

Abstract
Impaired clearance of amyloid-β (Aβ) is a major pathogenic event for Alzheimer's disease (AD). Aβ depositions in brain parenchyma as senile plaques and along cerebrovasculature as cerebral amyloid angiopathy (CAA) are hallmarks of AD. A major pathway that mediates brain Aβ clearance is the cerebrovascular system where Aβ is eliminated through the blood–brain barrier (BBB) and/or degraded by cerebrovascular cells along the interstitial fluid drainage pathway. An Aβ clearance receptor, the low-density lipoprotein receptor-related protein 1 (LRP1), is abundantly expressed in cerebrovasculature, in particular in vascular smooth muscle cells. Previous studies have indicated a role of LRP1 in endothelial cells in transcytosing Aβ out of the brain across the BBB; however, whether this represents a significant pathway for brain Aβ clearance remains controversial. Here, we demonstrate that Aβ can be cleared locally in the cerebrovasculature by an LRP1-dependent endocytic pathway in smooth muscle cells. The uptake and degradation of both endogenous and exogenous Aβ were significantly reduced in LRP1-suppressed human brain vascular smooth muscle cells. Conditional deletion of Lrp1 in vascular smooth muscle cell in amyloid model APP/PS1 mice accelerated brain Aβ accumulation and exacerbated Aβ deposition as amyloid plaques and CAA without affecting Aβ production. Our results demonstrate that LRP1 is a major Aβ clearance receptor in cerebral vascular smooth muscle cell and a disturbance of this pathway contributes to Aβ accumulation. These studies establish critical functions of the cerebrovasculature system in Aβ metabolism and identify a new pathway involved in the pathogenesis of both AD and CAA.
Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disorder and the most common cause of dementia in elderly population (Hardy and Selkoe, 2002; Goedert and Spillantini, 2006). The amyloid-β (Aβ) peptides are cleaved from amyloid precursor protein (Zheng and Koo, 2011). The imbalance between Aβ generation and clearance induces its accumulation, aggregation, and deposition in the brain, which is thought to be an early and main pathogenic event in AD (Hardy and Selkoe, 2002; Goedert and Spillantini, 2006; Koffie et al., 2011). Most cases of AD are sporadic and developed after the age of 65 years. Recent studies have indicated an overall impairment in Aβ clearance, rather than Aβ overproduction, in sporadic AD cases (Mawuenyega et al., 2010). Pathological studies of AD cases show that Aβ deposits in the vascular smooth muscle cell layer of the cerebral blood vessel as cerebral amyloid angiopathy (CAA) as well as senile plaques (Rensink et al., 2003; Revesz et al., 2003). At least, 83% of autopsy-confirmed AD patients have some degrees of CAA (Ellis et al., 1996). CAA is a major trigger of intracranial hemorrhage (Ellis et al., 1996; Maia et al., 2007) and progressive dementia in the elderly (Rensink et al., 2003; Revesz et al., 2003). Aβ drains through interstitial fluid (ISF) drainage pathways along the vascular smooth muscle cell layer of small and medium-sized arteries, arterioles of the cerebral cortex, and leptomeninges (Weller et al., 1998; Revesz et al., 2003; Thal et al., 2008), where CAA is abundantly detected. Despite increasing interests in understanding the mechanism of Aβ clearance from the brain, it remains unclear whether Aβ can be cleared locally by the vascular smooth muscle cells while Aβ flows along the ISF drainage pathway.
The low-density lipoprotein receptor-related protein 1 (LRP1), which is a highly efficient endocytic receptor that binds multiple ligands (Herz and Strickland, 2001; Bu, 2009), mediates Aβ clearance in the brain (Shibata et al., 2000; Deane et al., 2004). LRP1 is abundantly expressed in several different cell types in the brain including neurons (Qiu et al., 1999; Kanekiyo et al., 2011), glia (Wyss-Coray et al., 2003), and vascular smooth muscle cells (Urmoneit et al., 1997; Wilhelmus et al., 2007; Ruzali et al., 2012). Several Aβ-binding proteins including apolipoprotein E (apoE), α2-macroglobulin, and receptor-associated protein (RAP) are ligands of LRP1 (Herz and Strickland, 2001; Bu et al., 2006; Kanekiyo and Bu, 2009). Previous studies have suggested a role for LRP1 in mediating Aβ transcytosis across the blood–brain barrier (Shibata et al., 2000); however, whether LRP1 is sufficiently expressed in the endothelial cells to mediate Aβ transcytosis remains controversial. Given LRP1 is highly expressed in vascular smooth muscle cell layer of the cerebral blood vessels (Zlokovic, 2011) and its expression is vastly decreased in the vasculature of AD brains with CAA (Bell et al., 2009), we sought to examine whether LRP1 in vascular smooth muscle cells mediates local clearance of Aβ, and whether a disturbance of this pathway would exacerbate the formation of CAA.
Materials and Methods
Reagents.
Aβ40, Aβ42, 5(6)-carboxyfluorescein (FAM)-Aβ40, and FAM-Aβ42 were purchased from AnaSpec. Dry peptide was pretreated with neat trifluoroacetic acid, distilled under nitrogen, washed with 1,1,1,3,3,3-hexafluoro-2-propanol, distilled under nitrogen, and stored at −20°C. Aβ peptides were freshly dissolved in dimethyl sulfoxide at 200 μm for each experiment. Rabbit polyclonal anti-LRP1 antibody was produced in our laboratory (Liu et al., 2007). Anti-APP C-terminal, sAPPα, and sAPPβ antibodies were purchased from IBL. DAPT, pepstatin, leupeptin, and E-64d were purchased from Sigma-Aldrich.
Cell culture and LRP1 knockdown by siRNA.
Human brain vascular smooth muscle cells (HBVSMCs) were purchased from ScienCell and cultured under standard culture conditions according to the manufacturer's protocol. Knockdown of LRP1 was performed as described previously (Li et al., 2003). Single-stranded, LRP1-specific, sense and antisense RNA oligonucleotides were synthesized by Ambion. Cells were transfected with siRNA (100 nm) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's specifications and used for analysis 48 h after transfection.
Animals and tissue preparation.
