of dlk-1(lf); rpm-1(lf) double mutants. rpm-1 mutants exhibit defects in motor neuron synapse development and in touch neuron axon growth (Schaefer et al., 2000; Zhen et al., 2000). Both synaptic and axonal rpm-1 defects are strongly suppressed by dlk-1(lf) (Nakata et al., 2005) (Figures 1C,D and S2A). Neuronal expression of a DLK-1L cDNA at low concentrations fully rescued the dlk-1(lf) suppression phenotype (Figures 1C,1D and S2A, juEx2789, juEx2521). Expression of a DLK-1 minigene that produces both DLK-1L and DLK-1S proteins at comparable levels (Figure S2B) also fully rescued dlk-1 suppression phenotype (Figure 1D, juEx3452). In contrast, expression of DLK-1S alone showed no rescuing activity (Figures 1C and 1D, juEx2791, juEx2523), consistent with the interpretation that DLK-1S cannot substitute for DLK-1L. Furthermore, the rescuing activity of DLK-1L was strongly attenuated by co-overexpression with DLK-1S (Figures 1C and 1D, juEx2802, juEx2813). This inhibitory effect of DLK-1S was eliminated when the LZ domain was deleted from DLK-1S (Figure S2C). However, expression of a kinase-dead mutant DLK-1S(K162A), in which the Lys162 at the ATP binding site of the kinase domain was mutated to Ala (Nakata et al., 2005), inhibited DLK-1L to a similar degree as did wild type DLK-1S (Figure S2C). These data suggest that the ability of DLK-1S to inhibit DLK-1L requires its LZ domain but not its kinase activity. As further test for the role of DLK-1S, we expressed various DLK-1 constructs in the wild type background (Figure S2D). Expression of DLK-1L alone caused abnormal neuronal development, whereas expression of DLK-1(mini) gene had much weaker effect. Removing intron 7 from DLK-1(mini), which would prevent production of DLK-1S, resulted in gain-of-function effects similar to DLK-1(L). Finally, to address whether transgenically expressed DLK-1S could interfere with endogenous DLK-1L, we expressed DLK-1S in rpm-1(lf) single mutants, and observed significant suppression of rpm-1(lf) phenotypes (Figure S3A). Together, these analyses demonstrate that despite sharing identical kinase and LZ domains, DLK-1S is a potent inhibitor of DLK-1L function.
DLK-1L activity requires a conserved motif in its extreme C-terminus
If DLK-1S acts as an endogenous inhibitory isoform, how does DLK-1L become activated at all? Since DLK-1L and DLK-1S differ only in their C-termini, we hypothesized that the C-terminus of DLK-1L may contain elements important for its kinase activation, and that DLK-1S may act by preventing the interactions between such elements and the kinase domain. To test this idea, we generated a series of DLK-1L variants in which the C-terminus was either truncated or contained internal deletions (Supplemental Experimental Procedures), and assayed rescuing activity of these constructs in the dlk-1(lf); rpm-1(lf) double mutant strain (Figure 2, Table S2). We found that a region of 25 amino acids from residues 856 to 881 in the DLK-1L C-terminus was necessary for DLK-1L activity (Figure 2, juEx3586). Remarkably, a construct lacking all of the DLK-1L C-terminus except for aa 856-881 recapitulated the activity of the full length DLK-1L (Figure 2, juEx3657), suggesting this region is sufficient for DLK-1L regulation. Upon closer inspection of the amino acid sequences, we found a six-residue motif SDGLSD (aa 874-879, hereby referred to as the hexapeptide) that is completely conserved between C. elegans DLK-1 and vertebrate MAP3K13/LZK (Figure 3A); the remainder of the C-termini of these kinases show little sequence conservation. Moreover, dlk-1(ju620), a strong loss of function mutation, results in a missense alteration (G870E) adjacent to this hexapeptide. We expressed a DLK-1L(G870E) mutant cDNA and observed little rescuing activity (Figure 2, juEx3659). Deleting the conserved hexapeptide in DLK-1L also completely abolished rescuing activity (Figure 2, juEx4098). Together these results identify the conserved C-terminal hexapeptide as critical for DLK-1L function.
Figure 2. A small domain of the DLK-1L C-terminus is required for DLK-1 activity.
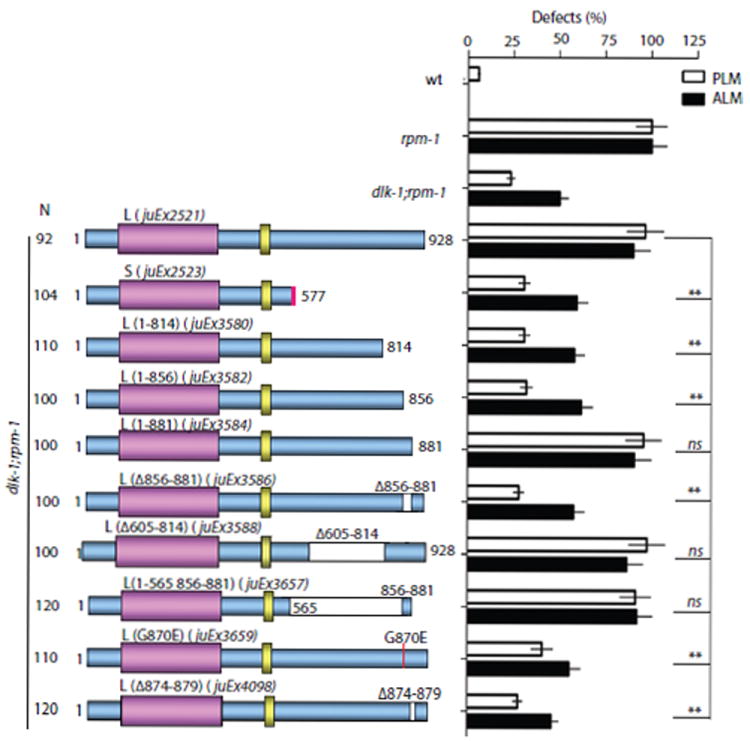
Transgenic expression of C-terminal truncated DLK-1L variants in dlk-1(ju476);rpm-1(ju44) double mutants identifies a small region (aa 856-881) and a conserved hexapeptide (aa 874-879) as required for DLK-1L activity. Data analysis and statistics are the same as in Figure 1.
Figure 3. DLK-1L can bind to itself or to DLK-1S, and the interactions are regulated by a C-terminal conserved hexapeptide.
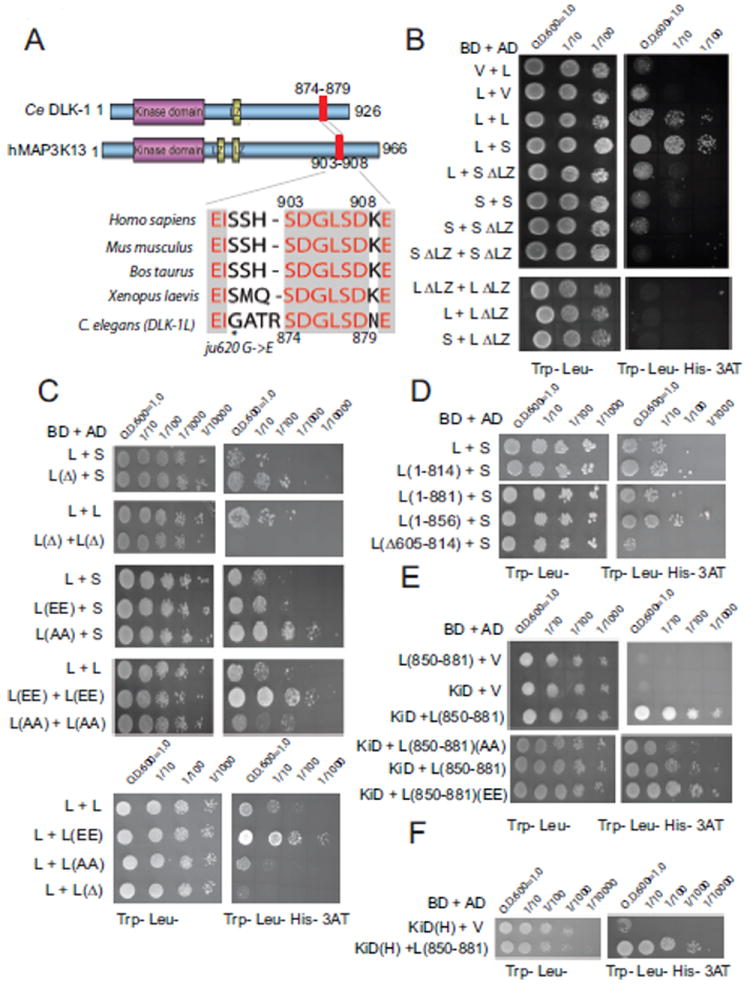
(A) Sequence conservation of the C-terminal peptide of DLK-1L (aa 856-881) with human MAP3K13/LZK (accession# EAW78225.1), mice (accession# NM_172821.3), Bos taurus (accession# NM_001101853.1) and Xenopus laevis (accession# NM_001172189.1). * marks dlk-1(ju620) mutation. Sequence alignments used ClustW. Protein phosphorylation sites were predicted by the NetPhos program (http://www.cbs.dtu.dk/services/NetPhos/).
(B-E) Yeast cells were plated in a dilution series on −Trp −Leu media (left). Histidine (His) selection plates contained 10 mM 3-AT (right). (B) DLK-1S binds to DLK-1L. BD, GAL4 binding domain; AD, GAL4 activation domain; V, empty vector; L, DLK-1L; S, DLK-1S; ΔLZ, Leucine zipper domain deletion.
(C) The C-terminal hexapeptide regulates DLK-1 interactions. L(Δ), DLK-1L(Δ874-879); L(EE), DLK-1(S874E, S878E); L(AA), DLK-1(S874A, S878A).
(D) aa 605-814 of DLK-1L are also required for the interactions between DLK-1L and DLK-1S.
(E) aa 850-881 of DLK-1L bind to the kinase domain (KiD). The interaction is enhanced by phosphomimetic mutations (EE) and reduced by non-phosphorylatable mutations (AA) of the hexapeptide.
(F) The DLK-1L C-terminal aa 850-881 interacts with the kinase domain of human MAP3K13 (KiD(H)).
The regulatory roles of the C-terminal hexapeptide in DLK-1 isoform-specific interactions
To determine how DLK-1S interacts with DLK-1L and how the C-terminal hexapeptide regulates their interaction, we next performed protein interaction studies using yeast two hybrid assays. We found that full-length DLK-1L interacted with itself and also with DLK-1S (Figure 3B). Removal of the LZ domain in DLK-1L or DLK-1S eliminated these interactions. Unexpectedly, despite containing the LZ domain, DLK-1S did not show interaction with itself, suggesting that the LZ domain is not sufficient for DLK-1 dimerization or oligomerization. To test the role of the C-terminal hexapeptide SDGLSD in the interactions between DLK-1 isoforms, we deleted it from DLK-1L. We found that a DLK-1L construct lacking the hexapeptide failed to show any homomeric interaction, and instead, displayed an enhanced heteromeric interaction with DLK-1S (Figure 3C). These results suggest that the C-terminal hexapeptide plays a critical regulatory role in DLK-1 isoform-specific interactions.
Since the C-terminal aa 856-881region can endow a truncated DLK-1(kinase+LZ) with complete function (Figure 2, juEx3588), we tested whether this domain might interact with the kinase domain. In yeast two hybrid assays, we observed that the aa 850-881 region interacted with the kinase domain of DLK-1 (Figure 3E). The hexapeptide SDGLSD contains two potential phosphorylation sites (Ser 874 and Ser878, Figure 3A). We addressed whether these Serines were sites of regulation by generating phosphomimetic and non-phosphorylatable forms of the hexapeptide. We found that full-length DLK-1L containing phosphomimetic (S874E, S878E) hexapeptide showed stronger binding to itself (Figure 3C). Conversely, full length DLK-1L containing non-phosphorylatable (S874A, S878A) hexapeptide showed an enhanced interaction with DLK-1S (Figure 3C). The phosphomimetic C-terminal aa 850-881 region also showed stronger binding to the kinase domain of DLK-1 (Figure 3E). The C-terminal domain alone did not interact with itself in yeast two hybrid assays (not shown), although a region of 209 amino acids between LZ domain and the hexapeptide was necessary for DLK-1L to interact with DLK-1S (Figure 3D). Taken together, the results from yeast two hybrid interaction assays suggest that phosphorylation of the DLK-1L hexapeptide could regulate the balance between active DLK-1L homomers and inactive DLK-1L/S heteromers.
To address the in vivo importance of DLK-1 C-terminal hexapeptide phosphorylation, we turned to transgenic expression. DLK-1L with a non-phosphorylatable hexapeptide (S874A, S878A) was expressed normally (Figure S4), but lacked rescuing activity (Figure 4A, juEx4708). However, co-expression of the phosphomimetic DLK-1L(S874E, S878E) overcame the inhibitory effects of DLK-1S (Figure 4A, juEx4694). Strikingly, co-expression of DLK-1S with the DLK-1L C-terminal 328 aa or the aa 850-881 fragment in dlk-1; rpm-1 mutants significantly rescued the suppression effects of dlk-1(lf) (Figure 4A, juEx3661, juEx3729). These results suggest that the C-terminus of DLK-1L can activate DLK-1 in trans.
Figure 4. Effects of the C-terminal hexapeptide on DLK-1 activation and functional conservation of human MAP3K13.
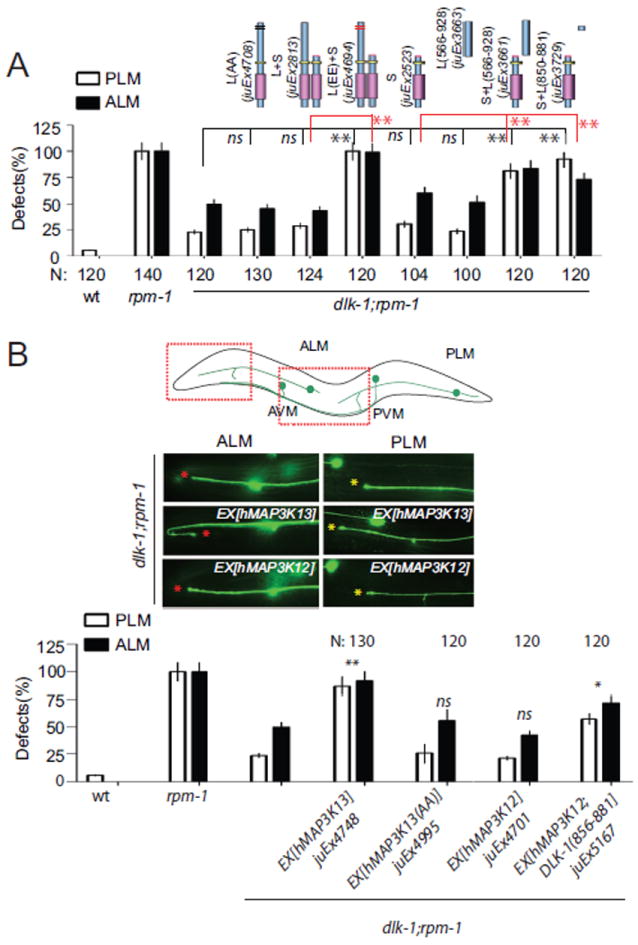
(A) Coexpressing the DLK-1L C terminus (aa 566-928) or the conserved domain (aa 850-881) with DLK-1S shows comparable rescuing activity to DLK-1L. DLK-1L containing non-phosphorylatable mutations of the hexapeptide does not rescue the suppression effects of dlk-1(ju476). DLK-1L containing the phospho-mimetic mutations of the hexapeptide overcomes the inhibition of DLK-1S.
(B) Pan-neural expression of human MAP3K13, but not MAP3K12, significantly rescues the suppression effects of dlk-1(ju476). Coexpression of MAP3K12 with the DLK-1 C-terminal hexapeptide partially rescues the suppression effects of dlk-1(ju476). Data analysis and statistics of Figure 4 are the same as in Figure 1.
Vertebrate MAP3K13/LZK proteins contain C-terminal hexapeptides identical to that of DLK-1L (Figure 3A). We therefore tested whether the function of DLK-1L was conserved with human MAP3K13. The kinase domain of MAP3K13 is 60% identical to that of DLK-1 (Figure S1B). We found that DLK-1L(aa 850-881) could bind to the kinase domain of human MAP3K13 in the yeast two hybrid assay (Figure 3F). We then expressed the human MAP3K13 cDNA under a pan-neural promoter in dlk-1(lf); rpm-1(lf) animals (Supplemental Experimental Procedures), and observed a significant rescue of dlk-1(lf) phenotypes (Figures 4B, S3, juEx4748). In contrast, expression of a mutant MAP3K13 containing Ala mutations in the hexapeptide (S903A, S907A) did not rescue dlk-1(lf) (Figure 4B, juEx4995). The MAP3K12/DLK shares an almost identical kinase domain with MAP3K13/LZK, but lacks the C-terminal hexapeptide. However, expression of MAP3K12/DLK alone failed to rescue dlk-1 phenotypes (Figure 4B, juEx4701). Interestingly, co-expression of MAP3K12/DLK with a fragment containing the DLK-1 C-terminal hexapeptide partially rescued dlk-1(lf) (Figure 4B juEx5167). These results show that human MAP3K13 complements dlk-1 function, and suggest that MAP3K13 can be activated by a similar mechanism involving the conserved hexapeptide.