Vascular smooth muscle cell-specific LRP1-KO mice (smLrp1−/−) were generated by breeding the Lrp1 floxed mice (Rohlmann et al., 1998) with sm22α-driven Cre recombinase mice (Holtwick et al., 2002). Littermates of male smLrp1−/− mice (LRP1flox/flox, sm22α-Cre+/−) or controls (LRP1flox/flox, sm22α-Cre−/−) were used. Furthermore, amyloid model APP/PS1 mice (Borchelt et al., 1997) were crossed with smLrp1−/− mice. Littermates of male APP/PS1-Tg mice (APP/PS1) and APP/PS1-Tg mice lacking LRP1 in vascular smooth muscle cells (APP/PS1; smLrp1−/−) were used for analysis. After perfusion with PBS, brain tissues were dissected and kept frozen at −80°C until further analysis. Some brain tissues were fixed in 10% neutralized formalin for histological analysis. All animal procedures were approved by the Animal Study Committee at Mayo Clinic and in accordance with the regulations of the American Association for the Accreditation of Laboratory Animal Care.
Isolation of brain microvessels.
Brain microvessels were isolated as previously described (Shinohara et al., 2010). In brief, mouse brains were homogenized in DMEM and resuspended with 17% dextran (Sigma-Aldrich). Following centrifugation at 10,000 × g for 15 min at 4°C, the pellets were suspended with DMEM containing 10% fetal bovine serum (FBS) and filtered through sterilized glass beads (425–600 μm, acid-washed; Sigma-Aldrich) on a 70 μm nylon cell strainer (BD Biosciences). After transfer onto a plastic dish with DMEM containing 10% FBS, dissociated vessels from glass beads were collected and lysed in RIPA buffer.
Confocal microscopy.
Cells were cultured on eight-well slides (Lab-Tek II Chamber SlideTM System; Nalge Nunc International) at 37°C for at least 24 h before experiments. After incubation with FAM-Aβ40 or FAM-Aβ42 (500 nm) at 37°C for 8 h, fluorescence associated with Aβ was observed by confocal laser-scanning fluorescence microscopy (model LSM 510 invert; Carl Zeiss). LysoTracker (50 nm; Invitrogen) was added 30 min before confocal imaging. Isolated mouse leptomeningeal arteries were placed on coverslip, fixed in 4% paraformaldehyde, permeabilized with PBS containing 0.2% Triton X-100, and incubated with rabbit polyclonal anti-LRP1 antibody in PBS containing 1% bovine serum albumin at 4°C for overnight followed by incubation with a goat anti-rabbit Alexa Fluor 488 secondary antibody (Invitrogen) and Alexa Fluor 568 phalloidin (F-actin marker; Invitrogen) for confocal microscopy analysis.
Fluorescence-activated cell sorter-based internalization assays.
Aβ internalization was analyzed by fluorescence-activated cell sorter (FACS) as previously described (Kanekiyo et al., 2011). Briefly, cells were incubated with FAM-Aβ40 (500 nm) or FAM-Aβ42 (500 nm) at 37°C for 24 h in DMEM with 10% FBS for internalization assay. Cells were removed from the plate using Cell Dissociation Solution (Sigma-Aldrich) and incubated with Pronase (0.5 mg/ml; Boehringer Mannheim) at 4°C for 20 min. Cells were then washed and resuspended in PBS containing 1.5% FBS, 1% sodium azide, and 1% paraformaldehyde. Cells (1 × 104) from each sample were analyzed for fluorescence on a BD FACSCalibur machine (BD Biosciences). Unstained cells without any exposure to fluorescence were used as a control for background fluorescence.
Aβ ELISA.
For measurements of endogenous Aβ from mouse brain, samples were homogenized in diethylamine (Shinohara et al., 2010). For measurements of Aβ in APP/PS1 mouse brain, samples were sequentially homogenized in Tris-buffered saline (TBS), TBS buffer containing 1% Triton X-100 and 5 m guanidine in 50 mm Tris-HCl, pH 8.0 (Youmans et al., 2011). The Aβ levels in culture media from HBVSMCs were directly analyzed. Cell-associated Aβ levels were analyzed after dissolved in guanidine. To assess human Aβ40 and human Aβ42, mouse monoclonal antibodies 2G3 recognizing human/mouse Aβ33–40 and 21F12 recognizing human/mouse Aβ33–42, respectively, were used as coating antibodies and a biotinylated human Aβ-specific antibody 3D6 recognizing human Aβ1–6 was used for detection. Mouse Aβ40 was assessed using a 266 antibody as a coating antibody and a biotinylated 2G3 antibody recognizing human/mouse Aβ13–28 as the detecting antibody. Mouse Aβ42 was assessed using a 266 antibody as a coating antibody and a biotinylated 21F12 antibody as the detecting antibody. After incubation with horseradish peroxidase streptavidin–streptavidin (Vector), the plate was developed with tetramethylbenzidine (Sigma-Aldrich) and read at 650 nm with a Bio-Tek 600 plate reader (Nalge Nunc International). Those anti-Aβ antibodies were kindly provided by Dr. Ronald B. DeMattos (Lilly Research Laboratories, Indianapolis, IN), and the ELISA systems have been described in previous publications (Zerbinatti et al., 2004; Liu et al., 2010).
Immunohistochemical imaging and image processing.
Paraffin-embedded sections were immunostained using pan-Aβ Ab 33.1.1 (human Aβ1–16 specific) or anti-glial fibrillary acidic protein (GFAP) (Sigma-Aldrich), and visualized through the Dako Envision Plus visualization system (Dako) (Chakrabarty et al., 2010). Immunohistochemically stained sections were captured using the Scanscope XT image scanner (Aperio Technologies) and analyzed using the ImageScope program (Chakrabarty et al., 2010). The final images and layouts were created using Photoshop CS2 (Adobe). Immunostained total Aβ plaque burdens in the cortex and CAA formation on leptomeningeal arteries were calculated using the Positive Pixel Count program available with the Imagescope software (Aperio Technologies). All of the above analyses were performed in a blinded manner.
Western blotting.
Samples were lysed in PBS containing 1% Triton X-100 and protease inhibitor mixture from Roche or RIPA buffer. Protein concentration was determined in each sample using a Protein Assay kit (Bio-Rad). An equal amount of sample protein was used for SDS-PAGE. Immunoreactive bands were detected and quantified using Odyssey Infrared Imaging System (LI-COR Biosciences).
Reverse transcription and PCR.
Total RNA was isolated from tissues or cells using RNeasy Mini Kit (QIAGEN) and subjected to DNase I digestion to remove contaminating genomic DNA. Total RNA was dissolved in nuclease-free water and stored at −80°C. Reverse transcription was performed using a SuperScript II RNase H-reverse transcriptase (Invitrogen), and the reaction mix was subjected to quantitative real-time PCR to detect levels of the corresponding mouse actin, neprilysin, IDE, MMP2, and MMP9. The set of actin primers (QIAGEN) was used as an internal control for each specific gene amplification. The relative levels of expression were quantified and analyzed by using Bio-Rad iCycler iQ software (Bio-Rad). The real-time value for each sample was averaged and compared using the CT method, where the amount of target RNA (2−ΔΔCT) was normalized to the endogenous actin reference (ΔCT) and related to the amount of target gene in tissue cells, which was set as the calibrator at 1.0. The primers used to amplify target genes by RT-PCR and quantitative PCR were as follows: mouse neprilysin-F (5′-GCA GCC TCA GCC GAA ACT AC-3′), mouse neprilysin-R (5′-CAC CGT CTC CAT GTT GCA GT-3′); mouse insulin-degrading enzyme (IDE)-F (5′-ACT AAC CTG GTG GTG AAG-3′), mouse IDE-R (5′-GGT CTG GTA TGG GAA ATG-3′); mouse metalloproteinase 2 (MMP2)-F (5′-GTC GCC CCT AAA ACA GAC AA-3′), mouse MMP2-R (5′-GGT CTC GAT GGT GTT CTG GT-3′); and mouse MMP9-F (5′-CGT CGT GAT CCC CAC TTA CT-3′), mouse MMP9-R (5′-AAC ACA CAG GGT TTG CCT TC-3′).
Statistical analysis.
All quantified data represent an average of samples. Statistical significance was determined by two-tailed paired Student's t test, and p < 0.05 was considered significant.
Results
LRP1 mediates Aβ metabolism in human brain vascular smooth muscle cell
To determine the roles of LRP1 in Aβ metabolism in vascular smooth muscle cells, we first examined whether LRP1 knockdown by siRNA (Fig. 1A) affects the endogenous Aβ levels produced by HBVSMCs, which express an abundance of APP (Fig. 1B) and produce Aβ (Fig. 1C). ELISA revealed that LRP1 knockdown significantly increased endogenous Aβ40 and Aβ42 levels in the media compared with controls (Fig. 1D) without affecting APP processing (Fig. 1E). We sought to further assess whether LRP1 mediates cellular Aβ uptake and degradation by incubating LRP1-suppressed HBVSMCs with FAM-labeled Aβ40 and Aβ42. The amounts of cell-associated Aβ were detected by FACS (Fig. 2A,B). The results showed that cell-associated Aβ40 and Aβ42 levels in HBVSMCs were increased in a time-dependent manner, and that knockdown of LRP1 expression significantly decreased Aβ uptake following 4 and 8 h of Aβ incubation (Fig. 2A,B). Similar results were obtained using unlabeled Aβ and detection by ELISA (Fig. 2D). Consistent with our FACS and ELISA results, we observed less internalized Aβ in LRP1-suppressed HBVSMCs by confocal microscopy (Fig. 2C). As internalized Aβ was detected primarily in the lysosomal compartments (Fig. 2C), we examined whether Aβ is degraded via the lysosomal pathway. HBVSMCs were incubated with Aβ40 for 1 h, followed by an additional incubation of 4 h in the presence or absence of lysosomal inhibitors without Aβ. When disappearance of cell-associated Aβ detected by ELISA was defined as degraded Aβ, LRP1 deletion, as well as lysosomal inhibitors, significantly suppressed Aβ degradation in HBVSMCs (Fig. 2E). However, Aβ degradation was not suppressed by lysosomal inhibitors upon LRP1 deletion (Fig. 2E), suggesting that Aβ clearance in HBVSMCs by the lysosomes depends on the presence of LRP1. These results suggest that LRP1 likely mediates Aβ clearance through the lysosomal degradation pathway in HBVSMCs.
Figure 1.

LRP1 mediates Aβ clearance in HBVSMCs. A, HBVSMCs were transfected with control or LRP1-siRNA and used for analysis 48 h after transfection. Western blotting showed that LRP1 expression was suppressed by LRP1-siRNA. B, HBVSMCs were incubated with or without γ-secretase inhibitor DAPT (10 μm) for 24 h, and the levels of full-length APP (FL-APP) and C-terminal fragments of APP (CTFs) were analyzed by Western blot. C, HBVSMCs were incubated for 4 d in the absence or presence of DAPT (10 μm), and the concentrations of Aβ40 in the media were analyzed by ELISA. D, Control and LRP1-suppressed HBVSMCs were incubated for 4 d, and the concentrations of endogenous Aβ40 and Aβ42 in the media were quantified by ELISA. E, To examine whether LRP1 knockdown in HBVSMCs affects APP processing, CTFs were analyzed by Western blot and quantified. Data are plotted as mean ± SD (n = 3). **p < 0.01. N.S., Not significant.
Figure 2.