DLK-1L and DLK-1S co-localize at synapses and in axons
Previous studies have shown DLK-1L is predominantly localized at synapses and detectable along axons (Abrams et al., 2008; Nakata et al., 2005). We next investigated where the DLK-1 isoform interactions could occur in neurons. We expressed functional XFP-DLK-1 fusion proteins in motor neurons and touch neurons (Table S2). Co-expressed YFP-DLK-1L and CFP-DLK-1S showed punctate colocalization patterns at motor neuron synapses and in touch neuron axons (Figures 5A and 5B). When expressed separately, GFP-DLK-1L showed punctate patterns in both wild type and dlk-1 mutants (Figure 5C). GFP-DLK-1S showed a similar punctate pattern in wild type animals, but became diffuse in dlk-1(tm4024) mutants, which lacks both DLK-1L and DLK-1S, or in dlk-1(ju591) mutants, in which the conserved Leu in the LZ domain of both DLK-1L and DLK-1S is mutated (Figures 1B, 5C). Moreover, removal of the LZ domain caused GFP-DLK-1S(ΔLZ) to be diffuse. These results are consistent with the DLK-1L and DLK-1S interaction occurring in vivo and show that the axonal localization of DLK-1S relies on its binding to DLK-1L through the LZ domain. Our previous studies showed that inactive DLK-1L(K162A) protein is more stable than wild type DLK-1L (Abrams et al., 2008). We found that overexpression of DLK-1S resulted in significant increase of GFP-DLK-1L expression (Figure S3B). In rpm-1(lf) mutants, the expression level of GFP-DLK-1S was not altered (Figure S3C). These data are consistent with the conclusion that binding of DLK-1S to DLK-1L keeps DLK-1L inactive. We further found that GFP-tagged C-terminus (aa 566-928) of DLK-1L recapitulated full-length DLK-1L localization (Figure 5C). However, phosphomimetic or nonphosphorylatable mutations of the hexapeptide did not change DLK-1L localization (Figure S4). We conclude that the C-terminus of DLK-1L is not only required for DLK-1L activity but also necessary for its subcellular localization in neurons.
Figure 5. DLK-1S co-localizes with DLK-1L in neurons, dependent on the LZ domain.
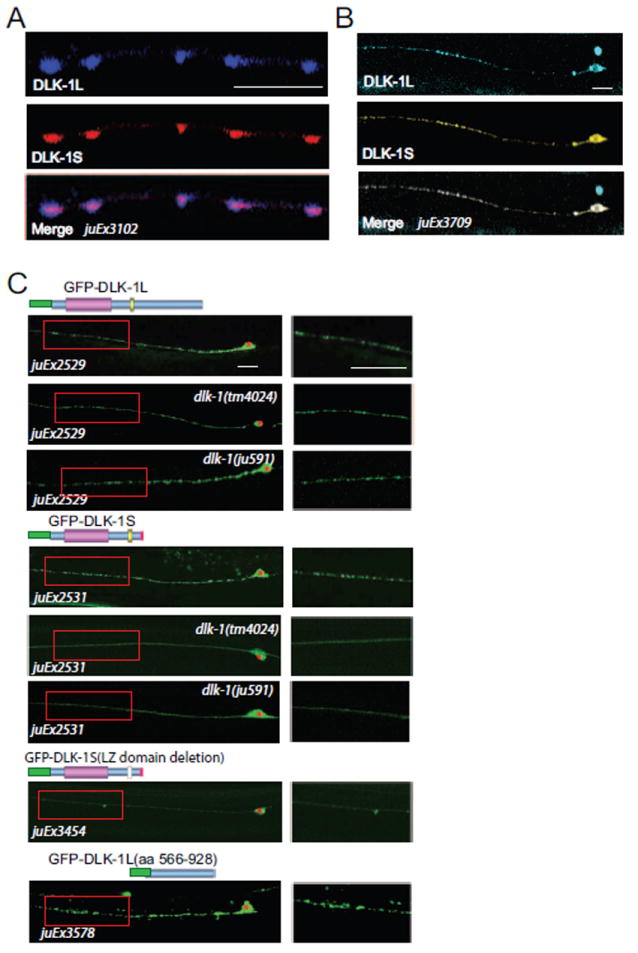
(A) Confocal stack images of functional CFP-DLK-1L and mCherry-DLK-1S (juEx3102) coexpressed in GABAergic motor neurons show that DLK-1L and DLK-1S are co-localized at synapses. Scale bar, 10 μm.
(B) Confocal stack images of functional CFP-DLK-1L and YFP-DLK-1S (juEx3709) coexpressed in touch neuron axons. Scale bar, 10 μm.
(C) Axonal punctate localization of DLK-1S requires its binding to DLK-1L through the LZ domain. The DLK-1L specific C-terminus is sufficient for its axonal localization. Shown are representative confocal images of functional GFP-DLK-1L and GFP-DLK-1S. DLK-1S without LZ domain is diffuse in axons, DLK-1S loses axonal punctate localization in dlk-1(tm4024) or dlk-1(ju591), a leucine zipper mutation. Cell bodies are marked by *. Enlarged axonal pictures are shown on right. Scale bars,10 μm.
Isoform-dependent regulation of DLK-1 activity is critical for axon regeneration
How might the isoform-dependent interaction of DLK-1 be regulated in vivo? To address this question, we focused on the function of DLK-1 in adult neurons. In dlk-1(lf) adult animals, injured axons fail to regrow (Hammarlund et al., 2009; Yan et al., 2009). Overexpression of DLK-1(mini) completely rescued the regeneration failure of injured PLM axons in dlk-1(tm4024) mutants (Figures 6A and 6B, juEx3444). Overexpression of DLK-1L not only rescued the regeneration failure, but also greatly enhanced the overall extent of axon regrowth (juEx2789) (Hammarlund et al., 2009; Yan et al., 2009). Paralleling our observations in developing neurons, overexpression of DLK-1S did not rescue the regeneration failure of dlk-1(lf) mutants (juEx2791), and blocked the regrowth enhancing effects of DLK-1L (juEx2815). The inhibitory activity of DLK-1S required its LZ domain (juEx2881). However, DLK-1L(EE) with C-terminal phosphomimetic mutations showed regrowth-enhancing effects when co-expressed with DLK-1S (juEx4694), suggesting that DLK-1S is less able to inhibit DLK-1L whose hexapeptide is phosphorylated.
Figure 6. DLK-1S negatively regulates DLK-1L function in adult PLM axon regeneration.
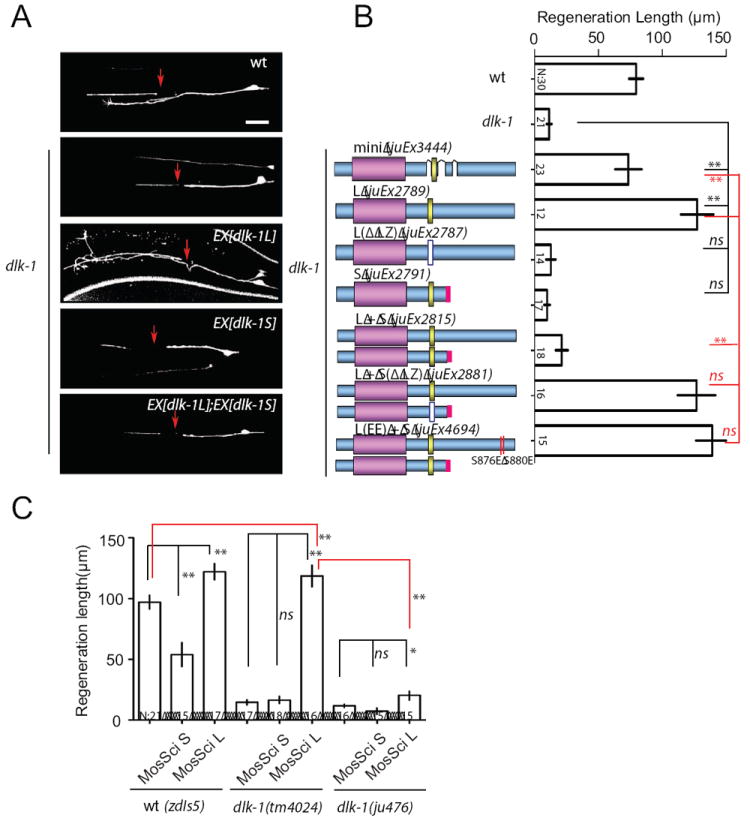
(A) Images are confocal micrographs of PLM neurons expressing different DLK-1 isoforms at 24 h after axotomy.
(B) Quantitation of PLM regrowth at 24 h after axotomy. Expression of DLK-1L restores axon regeneration ability in dlk-1(tm4024). Coexpressing DLK-1S, but not DLK-1S(ΔLZ), attenuates DLK-1L rescue ability. Expression of DLK-1L(EE) overcomes the inhibitory effects of DLK-1S.
(C) Effects of single-copy insertion transgenes of DLK-1 isoforms expressed under the pan-neural rgef-1 promoter. Single copy insertion of DLK-1S (juSi46) is sufficient to inhibit regeneration in wild type. Single copy expression of DLK-1L (juSi50) increases regeneration length in dlk-1(tm4024), but not in dlk-1(ju476) that only expresses DLK-1S. zdIs5 (Pmec-4-GFP) was used to visualize axons for axotomy. n, # of animals. Bar chart shows mean ± SEM; statistics, Anova. Scale bar, 10 μm.