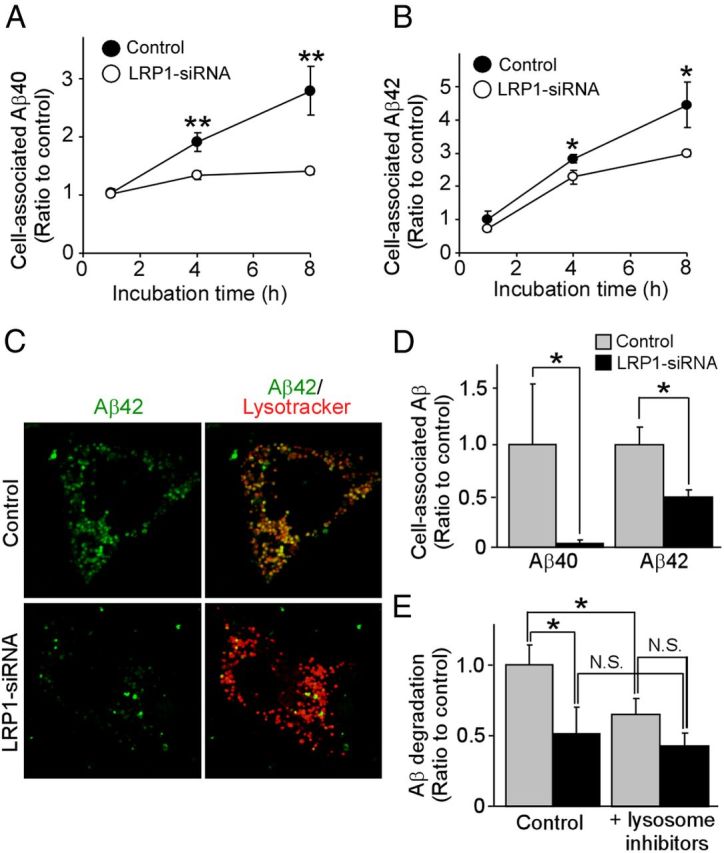
LRP1 mediates Aβ uptake and cellular clearance in HBVSMCs. A, B, The levels of cell-internalized Aβ40 (A) and Aβ42 (B) were analyzed by FACS in control and LRP1-suppressed HBVSMCs after incubation with FAM-labeled Aβ (500 nm) for 1, 4, and 8 h. C, Subcellular localization of internalized FAM-Aβ42 and its colocalization with lysosomal marker were observed by confocal microscopy in control and LRP1-suppressed HBVSMCs. D, Control and LRP1-suppressed HBVSMCs were incubated with unlabeled Aβ40 or Aβ42 (10 nm each) for 4 h, and cell-associated Aβ was quantified by ELISA. E, Control and LRP1-suppressed HBVSMCs were incubated with 1 μm Aβ40 (1 μm) for 1 h followed by additional incubation for 4 h in the presence or absence of lysosomal inhibitors (pepstatin, 1 μm; leupeptin, 50 μm; E-64d, 15 μm) without Aβ. Disappearance of cell-associated Aβ detected by ELISA was defined as degraded Aβ. Data are plotted as mean ± SD (n = 3). *p < 0.05; **p < 0.01. N.S., Not significant.
LRP1 deletion in vascular smooth muscle cells in mice leads to Aβ accumulation
To examine the effect of LRP1 deletion in vascular smooth muscle cells on Aβ clearance in vivo, we generated vascular smooth muscle cell-specific LRP1-KO mice (smLrp1−/−) by breeding the Lrp1 floxed mice (Rohlmann et al., 1998) with sm22α-driven Cre recombinase mice (Holtwick et al., 2002). The leptomeningeal artery isolated from 4-month-old smLrp1−/− (Lrp1flox/flox, sm22α-Cre+/−) mice or control (Lrp1flox/flox, sm22α-Cre−/−) mice were stained with LRP1-antibody and phalloidin (F-actin marker) and analyzed by confocal microscopy. Immunostaining clearly showed that the expression of LRP1 was deleted in leptomeningeal artery from smLrp1−/− mice (Fig. 3A). Western blotting also confirmed that LRP1 level in isolated brain vessels from smLrp1−/− mice was mostly deleted, while there was no difference in the LRP1 levels in cortex between control and smLrp1−/− mice (Fig. 3B). ELISA showed that endogenous levels of both Aβ40 and Aβ42 are significantly increased in the brains of smLrp1−/− mice compared with their littermate controls (Fig. 3C). These results indicate that LRP1 in vascular smooth muscle cells mediates Aβ metabolism, and a disturbance of this pathway is sufficient to induce Aβ accumulation in the brain.
Figure 3.

LRP1 deletion in vascular smooth muscle cells in mouse brains leads to increased endogenous Aβ levels. A, Leptomeningeal arteries isolated from control and smLrp1−/− mice were stained with LRP1 antibody and phalloidin (F-actin marker). Scale bar, 50 μm. B, LRP1 expression in isolated vessels and cortex from control and smLrp1−/− mice (4–5 months of age) detected by Western blot. C, Mouse endogenous Aβ40 and Aβ42 levels in the brains of control and smLrp1−/− mice analyzed by ELISA. Data are plotted as mean ± SEM (n = 6). *p < 0.05.
LRP1 deletion in vascular smooth muscle cells in amyloid model mice exacerbates Aβ deposition
To further confirm the role for LRP1 in vascular smooth muscle cells, Aβ metabolism, we crossed smLrp1−/− mice with amyloid model APP/PS1 mice (Borchelt et al., 1997). We compared amyloid plaque deposition and CAA by immunostaining for Aβ between littermates of APP/PS1 mice without (APP/PS1) or with LRP1 deletion (APP/PS1; smLrp1−/−) in vascular smooth muscle cells. We found that Aβ deposition in the brains of APP/PS1; smLrp1−/− mice was significantly higher than that of control APP/PS1 mice at 13–14 months of age (Fig. 4A,B). In addition to the cortical parenchyma, Aβ deposition manifested in cortical vessels as CAA, which was also increased in the APP/PS1; smLrp1−/− mice (Fig. 4C,D). Consistent with increased Aβ deposition, the concentrations of insoluble Aβ40 and Aβ42 in the guanidine fractions were significantly higher in the APP/PS1; smLrp1−/− mice (Fig. 4E). The effects of LRP1 deletion on Aβ accumulation is likely age dependent as we found no significant differences in the concentrations of insoluble Aβ40 (582 ± 68 vs 558 ± 99 pg/mg protein) and Aβ42 (4.83 ± 0.71 vs 4.07 ± 1.40 ng/mg protein) (mean ± SEM; n = 4) between control APP/PS1 and APP/PS1; smLrp1−/− mice at 3 months of age.
Figure 4.

LRP1 deletion in vascular smooth muscle cells in APP/PS1 amyloid model mice exacerbates Aβ deposition as amyloid plaques and CAA. A, Aβ depositions in brain sections from control APP/PS1 and APP/PS1; smLrp1−/− mice (13–14 months of age) were immunostained with a pan-Aβ antibody. Scale bar, 1 mm. B, Immunostaining of Aβ deposition along leptomeningeal arteries as CAA in control APP/PS1 and APP/PS1; smLrp1−/− mice. Scale bar, 20 μm. C, D, Amyloid plaque burdens in cortex (C) and CAA formation in leptomeningeal arteries (D) were quantified after scanning Aβ immunostaining by the Positive Pixel Count program (Aperio Technologies). E, The concentrations of insoluble Aβ40 and Aβ42 levels in the cortex from control APP/PS1 and APP/PS1; smLrp1−/− mice analyzed by ELISA at 13–14 months of age. Data are plotted as mean ± SEM (n = 5). *p < 0.05; **p < 0.01.
To examine whether LRP1 deletion in vascular smooth muscle cells affects APP processing, APP derivatives in both APP/PS1 and APP/PS1; smLrp1−/− mice at 13–14 months of age were analyzed by Western blotting (Fig. 5A). There were no significant differences in the level of full-length APP (Fig. 5B), CTFα (Fig. 5C), CTFβ (Fig. 5D), sAPPα (Fig. 5E), and sAPPβ (Fig. 5F) between APP/PS1 and APP/PS1; smLrp1−/− mice. Next, we analyzed the mRNA levels of neprilysin (Fig. 6A), IDE (Fig. 6B), MMP2 (Fig. 6C), and MMP9 (Fig. 6D), which are major Aβ degrading enzymes in the brain, in the cortex of control and smLrp1−/− mice. RT-PCR showed no significant differences in the mRNA levels of these enzymes in our experimental mice. Together, these results indicate that LRP1 deletion in vascular smooth muscle cells exacerbates Aβ deposition through a disturbance of lysosomal-mediated Aβ clearance along brain vasculature without affecting APP processing or the expression levels of major Aβ-degrading enzymes in amyloid model mice.
Figure 5.

LRP1 deletion in vascular smooth muscle cells does not affect APP processing in APP/PS1 mice. APP and its processing products in both APP/PS1 and APP/PS1; smLrp1−/− mice at the age of 13–14 months were analyzed by Western blot (A). There were no significant differences in the levels of full-length APP (B), CTFα (C), CTFβ (D), sAPPα (E), and sAPPβ (F) between the two groups of mice. Data are plotted as mean ± SEM (n = 5). N.S., Not significant.
Figure 6.

LRP1 deletion in vascular smooth muscle cells does not affect the expression levels of major Aβ-degrading enzymes. The mRNA levels of neprilysin (A), IDE (B), MMP2 (C), and MMP9 (D) in the cortex from control and smLrp1−/− mice were quantified by RT-PCR at the age of 12 months. Data are plotted as mean ± SEM (n = 6). N.S., Not significant.
LRP1 deletion in vascular smooth muscle cells in amyloid model mice exacerbates astrogliosis
Abnormal activation of astrocytes is observed in the brains of AD patients and APP transgenic mouse models (Wyss-Coray, 2006). To examine the extent of astrogliosis, brain sections were stained with anti-GFAP antibody to quantify the extent of astrogliosis (Fig. 7A). The immunostaining for GFAP clearly demonstrated that activation of astrocytes was enhanced in the brains of APP/PS1; smLrp1−/− mice compared with those of APP/PS1 mice at the age of 1 year (Fig. 7A). When GFAP levels in cortex (Fig. 7B) of those mice were analyzed by Western blotting, GFAP levels were also significantly increased in APP/PS1; smLrp1−/− mice. These findings demonstrate that the exacerbated Aβ deposition by LRP1 deletion in vascular smooth muscle cells leads to accelerate activation of astrocytes.
Figure 7.
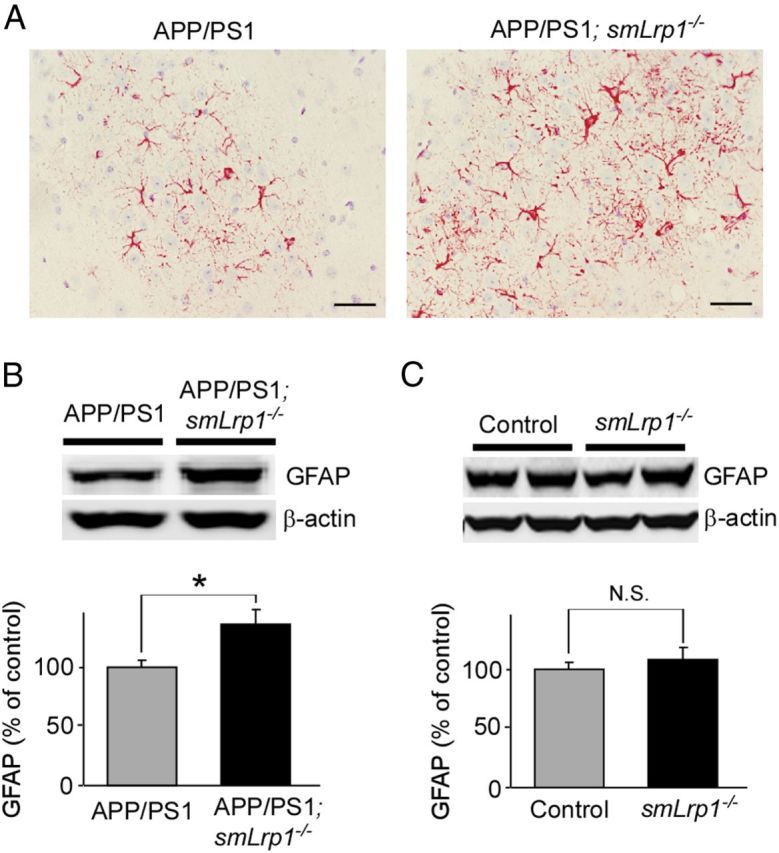
LRP1 deletion in vascular smooth muscle cells in APP/PS1 mice exacerbates astrogliosis. A, Astrogliosis in brain sections from control APP/PS1 and APP/PS1; smLrp1−/− mice (13–14 months of age) detected with a GFAP antibody. Scale bar, 50 μm. B, GFAP levels in the cortices of control APP/PS1 and APP/PS1; smLrp1−/− mice quantified by Western blotting at 13–14 months of age. The activation of astrocytes is enhanced in the cortex of APP/PS1; smLrp1−/− mice compared with control APP/PS1 mice. C, When GFAP levels in cortices of control and smLrp1−/− mice were analyzed at the age of 12 months, there was no significant difference between those mice. Data are plotted as mean ± SEM (n = 5). *p < 0.05. N.S., Not significant.
Discussion
In this study, we have demonstrated a critical role of LRP1 in vascular smooth muscle cells in mediating brain Aβ clearance. Importantly, we showed that deletion of a single Aβ metabolic receptor in vascular smooth muscle cells is sufficient to impair brain Aβ clearance in an amyloid mouse model. We also report for the first time, to our knowledge, that vascular smooth muscle cells in the brain are capable of mediating local clearance of Aβ.