As axon regeneration is highly sensitive to the dosage of DLK-1, we were concerned that the observed effects could be confounded by the variable levels of overexpression associated with multicopy extrachromosomal transgenes. We therefore generated single-copy transgenic expression of DLK-1S or DLK-1L driven by the rgef-1 pan-neural promoter, using the Mos-SCI technique (Frokjaer-Jensen et al., 2008) (Experimental Procedures). Single-copy expression of DLK-1S (juSi46) in wild type animals significantly impaired PLM neuron axon regeneration, while single-copy expression of DLK-1L (juSi50) strongly enhanced regeneration in wild type and in dlk-1(tm4024) mutants that lack both DLK-1L and DLK-1S (Figure 6C). Importantly, expression of DLK-1L from juSi50 showed weak rescue of the failure of axon regeneration in dlk-1(ju476) mutants, which express intact DLK-1S (Figures S1D, 6C). These results not only re-affirm that DLK-1S has potent and specific antagonistic effects on DLK-1L in axon regeneration, but also suggest that axonal injury can trigger DLK-1L activation by releasing the endogenous inhibition imposed by DLK-1S. The similar enhanced regeneration by expression of DLK-1L from single-copy transgene in both wild type and dlk-1(tm4024) implies that the formation of active DLK-1L/L homomeric protein complexes is under the control of injury signals.
Axotomy promotes accumulation of DLK-1L at the injury sites
We next investigated how axonal injury might modulate the association of DLK-1L and DLK-1S. We performed live imaging in PLM neurons expressing GFP-DLK-1L or GFP-DLK-1S, after laser axotomy of PLM in L4 animals as described (Wu et al., 2007). Within seconds after axotomy GFP-DLK-1L visibly accumulated at cut sites and continued to increase over the recording time (5-7 mins) (Figure 7 and Experimental Procedure). In contrast, GFP-DLK-1S showed no obvious changes at cut sites; cytosolic GFP decreased in intensity during the same period of imaging (Figure 7). Since DLK-1L(S874A, S878A) lacked activity and could bind to DLK-1S more strongly than to wild type DLK-1L (Figure 3C), we imaged its dynamics and found that GFP-DLK-1L(S874A, S878A) did not show significant changes immediately after axotomy (Figure 7B). The differential localization of DLK-1L and DLK-1L(AA) upon axonal injury are consistent with DLK-1 becoming dissociated from DLK-1S at the cut site in response to injury.
Figure 7. Axotomy triggers local accumulation of DLK-1L but not DLK-1S at the injury site.
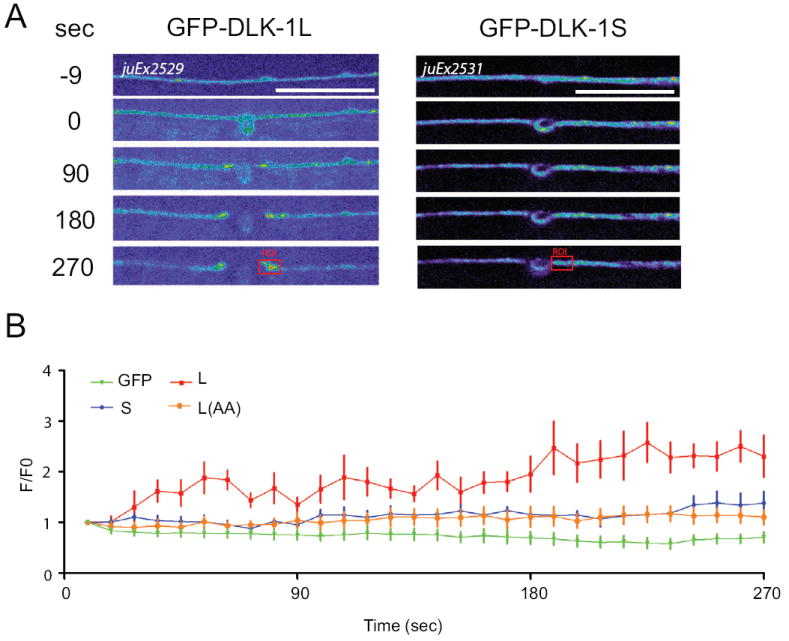
(A) Local accumulation of GFP-DLK-1L at severed ends of axons within minutes of damage. Time 0 refers to laser axotomy. Note accumulation both in proximal (right) and distal (left) axon fragments. Images were taken every 9 seconds for 5 mins using spinning disk confocal microscopy. In order to visualize dynamics of GFP-DLK-1 in axons, we used higher imaging laser energy and longer exposure time than those in Figure 5A.
(B) Normalized intensity of GFP fluorescence in a 3 μm region of the proximal axon near cut sites (Region of interest (ROI), red rectangles) at different time points after axotomy. DLK-1L, DLK-1S and DLK-1L(AA) fluorescent intensity change (F/F0) differed (P<0.05) observed from 36 sec post-axotomy to the end of the experiments. Cytosolic GFP in muIs32 was used as a negative control. Data shown as mean ± SEM; statistics, t-test. n >=10 from two independent recordings. Scale bars, 10 μm.
High concentration of Ca2+ can trigger the dissociation of DLK-1L/DLK-1S
We next addressed the mechanism by which axotomy might regulate DLK-1 isoform-mediated activation. Axotomy causes a wide range of changes in axons, including membrane breakage, disruption of cytoskeleton and organelle trafficking, and transient increases in Ca2+ (Barron, 2004; Stirling and Stys, 2010; Wang and Jin, 2011). Among these, Ca2+ increase is one of the earliest events, and previous studies have shown that increasing Ca2+ levels can promote axon regeneration in a DLK-1 dependent manner (Ghosh-Roy et al., 2010). To test whether Ca2+ can influence DLK-1 isoform interactions, we first turned to heterologous expression in cultured cells, which allowed us to detect DLK-1 protein interactions after acute manipulation of Ca2+. We co-expressed FLAG-DLK-1L, HA-DLK-1L and HA-DLK-1S in HEK 293 cells, and stimulated the cells using the Ca2+ ionophore ionomycin, with or without the Ca2+ chelator BAPTA-AM (see Experimental Procedure). We then immunoprecipitated FLAG-DLK-1L and quantitated the amount of co-immunoprecipitated HA-DLK-1L and HA-DLK-1S by western blotting. Without ionomycin stimulation DLK-1L was predominantly bound to DLK-1S (Figure 8A). Ionomycin treatment led to a two-fold decrease in the amount of co-immunoprecipitated DLK-1S, accompanied with a two-fold increase of co-immunoprecipitated DLK-1L (Figures 8A and 8B). These results support our two hybrid interaction studies. Ionomycin treatment at different concentrations did not affect the co-immunoprecipitation pattern of DLK-1L and DLK-1S (Figure S5B). Incubation with BAPTA-AM blocked the effect of ionomycin treatment (Figures 8A and 8B), indicating that the change in association between DLK-1L with DLK-1L or DLK-1S induced by ionomycin treatment is likely due to the transient increase of intracellular Ca2+ levels.
Figure 8. High concentrations of intracellular Ca2+ can trigger dissociation of DLK-1L/DLK-1S and promote binding between DLK-1L.
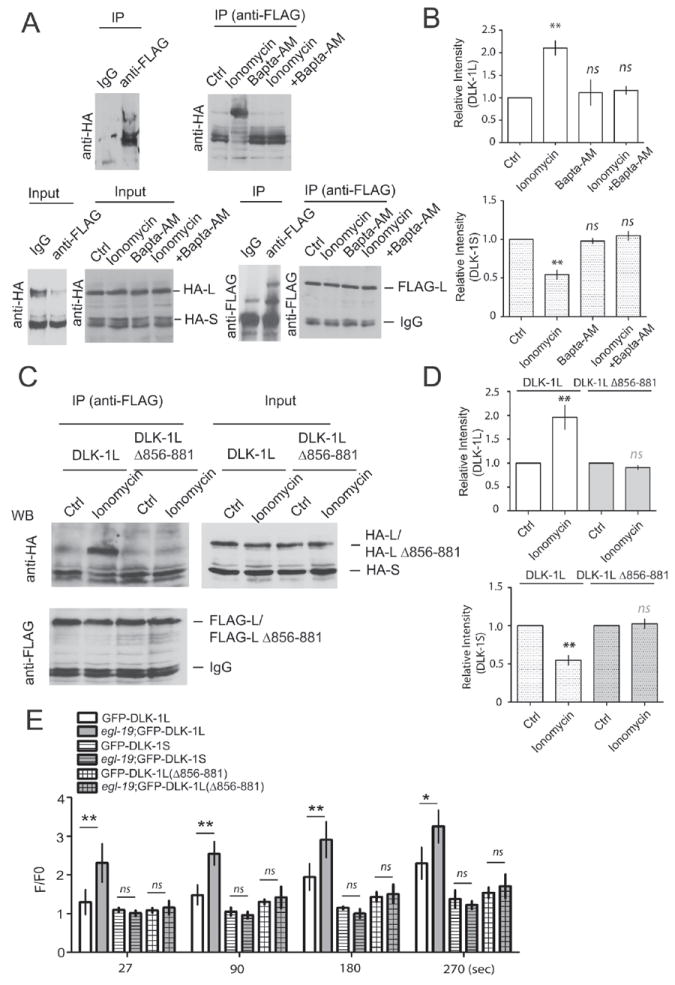
(A-B) Increasing intracellular Ca2+ concentration alters binding interactions between DLK-1L and DLK-1S. FLAG-DLK-1L, HA-DLK-1L and HA-DLK-1S were coexpressed in HEK293 cells at ratio of 1:1:2. Cells were treated with 3 μM ionomycin, with or without 10 μM BAPTA-AM for 15 mins. Rabbit anti-FLAG antibodies were used for immunoprecipitation of DLK-1L, and 7% SDS-PAGE gels were used to separate proteins of different molecular weights. Mouse anti-HA antibodies were used for western blot to detect co-immunoprecipitated HA-DLK-1L or HA-DLK-1S. Rabbit anti-FLAG antibodies were used to show immunoprecipitated FLAG-DLK-1L. 5% protein lysis before IP was shown as input. (A) Images of western blots. (B) Quantification of immunoprecipitated HA-DLK-1L and HA-DLK-1S under different treatments. The intensities of bands were first normalized to the input, and then normalized by co-immunoprecipitated DLK-1L or DLK-1S in untreated groups. The amount of co-immunoprecipitated proteins bound to DLK-1L under different treatments is shown as mean ± SEM. DLK-1L (up), DLK-1S (bottom). Statistics, t-test. **P<0.01, n=5.
(C-D) DLK-1L C-terminal conserved domain is important for Ca2+ regulation. Full length DLK-1L or DLK-1L(Δ856-881) was coexpressed with DLK-1S in HEK293 cells in the same ratio as in (A-B). Cells were treated with ionomycin. IP experiments were performed as in (A-B). Deletion of aa 856-881 in DLK-1L abolishes the effect of ionomycin. (C) Images of western blots. (D) Quantification data. n = 4. Data shown as mean ± SEM; statistics, t-test. **P<0.01, n=5
(E) Increase of Ca2+ influx by an egl-19(gf) mutation further enhances accumulation of GFP-DLK-1L(juEx2529) but not DLK-1S(juEx2531) or DLK-1L(Δ856-881)(juEx3813) at cut sites after axotomy. Experimental conditions and data analysis as in Figure 7. Data shown as mean ± SEM; statistics, t-test, **P<0.01, *P<0.05; n>=10 from two independent recordings.
Both the yeast two hybrid protein interaction studies and in vivo transgene expression have shown that the DLK-1L C-terminal aa 856-881 region regulates the interactions between DLK-1 isoforms (Figures 2 and 3C). We wondered whether this region could be involved in Ca2+ dependent modulation of DLK-1 isoform specific interactions. We found that DLK-1L and the mutant DLK-1L(Δ856-881), DLK-1L(Δ874-879) and DLK-1(S874A, S878A) bound to DLK-1S to a similar degree under normal culture condition (Figures 8C,8D and S5A), consistent with the yeast two hybrid interactions. However, ionomycin treatment did not cause detectable binding partner changes of the mutant DLK-1L(Δ856-881), DLK-1L(Δ874-879), and DLK-1(S874A, S878A). We also found that DLK-1(S874E, S878E) showed strong binding to itself even without ionomycin treatment (Figure S5A). These results support the idea that C-terminal of DLK-1L is required for the dissociation of DLK-1L/S heteromeric complexes caused by increasing Ca2+ levels.
To test whether Ca2+ played a regulatory role in vivo, we next analyzed GFP-DLK-1L dynamics in egl-19(ad695gf) animals, which is a gain of function mutation in the Ca2+-channel (Kerr et al., 2000; Lee et al., 1997). Previous studies have shown that egl-19(gf) enhances Ca2+ influx in PLM neurons after axotomy (Ghosh-Roy et al., 2010). We found that egl-19(gf) mutants also displayed significantly increased accumulation of GFP-DLK-1L at cut sites, comparing to wild type (Figure 8E, juEx2529). In contrast, neither DLK-1S nor DLK-1L(Δ856-881) showed local changes upon immediate axonal injury in egl-19(gf) or wild type (Figure 8E, juEx2531, juEx3823). These data suggest that a transient increase in Ca2+ levels, as caused by axonal injury or synaptic activity, can trigger the release of DLK-1L from inhibition by DLK-1S, and that this dissociation may be influenced by the phosphorylation state of the C-terminal hexapeptide.
Discussion
The DLK kinases play key roles in synapse and axon development and axon regeneration (Chen et al., 2011; Collins et al., 2006; Hammarlund et al., 2009; Itoh et al., 2009; Lewcock et al., 2007; Nakata et al., 2005; Xiong et al., 2010; Yan et al., 2009; Shin et al., 2012). In particular, timely activation of DLK kinases is critical for early responses to axonal injury (Chen et al., 2011; Hammarlund et al., 2009). In this study, we have uncovered a novel regulatory mechanism that endows C. elegans DLK-1 kinase with the ability to be rapidly activated by axon injury. We find that the newly identified short isoform, DLK-1S, acts as an endogenous inhibitor of the active long isoform DLK-1L. Our data support a model in which the balance between the active DLK-1L homomeric complexes and inactive DLK-1L/S heteromeric complexes can be spatially and temporally regulated by the conserved hexapeptide in a stimulus- or Ca2+-dependent manner (Figure S6). This regulation is mediated via a C-terminal hexapeptide that is highly conserved in the DLK-1 and MAP3K13 family. Our observation that human MAP3K13 can functionally complement C. elegans DLK-1 suggests unexplored roles of MAP3K13 in axon injury and hints at the possibility that similar kinase activation mechanisms may apply to vertebrate neurons.
DLK-1 activity and spatial control of signal transduction in neurons
Our observation that the inhibitory DLK-1S co-localizes with DLK-1L in axons and synapses in a manner dependent on its binding to DLK-1L supports a conclusion that most DLK-1 proteins are maintained in an inactive state in uninjured axons (Figure S6A). We also find that axotomy promotes accumulation of DLK-1L, but not of DLK-1S, at the tips of severed axons and that increasing intracellular Ca2+ can abrogate DLK-1L/DLK-1S heteromeric interaction. Injury triggers rapid Ca2+ transients, which can promote axon regenerative response (Ghosh-Roy et al., 2010). We find that egl-19(gf) further enhances DLK-1L accumulation at the cut site. Thus, we speculate that such Ca2+ transients may contribute to the dissociation of DLK-1L from DLK-1S at the injury site. In developing neurons DLK proteins are enriched at synaptic terminals. Overactivation of DLK kinases disrupts synapses and axon growth and termination (Nakata et al., 2005; Yan et al., 2009). The PHR E3 ligases, which are localized adjacent to DLKs at synapses, provide one level of control of signal transduction through ubiquitin-mediated protein degradation of the activated kinases (Abrams et al., 2008; Nakata et al., 2005). Synaptic activity triggers Ca2+ transients, and therefore could also locally activate DLK kinases, in a similar manner to axon injury. Indeed, it was reported that depolarization, which is coupled with changes in Ca2+ levels, can activate mouse DLK in cell lines (Mata et al., 1996). The isoform-specific regulation of DLK-1 activity by Ca2+ reported here has further advanced our understanding of how developing synapses can be regulated in an activity-dependent manner. Together, our data have revealed an unexpected mode of MAP kinase activation that is ideally suited for spatial and temporal control of DLK signal transduction in neurons.
Activation mechanism of DLK-1 as MAPKKKs
A striking observation in our study is that the inhibitory effect of DLK-1S does not depend on its kinase activity. This implies that DLK-1S binding sterically hinders DLK-1L activity. We find that the C-terminus of DLK-1L is necessary for its activity and for its localization. Within the C-terminus, we have identified a domain that can bind the kinase domain and influence DLK-1L and DLK-1S heteromeric interactions. Remarkably, the conserved core of this domain, the SDGLSD hexapeptide, is completely conserved from C. elegans DLK-1 to human MAP3K13. This hexapeptide does not match known Ca2+ binding sites, nor known phosphorylation consensus sites. However, both the hexapeptide and neighboring sequences are rich in charged amino acid residues, suggesting a possible role in sensing ionic changes. Our phosphomimetic manipulations suggest that the charge state of this hexapeptide can tip the balance of DLK-1 homo- and hetero-meric interactions. These observations imply involvement of Ca2+ or injury triggered kinases, the identity of which remains to be addressed in the future.
MAPKKKs provide stimulus specificity in signal transduction cascades, and must be maintained in inactive states under basal conditions (Craig et al., 2008; L’Allemain, 1994). Most MAPKKKs contain regulatory domains in addition to the kinase domain. A well-known example is Raf MAPKKK (Chong et al., 2003), which is maintained in an inactive state by binding of an N-terminal regulatory region to its kinase domain (Pumiglia et al., 1995). Release of this autoinhibition involves several proteins that bind to various regions of Raf, culminating with kinase activation by homo- or hetero-dimerization with its activation partner KSR (Fantl et al., 1994; Freed et al., 1994; Xing et al., 1997). Similarly, the N-terminus of MTK1/MEKK4 MAPKKK binds to and inhibits its kinase domain; and stress signals activate GADD45 proteins that bind to the N-terminus causing dissociation of the kinase domain and activation of MTK1 through protein dimerization (Mita et al., 2002; Miyake et al., 2007; Takekawa and Saito, 1998).