One of the major Aβ clearance pathways in the brain is through proteolytic degradation by Aβ-degrading enzymes including neprilysin, IDE, and MMPs (Bu, 2009). Neprilysin levels are decreased in vascular smooth muscle cells in AD patients, which are correlated with CAA formation (Miners et al., 2006). Cellular Aβ clearance is also a major Aβ clearance pathway. For example, astrocytes (Wyss-Coray et al., 2003; Koistinaho et al., 2004) and microglia (Wyss-Coray et al., 2001; El Khoury and Luster, 2008; Luo et al., 2010) can degrade Aβ to eliminate it from the brain. In addition to brain-resident cells, perivascular macrophages also appear to play a role in the degradation of Aβ and CAA formation (Hawkes and McLaurin, 2009). Aβ is also directly cleared to peripheral through the blood–brain barrier (Bell et al., 2007; Zlokovic, 2011). Despite the facts that some portions of Aβ are produced in vascular smooth muscle cells and that Aβ is drained along ISF drainage pathways from brain parenchyma into vascular smooth muscle cell layers of cerebral vasculatures (Weller et al., 1998; Revesz et al., 2003; Thal et al., 2008), Aβ clearance through vascular smooth muscle cells has received little attention compared with other brain cell types. If these vascular smooth muscle cell functions against Aβ were interfered, Aβ might accumulate in the brain, resulting in its deposition as senile plaques and CAA. In fact, risk factors of vascular diseases including diabetes mellitus, atherosclerosis, stroke, hypertension, transient ischemic attacks, and microvessel pathology are closely related to AD (de la Torre, 2002). Four risk factors (diabetes mellitus, hypertension, heart disease, and current smoking) have been reported to be associated with a higher risk of AD among individuals without dementia upon monitoring for a mean of 5.5 years (Luchsinger et al., 2005). Importantly, the risk of AD was significantly increased depending on the numbers of those four factors, where hazards ratios of probable and possible AD for the presence of one, two, three, or more risk factors were 1.7, 2.5, and 3.4 compared with no risk factors, respectively (Luchsinger et al., 2005). It is likely that impaired vascular function under these conditions further exacerbates amyloid and CAA pathology by reducing the efficiency of Aβ clearance through LRP1-mediated pathway in brain vascular smooth muscle cells.
LRP1 is a multifunctional endocytic receptor involved in the metabolism of various extracellular ligands (Herz and Strickland, 2001; Bu, 2009). LRP1 is abundantly expressed in cerebrovascular cells, in particular in vascular smooth muscle cells, as well as astrocytes, microglia (Marzolo et al., 2000), and neurons (Bu et al., 1994). Although it is still unclear whether LRP1 directly or indirectly mediates Aβ clearance, the involvement of LRP1 in Aβ metabolism in the brain is supported by findings that Aβ clearance from the brain is inhibited by LRP1 antagonist, RAP, or antibodies against LRP1 in mice (Shibata et al., 2000). While several in vitro studies have shown that LRP1 in endothelial cells could mediate Aβ transcytosis (Yamada et al., 2008; Pflanzner et al., 2011), there is no direct evidence to demonstrate that LRP1 in endothelial cells controls the elimination of Aβ at the blood–brain barrier in vivo. Here, in this report, we demonstrated directly that a deletion of LRP1 in vascular smooth muscle cells disturbs its ability to mediate Aβ clearance through cellular uptake and degradation using both in vitro and in vivo models, suggesting that LRP1-mediated Aβ clearance in vascular smooth muscle cells is critical for overall brain Aβ clearance by the cerebrovascular system. Consistent with our findings, Bell et al. (2009) have demonstrated that overexpression of serum response factor and myocardin in vascular smooth muscle cells suppressed Aβ clearance possibly through downregulation of LRP1.
LRP1 in vascular smooth muscle cells controls cellular uptake of Aβ and its subsequent trafficking to lysosome. Lysosomes contain a variety of acid hydrolases including cathepsin B and cathepsin D, which can efficiently degrade Aβ (Nixon et al., 2001); however, when the lysosomal function is interfered or overwhelmed, Aβ accumulation and aggregation are likely induced both in intracellular and extracellular space. Indeed, accumulation of Aβ beyond the capacity of lysosomal function results in Aβ aggregation in the lysosomes, which can provide seeding for further Aβ aggregation (Hu et al., 2009). Aβ might be accumulated more in perivascular region if its drainage along the vasculature stagnates (Weller et al., 1998; Revesz et al., 2003; Thal et al., 2008). In general, the cerebroarterial pulsations are thought to mainly provide the driving force for drainage of ISF along the cerebrovasculature (Schley et al., 2006). Because LRP1 can function as a signal transducing receptor through coupling with other receptors (Herz and Strickland, 2001), it might be involved in controlling the homeostasis of cerebrovasculature. Thus, deletion of LRP1 in vascular smooth muscle cells may lead to Aβ accumulation due to both a direct impairment of cellular Aβ uptake and lysosomal degradation and potential changes in overall vascular functions. Further studies are needed to address these possibilities.
Several pathological findings indicate that LRP1 levels are significantly decreased during aging and AD (Kang et al., 2000), in particular in brain vasculature (Bell et al., 2009). Hypoxia is one of the factors that suppress LRP1 expression in cerebral vessels (Bell et al., 2009). AD brains might be exposed to hypoxia due to decreased cerebral blood flow seen in AD patients. Toward this, we also found that hydrogen peroxide, a major reactive oxygen species, suppresses LRP1 expression in HBVSMCs (Fig. 8). Reactive oxygen species are excessively produced during aging, by inflammation, or upon Aβ toxicity (Dumont and Beal, 2011). Therefore, aging or vascular damages may impair Aβ clearance by perturbing LRP1 expression and function in vascular smooth muscle cells, which leads to eventual Aβ accumulation and aggregation as senile plaques and CAA. Aging of cerebral arteries, CAA, and apoE4 also impede perivascular drainage of solutes from the brain, which might further impair Aβ clearance (Hawkes et al., 2011, 2012), although it is currently unclear whether LRP1 is involved in these processes.
Figure 8.

Hydrogen peroxide downregulates LRP1 levels in HBVSMCs. LRP1 expression levels in HBVSMCs after incubation with increasing concentrations of hydrogen peroxide (H2O2) for 24 h and analyzed by Western blot. H2O2 significantly suppressed LRP1 levels in a concentration-dependent manner. Data are plotted as mean ± SD (n = 3). *p < 0.05; **p < 0.01.
Together, our findings provide novel insights into the molecular mechanisms of AD and CAA, and suggest that restoring LRP1 expression in the brain vasculature in patients with AD and CAA could be explored for therapy. Thus, it might be possible to screen for chemical compounds that can increase LRP1 expression and/or function in vascular smooth muscle cells. In fact, several compounds including rifampicin and caffeine seem to upregulate LRP1 expression and accelerate Aβ clearance in cerebrovasculature in mice (Qosa et al., 2012). These compounds can be explored as novel therapies for both AD and CAA.