To our knowledge there is no exact precedent among MAPKKKs for the mechanism of DLK-1 activation. Although many MAP kinases are known to self-activate through kinase dimerization (Mita et al., 2002; Miyake et al., 2007; Takekawa and Saito, 1998), DLK-1L/S heteromeric binding does not activate DLK-1L. Notably, presence of the C-terminal domain in the DLK-1L/S heteromeric state is not sufficient to trigger DLK-1L activation. We envision one possible activation mode that may involve conformational changes of the homomeric kinase domain mediated by an intermolecular interaction between the kinase domain and the C-terminal activation domain (Figure S6B). In neurons, regulation of kinase activity by Ca2+ is well known. However, most Ca2+-dependent kinases either bind Ca2+ directly, or are regulated by Ca2+-binding proteins, as in the case of CamKII (Meador et al., 1993). Ca2+/calmodulin binding causes phosphorylation of the internal CamKII peptide and changes the holoenzyme conformation, leading to kinase activation (Yang and Schulman, 1999; Chao et al., 2011). Although several DLK kinase binding proteins have been reported (Fukuyama et al., 2000; Horiuchi et al., 2007; Ghosh et al., 2011; Whitmarsh et al., 1998), none has been associated with Ca2+. Our studies therefore provide new insights for a structural understanding of DLK kinases.
Implications for function conservation of the DLK family of proteins
The C. elegans and Drosophila DLK kinases are orthologous to two closely related vertebrate MAPKKKs, DLK/MUK/ZPK/MAP3K12 and LZK/MAP3K13 (Holzman et al., 1994; Sakuma et al., 1997). MAP3K12 and MAP3K13 display >95% sequence identity in their kinase domain, but their C-termini diverge significantly. It has been shown that the LZ domain of MAP3K13 can mediate dimerization, but is not sufficient to activate MAP3K13 (Ikeda et al., 2001). MAP3K12 and MAP3K13 are known to activate different downstream kinases in cultured cells (Ikeda et al., 2001; Nihalani et al., 2000), and may have opposite roles in regulating neurite outgrowth (Dickson et al., 2010; Eto et al., 2009).
We have shown that the C-terminal aa 856-881 domain of DLK-1L can bind to the kinase domain of both human MAP3K13 and C. elegans DLK-1. Expression of human MAP3K13 in C. elegans neurons can functionally complement dlk-1. The DLK-1L hexapeptide is completely conserved in MAP3K13 but not in MAP3K12. Short isoforms for MAP3K12 are detected as ESTs; MAP3K12 and MAP3K13 can form homomers or heteromers (Ikeda et al., 2001; Nihalani et al., 2000). Thus far, most reported studies have focused on the MAP3K12/DLK, and different types of neurons lacking DLK show a range of phenotypes from neurite regeneration to axon degeneration and to neuronal death (Ghosh et al.,2011; Itoh et al., 2009; Miller et al., 2009; Itoh et al., 2011). At present, much less is known about MAP3K13/LZK, although it has been implicated in neurite outgrowth, and may interact with the regrowth inhibitor Nogo (Dickson et al., 2010).
Our discovery of antagonistic C. elegans DLK-1 isoforms, together with our demonstration of functional conservation of DLK-1L and MAP3K13/LZK, suggest that the activation mechanisms of DLK family MAPKKKs in neurons may be conserved. Specifically, we speculate that MAP3K13 could also be kept in an inhibited state by an endogenous inhibitory isoform. As MAP3K12 and MAP3K13 are almost identical in their kinase and LZ domains, MAP3K12 isoforms could provide this inhibitory function. Clearly it will be informative to assess the role of MAP3K13 in synaptic development and axon regrowth and its possible cross-talk with MAP3K12.
Experimental Procedures
C. elegans genetics
We maintained C. elegans strains on NGM plates at 20 - 22.5°C as described by Brenner (1974). The dlk-1 mutations are listed in Supplemental Table 1; mutations affecting the kinase domain are shown in Supplemental Figure 1B. juIs1[Punc-25-SNB-1∷GFP] for GABA motor neuron synapses (Hallam and Jin, 1998); muIs32[Pmec-7-GFP] (Ch’ng et al., 2003) for touch neuron morphology and axon regeneration studies. Other transgenes and strains are described in Supplemental Table 2.
Fluorescence microscopy
We scored fluorescent reporters in live animals using a Zeiss Axioplan 2 microscope equipped with Chroma HQ filters. For quantification of touch neuron morphology using muIs32, 100-150 one-day old adults were analyzed. For quantification of GABAergic motor neuron synapse morphology using juIs1, Confocal images of dorsal cords in the midbody were collected on one-day old adults immobilized in 1% 1-phenoxy-2-propanol (TCI America, Portland, OR) in M9 buffer. For GFP-DLK-1L, GFP-DLK-1S and CFP-DLK-1L/YFP-DLK-1S, images were collected from one-day old adults using a Zeiss LSM510 confocal microscope. For synapse morphology and DLK-1L/S localization, z-stack images (1 μm/section) were shown in the figures.
Laser axotomy
We cut PLM axons in anesthetized L4 larvae using a near-infrared Ti-Sapphire laser (KMLabs, Boulder, CO) as described (Wu et al., 2007). To image the dynamics of GFP-DLK-1L/S in Figure 7, animals were immobilized using 6 mM levamisole (Sigma) in M9 buffer as described (Ou et al., 2010), and pictures were taken using a Yokogawa CSU-XA1 spinning-disk confocal microscope with a Photometrics Cascade II EMCCD camera (1024 × 1024), controlled by μManager (www.micro-manager.org). One image was taken right before axotomy, and images were collected every 9 seconds for 7 minutes post-axotomy. To visualize the axons for laser axotomy in GFP DLK-1L/S dynamic experiments we used 0.5 sec exposure time, which was much longer than in the localization analysis in Figure 5. The fluorescence intensity of the first 3μm axon fragments near cut sites was measured using MetaMorph (Molecular Devices). A comparable non-axonal region of interest was also measured as background. For the quantification of GFP DLK-1L/S protein level after axotomy, the intensity of each time point (Ft=Fi-Fb; Ft: fluorescence intensity at time point t; Fi : fluorescence intensity of the region of interest; Fb: intensity of the background) was normalized by the intensity at axotomy (F0).
Statistical analysis
In comparisons of measurement of axonal regrowth in Figure 4 we used one - tailed Student’s t test. Comparisons involving multiple groups used one-way Anova and Bonferroni post tests in Graphpad Prism (GraphPad Software, La Jolla, CA). To compare variables such as axon termination proportions in Figure 1, 2 and 3 we used the Fisher exact test.
Protein analysis and yeast two hybrid assay
For expression studies in 293T cells, full-length dlk-1L and dlk-1S cDNAs were cloned into pcDNA3-HA or pcDNA-FLAG to generate pCZGY1711(FLAG-DLK-1 L), pCZGY1710(HA-DLK1L), pCZGY1709(HA-DLK-1S), pCZGY1708(FLAG-DLK-1L(Δ856-881)) and pCZGY1707(HA-DLK-1L(Δ856-881)). Cells were cultured using standard procedures in DMEM medium (Nakata et al., 2005). Lipofectamine 2000 (Invitrogen) was used in cell transfection. One day after transfection, cells were treated with 3 μM Ionomycin (Cell Signaling Technology #9995) with or without BAPTA-AM (10 mM) (Sigma #126150-97-8) for 15 min and lysed using radioimmunoprecipitation buffer (25 mM Tris-HCl, pH 7.4, 150 mM KCl, 5 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS, and protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN). Equivalent amounts of lysates were incubated with rabbit anti-FLAG (Sigma F7425) for 6-8 hours at 4°C. Immune complexes were precipitated with protein A agarose (GE Healthcare, Piscataway, NJ) for 1 hour at 4°C, washed four times with lysis buffer, and eluted by heating to 95°C for 5 min in SDS sample buffer containing 1mM DTT. Blots were probed with rabbit anti-Flag antibodies (Sigma F7425), or a mouse anti-HA monoclonal antibody (Cell Signaling, #2367). The blot was visualized with Amersham HRP-conjugated anti-rabbit or anti-mouse secondary antibodies at 1:5000 (Amersham) using the SuperSignal West Femto kit (Pierce, Rockford, IL).
Yeast two-hybrid assays were performed using pACT2 and pBTM166 vectors (Clontech, Mountain View, CA). DLK-1 cDNAs encoding full length or fragment of the protein were fused to the GAL4 DNA binding domain in pACT2 or the GAL4 activation domain in pBTM166. Pairs of plasmids were co-transformed into yeast strain L40, and selected on −Leu −Trp plates to obtain double transformants. Single clone was picked from each transformation and cultured until OD600 = 1. Pellets of yeast cells were collected by centrifugation, washed 3 times and resuspended in water and plated in a dilution series of 10 to 1000 times by pipeting 5 μl per spot onto Histidine selection plates containing 10 mM 3-AT.
mRNA analysis
Total RNA was isolated using TRIzol (Invitrogen) from mixed stage worms cultured under identical conditions. 5 ug of total RNA was reverse transcribed into cDNA using an oligo dT primer (Invitrogen). Primers (F(YJ8377): 5’AATACAGAGGAAGCGGCATGAG; R(YJ8378): 5’CGGAAATCCCGTGGATAATG) were used to specifically detect DLK-1S transcript; ama-1 was used as internal control. To determine the 3’ ends of DLK-1L and DLK-1S, we used the 3’RACE kit (Invitrogen) and nestED PCR with the following primers: for DLK-1L, F1(YJ8379): 5’CAGAGGAAGCGGCATGAG; for DLK-1S, F2(YJ8380): 5’GAGCAGTGGCACAATCAGAAC. We obtained two DNA fragments and confirmed that their sequences corresponded to the two isoforms of DLK-1. We also analyzed two cDNA clones provided by Yuji Kohara (National Institute of Genetics, Mishima, Japan); yk826d12 contained 3’ sequences and UTR matching DLK-1S, and yk674b2 contained 3’ sequences and UTR matching DLK-1L. Northern blotting was done following the protocol as described (Bagga et al., 2005). ~60 μg total RNA from mixed stage N2 was run for northern blot. Probes were made using the Prime-It II Random Primer Labeling Kit (Stratagene, #300385) with a template containing 1.65kb cDNA fragment that covered the entire common region for DLK-1L and DLK-1S.
DNA constructs and generation of transgenes
All DNA expression constructs were made using Gateway cloning technology (Invitrogen). Sequences of the final clones were confirmed. The information for each construct is in Supplemental Table 2. The primer sequences are included in supplemental materials or available upon request.
Transgenic animals were generated following standard procedures (Mello et al., 1991). In general, plasmid DNAs of interest were used at 1-50 ng/μl with the co-injection marker Pttx-3-RFP at 50 ng/μl. For each construct, 3 to 10 independent transgenic lines were analyzed. Supplemental Table 1 lists the genotypes and DNA constructs for the transgenes. Mos-SCI transgenic worms were generated at the Ch II ttTi5606 site as described (Frokjaer-Jensen et al., 2008). The rgef-1 promoter, dlk-1L/S cDNA and unc-54 3’UTR were recombined into pCFJ150 by 3 way LR reactions to generate Prgef-1-DLK-1L(pCZGY1705) and Prgef-1-DLK-1S(pCZGY1704) Mos-SCI plasmids. These plasmids were injected into EG6699 (ttTi5605 II; unc-119(ed3) III; oxEx1578) to generate single copy insertion. Primers (F (YJ8987): 5’GGAGTTCGGACAGAAAGAAG3’ and R (YJ8988): 5’AGCCATTCAAGTTCGGAGATAG3’) were used to distinguish Mos-SCI dlk-1 cDNA from genomic dlk-1.
Supplementary Material
Highlights.
The DLK-1 short isoform acts as an endogenous inhibitor of DLK-1 function.
A novel conserved C-terminal hexapeptide is required for DLK-1 activation.
Ca2+ regulates DLK-1 isoform-specific interactions.
Human MAP3K13 contains an identical hexapeptide and complements DLK-1 function in vivo.
Acknowledgments
We thank X. Huang, Y. Dai, K. Nakata and X.-M. Wang for initial identification of dlk-1 alleles, Y. Kohara for cDNAs, W. Xiong and U. Mueller for advice on cell culture, A. Pasquinelli for her generosity in sharing equipment and lab space, and Z. Kai, E. Finnegan, J. Broughton for their time, help and advice for the Northern blotting experiment. We thank A.D. Chisholm for advice on data interpretation, and our lab members for discussions. The dlk-1(tm4024) mutation was provided by Dr. S. Mitani (the Japanese National Bioresource Project). D.Y. was an Associate of the Howard Hughes Medical Institute, and is now supported by K99/R00 award K99NS076646. Y.J. is an Investigator of the Howard Hughes Medical Institute. This work was also supported by NIH R01 NS035546 to Y.J; and R01 NS057317 to Andrew D. Chisholm and Y.J.
Footnotes
Contributions
D.Y. and Y.J. designed the experiments. D.Y. performed the experiments. D.Y. and Y.J. interpreted the data and wrote the manuscript.
Competing financial interests
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrams B, Grill B, Huang X, Jin Y. Cellular and molecular determinants targeting the Caenorhabditis elegans PHR protein RPM-1 to perisynaptic regions. Dev Dyn. 2008;237:630–639. doi: 10.1002/dvdy.21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagga S, Bracht J, Hunter S, Massirer K, Holtz J, Eachus R, Pasquinelli AE. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell. 2005;122:553–563. doi: 10.1016/j.cell.2005.07.031. [DOI] [PubMed] [Google Scholar]
- Barron KD. The axotomy response. J Neurol Sci. 2004;220:119–121. doi: 10.1016/j.jns.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Blouin R, Beaudoin J, Bergeron P, Nadeau A, Grondin G. Cell-specific expression of the ZPK gene in adult mouse tissues. DNA Cell Biol. 1996;15:631–642. doi: 10.1089/dna.1996.15.631. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch’ng Q, Williams L, Lie YS, Sym M, Whangbo J, Kenyon C. Identification of genes that regulate a left-right asymmetric neuronal migration in Caenorhabditis elegans. Genetics. 2003;164:1355–1367. doi: 10.1093/genetics/164.4.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LH, Stratton MM, Lee IH, Rosenberg OS, Levitz J, Mandell DJ, Kortemme T, Groves JT, Schulman H, Kuriyan J. A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin-dependent kinase II holoenzyme. Cell. 2011;146:732–745. doi: 10.1016/j.cell.2011.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Chen L, Wang Z, Ghosh-Roy A, Hubert T, Yan D, O’Rourke S, Bowerman B, Wu Z, Jin Y, Chisholm AD. Axon regeneration pathways identified by systematic genetic screening in C. elegans. Neuron. 2011;71:1043–1057. doi: 10.1016/j.neuron.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong H, Vikis HG, Guan KL. Mechanisms of regulating the Raf kinase family. Cell Signal. 2003;15:463–469. doi: 10.1016/s0898-6568(02)00139-0. [DOI] [PubMed] [Google Scholar]
- Collins CA, Wairkar YP, Johnson SL, DiAntonio A. Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron. 2006;51:57–69. doi: 10.1016/j.neuron.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Craig EA, Stevens MV, Vaillancourt RR, Camenisch TD. MAP3Ks as central regulators of cell fate during development. Dev Dyn. 2008;237:3102–3114. doi: 10.1002/dvdy.21750. [DOI] [PubMed] [Google Scholar]
- Dickson HM, Zurawski J, Zhang H, Turner DL, Vojtek AB. POSH is an intracellular signal transducer for the axon outgrowth inhibitor Nogo66. J Neurosci. 2010;30:13319–13325. doi: 10.1523/JNEUROSCI.1324-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto K, Kawauchi T, Osawa M, Tabata H, Nakajima K. Role of dual leucine zipper-bearing kinase (DLK/MUK/ZPK) in axonal growth. Neurosci Res. 2010;66:37–45. doi: 10.1016/j.neures.2009.09.1708. [DOI] [PubMed] [Google Scholar]
- Fantl WJ, Muslin AJ, Kikuchi A, Martin JA, MacNicol AM, Gross RW, Williams LT. Activation of Raf-1 by 14-3-3 proteins. Nature. 1994;371:612–614. doi: 10.1038/371612a0. [DOI] [PubMed] [Google Scholar]
- Freed E, Symons M, Macdonald SG, McCormick F, Ruggieri R. Binding of 14-3-3 proteins to the protein kinase Raf and effects on its activation. Science. 1994;265:1713–1716. doi: 10.1126/science.8085158. [DOI] [PubMed] [Google Scholar]
- Frokjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen SP, Grunnet M, Jorgensen EM. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet. 2008;40:1375–1383. doi: 10.1038/ng.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuyama K, Yoshida M, Yamashita A, Deyama T, Baba M, Suzuki A, Mohri H, Ikezawa Z, Nakajima H, Hirai S, Ohno S. MAPK upstream kinase (MUK)-binding inhibitory protein, a negative regulator of MUK/dual leucine zipper-bearing kinase/leucine zipper protein kinase. J Biol Chem. 2000;275:21247–21254. doi: 10.1074/jbc.M001488200. [DOI] [PubMed] [Google Scholar]
- Ghosh AS, Wang B, Pozniak CD, Chen M, Watts RJ, Lewcock JW. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J Cell Biol. 2011;194:751–764. doi: 10.1083/jcb.201103153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh-Roy A, Wu Z, Goncharov A, Jin Y, Chisholm AD. Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J Neurosci. 2010;30:3175–3183. doi: 10.1523/JNEUROSCI.5464-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallam SJ, Jin Y. lin-14 regulates the timing of synaptic remodelling in Caenorhabditis elegans. Nature. 1998;395:78–82. doi: 10.1038/25757. [DOI] [PubMed] [Google Scholar]
- Hammarlund M, Nix P, Hauth L, Jorgensen EM, Bastiani M. Axon regeneration requires a conserved MAP kinase pathway. Science. 2009;323:802–806. doi: 10.1126/science.1165527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai S, Izawa M, Osada S, Spyrou G, Ohno S. Activation of the JNK pathway by distantly related protein kinases, MEKK and MUK. Oncogene. 1996;12:641–650. [PubMed] [Google Scholar]
- Holzman LB, Merritt SE, Fan G. Identification, molecular cloning, and characterization of dual leucine zipper bearing kinase. A novel serine/threonine protein kinase that defines a second subfamily of mixed lineage kinases. J Biol Chem. 1994;269:30808–30817. [PubMed] [Google Scholar]
- Horiuchi D, Collins CA, Bhat P, Barkus RV, Diantonio A, Saxton WM. Control of a kinesin-cargo linkage mechanism by JNK pathway kinases. Curr Biol. 2007;17:1313–1317. doi: 10.1016/j.cub.2007.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda A, Masaki M, Kozutsumi Y, Oka S, Kawasaki T. Identification and characterization of functional domains in a mixed lineage kinase LZK. FEBS Lett. 2001;488:190–195. doi: 10.1016/s0014-5793(00)02432-7. [DOI] [PubMed] [Google Scholar]