Footnotes
This work was supported by NIH Grants R01 AG027924, R01 AG035355, P01 AG030128, and P01 NS074969 (G.B.), a grant from the Alzheimer's Association (G.B.), a fellowship from the American Heart Association (T.K.), a fellowship from The Robert and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program (T.K.), Center of Regenerative Medicine Career Development Award in Mayo Clinic (T.K.), and NIH Alzheimer's Disease Research Center Pilot Grant P50 AG16574 (T.K.). We thank Monica Castanedes Casey, Linda Rousseau, Virginia Phillips, and Dr. Dennis Dickson for histology and immunohistochemistry. We also thank Aurelie N′songo and Caroline Stetler for technical assistance and careful readings of this manuscript.
References
- Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV. Transport pathways for clearance of human Alzheimer's amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab. 2007;27:909–918. doi: 10.1038/sj.jcbfm.9600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Deane R, Chow N, Long X, Sagare A, Singh I, Streb JW, Guo H, Rubio A, Van Nostrand W, Miano JM, Zlokovic BV. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat Cell Biol. 2009;11:143–153. doi: 10.1038/ncb1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Bu G. Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu G, Maksymovitch EA, Nerbonne JM, Schwartz AL. Expression and function of the low density lipoprotein receptor-related protein (LRP) in mammalian central neurons. J Biol Chem. 1994;269:18521–18528. [PubMed] [Google Scholar]
- Bu G, Cam J, Zerbinatti C. LRP in amyloid-beta production and metabolism. Ann N Y Acad Sci. 2006;1086:35–53. doi: 10.1196/annals.1377.005. [DOI] [PubMed] [Google Scholar]
- Chakrabarty P, Ceballos-Diaz C, Beccard A, Janus C, Dickson D, Golde TE, Das P. IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice. J Immunol. 2010;184:5333–5343. doi: 10.4049/jimmunol.0903382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- de la Torre JC. Alzheimer disease as a vascular disorder: nosological evidence. Stroke. 2002;33:1152–1162. doi: 10.1161/01.str.0000014421.15948.67. [DOI] [PubMed] [Google Scholar]
- Dumont M, Beal MF. Neuroprotective strategies involving ROS in Alzheimer disease. Free Radic Biol Med. 2011;51:1014–1026. doi: 10.1016/j.freeradbiomed.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury J, Luster AD. Mechanisms of microglia accumulation in Alzheimer's disease: therapeutic implications. Trends Pharmacol Sci. 2008;29:626–632. doi: 10.1016/j.tips.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Ellis RJ, Olichney JM, Thal LJ, Mirra SS, Morris JC, Beekly D, Heyman A. Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, Part XV. Neurology. 1996;46:1592–1596. doi: 10.1212/wnl.46.6.1592. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG. A century of Alzheimer's disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci U S A. 2009;106:1261–1266. doi: 10.1073/pnas.0805453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes CA, Härtig W, Kacza J, Schliebs R, Weller RO, Nicoll JA, Carare RO. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 2011;121:431–443. doi: 10.1007/s00401-011-0801-7. [DOI] [PubMed] [Google Scholar]
- Hawkes CA, Sullivan PM, Hands S, Weller RO, Nicoll JA, Carare RO. Disruption of arterial perivascular drainage of amyloid-beta from the brains of mice expressing the human APOE epsilon4 allele. PLoS One. 2012;7:e41636. doi: 10.1371/journal.pone.0041636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J Clin Invest. 2001;108:779–784. doi: 10.1172/JCI13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtwick R, Gotthardt M, Skryabin B, Steinmetz M, Potthast R, Zetsche B, Hammer RE, Herz J, Kuhn M. Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc Natl Acad Sci U S A. 2002;99:7142–7147. doi: 10.1073/pnas.102650499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Crick SL, Bu G, Frieden C, Pappu RV, Lee JM. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc Natl Acad Sci U S A. 2009;106:20324–20329. doi: 10.1073/pnas.0911281106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo T, Bu G. Receptor-associated protein interacts with amyloid-beta peptide and promotes its cellular uptake. J Biol Chem. 2009;284:33352–33359. doi: 10.1074/jbc.M109.015032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo T, Zhang J, Liu Q, Liu CC, Zhang L, Bu G. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-β uptake. J Neurosci. 2011;31:1644–1651. doi: 10.1523/JNEUROSCI.5491-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DE, Pietrzik CU, Baum L, Chevallier N, Merriam DE, Kounnas MZ, Wagner SL, Troncoso JC, Kawas CH, Katzman R, Koo EH. Modulation of amyloid beta-protein clearance and Alzheimer's disease susceptibility by the LDL receptor-related protein pathway. J Clin Invest. 2000;106:1159–1166. doi: 10.1172/JCI11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Hyman BT, Spires-Jones TL. Alzheimer's disease: synapses gone cold. Mol Neurodegener. 2011;6:63. doi: 10.1186/1750-1326-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med. 2004;10:719–726. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- Li Y, Lu W, Bu G. Essential role of the low density lipoprotein receptor-related protein in vascular smooth muscle cell migration. FEBS Lett. 2003;555:346–350. doi: 10.1016/s0014-5793(03)01272-9. [DOI] [PubMed] [Google Scholar]
- Liu Q, Zerbinatti CV, Zhang J, Hoe HS, Wang B, Cole SL, Herz J, Muglia L, Bu G. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron. 2007;56:66–78. doi: 10.1016/j.neuron.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Trotter J, Zhang J, Peters MM, Cheng H, Bao J, Han X, Weeber EJ, Bu G. Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J Neurosci. 2010;30:17068–17078. doi: 10.1523/JNEUROSCI.4067-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchsinger JA, Reitz C, Honig LS, Tang MX, Shea S, Mayeux R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology. 2005;65:545–551. doi: 10.1212/01.wnl.0000172914.08967.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo XG, Ding JQ, Chen SD. Microglia in the aging brain: relevance to neurodegeneration. Mol Neurodegener. 2010;5:12. doi: 10.1186/1750-1326-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia LF, Mackenzie IR, Feldman HH. Clinical phenotypes of cerebral amyloid angiopathy. J Neurol Sci. 2007;257:23–30. doi: 10.1016/j.jns.2007.01.054. [DOI] [PubMed] [Google Scholar]