- Itoh A, Horiuchi M, Bannerman P, Pleasure D, Itoh T. Impaired regenerative response of primary sensory neurons in ZPK/DLK gene-trap mice. Biochem Biophys Res Commun. 2009;383:258–262. doi: 10.1016/j.bbrc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Itoh A, Horiuchi M, Wakayama K, Xu J, Bannerman P, Pleasure D, Itoh T. ZPK/DLK, a mitogen-activated protein kinase kinase kinase, is a critical mediator of programmed cell death of motoneurons. J Neurosci. 2011;31:7223–7228. doi: 10.1523/JNEUROSCI.5947-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Gereau RWt, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60:135–148. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr R, Lev-Ram V, Baird G, Vincent P, Tsien RY, Schafer WR. Optical imaging of calcium transients in neurons and pharyngeal muscle of C. elegans. Neuron. 2000;26:583–594. doi: 10.1016/s0896-6273(00)81196-4. [DOI] [PubMed] [Google Scholar]
- L’Allemain G. Deciphering the MAP kinase pathway. Prog Growth Factor Res. 1994;5:291–334. doi: 10.1016/0955-2235(94)90011-6. [DOI] [PubMed] [Google Scholar]
- Lee RY, Lobel L, Hengartner M, Horvitz HR, Avery L. Mutations in the alpha1 subunit of an L-type voltage-activated Ca2+ channel cause myotonia in Caenorhabditis elegans. EMBO J. 1997;16:6066–6076. doi: 10.1093/emboj/16.20.6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewcock JW, Genoud N, Lettieri K, Pfaff SL. The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron. 2007;56:604–620. doi: 10.1016/j.neuron.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Mata M, Merritt SE, Fan G, Yu GG, Holzman LB. Characterization of dual leucine zipper-bearing kinase, a mixed lineage kinase present in synaptic terminals whose phosphorylation state is regulated by membrane depolarization via calcineurin. J Biol Chem. 1996;271:16888–16896. doi: 10.1074/jbc.271.28.16888. [DOI] [PubMed] [Google Scholar]
- Meador WE, Means AR, Quiocho FA. Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures. Science. 1993;262:1718–1721. doi: 10.1126/science.8259515. [DOI] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielke K, Herdegen T. JNK and p38 stresskinases--degenerative effectors of signal-transduction-cascades in the nervous system. Prog Neurobiol. 2000;61:45–60. doi: 10.1016/s0301-0082(99)00042-8. [DOI] [PubMed] [Google Scholar]
- Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, DiAntonio A. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat Neurosci. 2009;12:387–389. doi: 10.1038/nn.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming GL, Wong ST, Henley J, Yuan XB, Song HJ, Spitzer NC, Poo MM. Adaptation in the chemotactic guidance of nerve growth cones. Nature. 2002;417:411–418. doi: 10.1038/nature745. [DOI] [PubMed] [Google Scholar]
- Mita H, Tsutsui J, Takekawa M, Witten EA, Saito H. Regulation of MTK1/MEKK4 kinase activity by its N-terminal autoinhibitory domain and GADD45 binding. Mol Cell Biol. 2002;22:4544–4555. doi: 10.1128/MCB.22.13.4544-4555.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake Z, Takekawa M, Ge Q, Saito H. Activation of MTK1/MEKK4 by GADD45 through induced N-C dissociation and dimerization-mediated trans autophosphorylation of the MTK1 kinase domain. Mol Cell Biol. 2007;27:2765–2776. doi: 10.1128/MCB.01435-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata K, Abrams B, Grill B, Goncharov A, Huang X, Chisholm AD, Jin Y. Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell. 2005;120:407–420. doi: 10.1016/j.cell.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Nihalani D, Merritt S, Holzman LB. Identification of structural and functional domains in mixed lineage kinase dual leucine zipper-bearing kinase required for complex formation and stress-activated protein kinase activation. J Biol Chem. 2000;275:7273–7279. doi: 10.1074/jbc.275.10.7273. [DOI] [PubMed] [Google Scholar]
- Ou CY, Poon VY, Maeder CI, Watanabe S, Lehrman EK, Fu AK, Park M, Fu WY, Jorgensen EM, Ip NY, Shen K. Two cyclin-dependent kinase pathways are essential for polarized trafficking of presynaptic components. Cell. 2010;141:846–858. doi: 10.1016/j.cell.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlson E, Hanz S, Ben-Yaakov K, Segal-Ruder Y, Seger R, Fainzilber M. Vimentin-dependent spatial translocation of an activated MAP kinase in injured nerve. Neuron. 2005;45:715–726. doi: 10.1016/j.neuron.2005.01.023. [DOI] [PubMed] [Google Scholar]
- Piper M, Anderson R, Dwivedy A, Weinl C, van Horck F, Leung KM, Cogill E, Holt C. Signaling mechanisms underlying Slit2-induced collapse of Xenopus retinal growth cones. Neuron. 2006;49:215–228. doi: 10.1016/j.neuron.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pumiglia K, Chow YH, Fabian J, Morrison D, Decker S, Jove R. Raf-1 N-terminal sequences necessary for Ras-Raf interaction and signal transduction. Mol Cell Biol. 1995;15:398–406. doi: 10.1128/mcb.15.1.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma H, Ikeda A, Oka S, Kozutsumi Y, Zanetta JP, Kawasaki T. Molecular cloning and functional expression of a cDNA encoding a new member of mixed lineage protein kinase from human brain. J Biol Chem. 1997;272:28622–28629. doi: 10.1074/jbc.272.45.28622. [DOI] [PubMed] [Google Scholar]
- Samuels IS, Saitta SC, Landreth GE. MAP’ing CNS development and cognition: an ERKsome process. Neuron. 2009;61:160–167. doi: 10.1016/j.neuron.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer AM, Hadwiger GD, Nonet ML. rpm-1, a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron. 2000;26:345–356. doi: 10.1016/s0896-6273(00)81168-x. [DOI] [PubMed] [Google Scholar]
- Shin JE, Cho Y, Beirowski B, Milbrandt J, Cavalli V, Diantonio A. Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron. 2012;74:1015–1022. doi: 10.1016/j.neuron.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling DP, Stys PK. Mechanisms of axonal injury: internodal nanocomplexes and calcium deregulation. Trends Mol Med. 2010;16:160–170. doi: 10.1016/j.molmed.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S, Unsicker K. ERK and cell death: ERK1/2 in neuronal death. Febs J. 2010;277:22–29. doi: 10.1111/j.1742-4658.2009.07367.x. [DOI] [PubMed] [Google Scholar]
- Takekawa M, Saito H. A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAPKKK. Cell. 1998;95:521–530. doi: 10.1016/s0092-8674(00)81619-0. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Wang Z, Jin Y. Genetic dissection of axon regeneration. Curr Opin Neurobiol. 2011;21:189–196. doi: 10.1016/j.conb.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science. 1998;281:1671–1674. doi: 10.1126/science.281.5383.1671. [DOI] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW. Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nat Neurosci. 2001;4:151–158. doi: 10.1038/83976. [DOI] [PubMed] [Google Scholar]
- Wu Z, Ghosh-Roy A, Yanik MF, Zhang JZ, Jin Y, Chisholm AD. Caenorhabditis elegans neuronal regeneration is influenced by life stage, ephrin signaling, and synaptic branching. Proc Natl Acad Sci U S A. 2007;104:15132–15137. doi: 10.1073/pnas.0707001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing H, Kornfeld K, Muslin AJ. The protein kinase KSR interacts with 14-3-3 protein and Raf. Curr Biol. 1997;7:294–300. doi: 10.1016/s0960-9822(06)00152-7. [DOI] [PubMed] [Google Scholar]
- Xiong X, Collins CA. A conditioning lesion protects axons from degeneration via the Wallenda/DLK MAP kinase signaling cascade. J Neurosci. 2012;32:610–615. doi: 10.1523/JNEUROSCI.3586-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Wang X, Ewanek R, Bhat P, Diantonio A, Collins CA. Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J Cell Biol. 2010;191:211–223. doi: 10.1083/jcb.201006039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Wu Z, Chisholm AD, Jin Y. The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell. 2009;138:1005–1018. doi: 10.1016/j.cell.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E, Schulman H. Structural examination of autoregulation of multifunctional calcium/calmodulin-dependent protein kinase II. J Biol Chem. 1999;274:26199–26208. doi: 10.1074/jbc.274.37.26199. [DOI] [PubMed] [Google Scholar]
- Zhen M, Huang X, Bamber B, Jin Y. Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron. 2000;26:331–343. doi: 10.1016/s0896-6273(00)81167-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.