- Marzolo MP, von Bernhardi R, Bu G, Inestrosa NC. Expression of alpha(2)-macroglobulin receptor/low density lipoprotein receptor-related protein (LRP) in rat microglial cells. J Neurosci Res. 2000;60:401–411. doi: 10.1002/(SICI)1097-4547(20000501)60:3<401::AID-JNR15>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miners JS, Van Helmond Z, Chalmers K, Wilcock G, Love S, Kehoe PG. Decreased expression and activity of neprilysin in Alzheimer disease are associated with cerebral amyloid angiopathy. J Neuropathol Exp Neurol. 2006;65:1012–1021. doi: 10.1097/01.jnen.0000240463.87886.9a. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Mathews PM, Cataldo AM. The neuronal endosomal-lysosomal system in Alzheimer's disease. J Alzheimers Dis. 2001;3:97–107. doi: 10.3233/jad-2001-3114. [DOI] [PubMed] [Google Scholar]
- Pflanzner T, Janko MC, Andre-Dohmen B, Reuss S, Weggen S, Roebroek AJ, Kuhlmann CR, Pietrzik CU. LRP1 mediates bidirectional transcytosis of amyloid-beta across the blood-brain barrier. Neurobiol Aging. 2011;32:2323.e1–2323.e11. doi: 10.1016/j.neurobiolaging.2010.05.025. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Strickland DK, Hyman BT, Rebeck GW. Alpha2-macroglobulin enhances the clearance of endogenous soluble beta-amyloid peptide via low-density lipoprotein receptor-related protein in cortical neurons. J Neurochem. 1999;73:1393–1398. doi: 10.1046/j.1471-4159.1999.0731393.x. [DOI] [PubMed] [Google Scholar]
- Qosa H, Abuznait AH, Hill RA, Kaddoumi A. Enhanced brain amyloid-beta clearance by rifampicin and caffeine as a possible protective mechanism against Alzheimer's disease. J Alzheimers Dis. 2012;31:151–165. doi: 10.3233/JAD-2012-120319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rensink AA, de Waal RM, Kremer B, Verbeek MM. Pathogenesis of cerebral amyloid angiopathy. Brain Res Brain Res Rev. 2003;43:207–223. doi: 10.1016/j.brainresrev.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Revesz T, Ghiso J, Lashley T, Plant G, Rostagno A, Frangione B, Holton JL. Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol. 2003;62:885–898. doi: 10.1093/jnen/62.9.885. [DOI] [PubMed] [Google Scholar]
- Rohlmann A, Gotthardt M, Hammer RE, Herz J. Inducible inactivation of hepatic LRP gene by cre-mediated recombination confirms role of LRP in clearance of chylomicron remnants. J Clin Invest. 1998;101:689–695. doi: 10.1172/JCI1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzali WA, Kehoe PG, Love S. Influence of LRP-1 and apolipoprotein E on amyloid-beta uptake and toxicity to cerebrovascular smooth muscle cells. J Alzheimers Dis. 2012 doi: 10.3233/JAD-2012-121336. [DOI] [PubMed] [Google Scholar]
- Schley D, Carare-Nnadi R, Please CP, Perry VH, Weller RO. Mechanisms to explain the reverse perivascular transport of solutes out of the brain. J Theor Biol. 2006;238:962–974. doi: 10.1016/j.jtbi.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer's amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M, Sato N, Kurinami H, Takeuchi D, Takeda S, Shimamura M, Yamashita T, Uchiyama Y, Rakugi H, Morishita R. Reduction of brain beta-amyloid (Abeta) by fluvastatin, a hydroxymethylglutaryl-CoA reductase inhibitor, through increase in degradation of amyloid precursor protein C-terminal fragments (APP-CTFs) and Abeta clearance. J Biol Chem. 2010;285:22091–22102. doi: 10.1074/jbc.M110.102277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Griffin WS, de Vos RA, Ghebremedhin E. Cerebral amyloid angiopathy and its relationship to Alzheimer's disease. Acta Neuropathol. 2008;115:599–609. doi: 10.1007/s00401-008-0366-2. [DOI] [PubMed] [Google Scholar]
- Urmoneit B, Prikulis I, Wihl G, D'Urso D, Frank R, Heeren J, Beisiegel U, Prior R. Cerebrovascular smooth muscle cells internalize Alzheimer amyloid beta protein via a lipoprotein pathway: implications for cerebral amyloid angiopathy. Lab Invest. 1997;77:157–166. [PubMed] [Google Scholar]
- Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE. Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer's disease. Am J Pathol. 1998;153:725–733. doi: 10.1016/s0002-9440(10)65616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmus MM, Otte-Höller I, van Triel JJ, Veerhuis R, Maat-Schieman ML, Bu G, de Waal RM, Verbeek MM. Lipoprotein receptor-related protein-1 mediates amyloid-beta-mediated cell death of cerebrovascular cells. Am J Pathol. 2007;171:1989–1999. doi: 10.2353/ajpath.2007.070050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, Masliah E, Mucke L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9:453–457. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- Yamada K, Hashimoto T, Yabuki C, Nagae Y, Tachikawa M, Strickland DK, Liu Q, Bu G, Basak JM, Holtzman DM, Ohtsuki S, Terasaki T, Iwatsubo T. The low density lipoprotein receptor-related protein 1 mediates uptake of amyloid beta peptides in an in vitro model of the blood-brain barrier cells. J Biol Chem. 2008;283:34554–34562. doi: 10.1074/jbc.M801487200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youmans KL, Leung S, Zhang J, Maus E, Baysac K, Bu G, Vassar R, Yu C, LaDu MJ. Amyloid-beta42 alters apolipoprotein E solubility in brains of mice with five familial AD mutations. J Neurosci Methods. 2011;196:51–59. doi: 10.1016/j.jneumeth.2010.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbinatti CV, Wozniak DF, Cirrito J, Cam JA, Osaka H, Bales KR, Zhuo M, Paul SM, Holtzman DM, Bu G. Increased soluble amyloid-beta peptide and memory deficits in amyloid model mice overexpressing the low-density lipoprotein receptor-related protein. Proc Natl Acad Sci U S A. 2004;101:1075–1080. doi: 10.1073/pnas.0305803101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Koo EH. Biology and pathophysiology of the amyloid precursor protein. Mol Neurodegener. 2011;6:27. doi: 10.1186/1750-1326-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12:723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]