

Abstract
Heterotopic ossification (HO) is a disabling condition associated with neurologic injury, inflammation, and overactive BMP signaling. The inductive factors involved in lesion formation are unknown. We found that the expression of the neuro-inflammatory factor Substance P (SP) is dramatically increased in early lesional tissue in patients who have either fibrodysplasia ossificans progressiva (FOP) or acquired HO, and in three independent mouse models of HO. In Nse-BMP4, a mouse model of HO, robust HO forms in response to tissue injury; however null mutations of the preprotachykinin gene encoding SP prevent HO. Importantly, ablation of SP+ sensory neurons, treatment with an antagonist of SP receptor NK1r, deletion of NK1r gene, or genetic down-regulation of NK1r-expressing mast cells also profoundly inhibits injury-induced HO. These observations establish a potent neuro-inflammatory induction and amplification circuit for BMP-dependent HO lesion formation, and identify novel molecular targets for prevention of HO.
Keywords: Heterotopic Ossification (HO), Fibrodysplasia Ossificans Progressiva (FOP), bone morphogenetic protein (BMP), Substance P (SP), Tachykinin Receptor 1 (NK1r), NK1r Antagonist, Mast Cells
Introduction
Heterotopic ossification (HO), the formation of extraskeletal bone, is a common and serious complication of soft tissue trauma.(1-3) In fibrodysplasia ossificans progressiva (FOP), a rare, life-threatening condition of progressive and episodic HO, mutations in a bone morphogenetic protein (BMP) type I receptor, ACVR1/ALK2, cause dysregulated BMP signaling.(4) Despite advances in understanding the genetics of HO, the cellular and molecular triggers of HO remain unclear.
A fundamental feature of all forms of HO is the requirement for an inflammatory trigger(3, 5-7). In a previous study we found that injury induced inflammation triggers HO in a unique transgenic mouse model with features of both sporadic HO and FOP in which BMP4 is driven by neuron-specific enolase (Nse) promoter(8). Detailed studies found that the transgene is expressed not only in neurons (including DRG neurons), but also in macrophages(8-10). The robust injury induced phenotype and the unique transgene expression pattern make this an attractive animal model for injury-induced sporadic HO as well as FOP,(11)and suggest that HO might be prevented if the inflammatory trigger could be inhibited. However, it is challenging to identify the specific loci that we can efficiently regulate the inflammation triggers, because it is well known that the inflammatory response is regulated by multiple cytokines(12) and also under complex neuro-endocrine control(13-17).
In this study, we reasoned that inflammatory neuropeptides might be ideal candidates to trigger inflammation and the HO, because they mediate neuro-inflammatory feedback loops both in physiological and pathophysiological conditions and are often dysregulated in trauma(18-20). Among pro-inflammatory neuropeptides, substance P (SP) (21-23) was identified in active areas of bone regeneration following fracture.(24, 25) Moreover, the SP receptor, neurokinin 1 (NK1r), was demonstrated on chondrocytes(26), osteocytes(27), osteoblasts(28), osteoclasts(29) and mast cells(30). SP is an undecapeptide expressed by subsets of neurons in the central and peripheral nervous systems(31-33) and also by non-neuronal cells including macrophages and T lymphocytes, cells involved at the earliest stages of pre-osseous fracture repair(34, 35). SP enhances lymphocyte proliferation and immunoglobulin production as well as cytokine secretion from lymphocytes, monocytes, macrophages, and mast cells.(36-38) By promoting vasodilatation, leukocyte chemotaxis, and leukocyte/endothelial cell adhesion, SP promotes the extravasation, migration, and accumulation of leukocytes at sites of tissue injury(21, 22, 39). In addition to immune modulation, SP also participates in injury-inducible mobilization of CD29+ mesenchymal stem/progenitor cells, a cell type that is involved in HO formation(40). Clinical studies indicate that SP is dysregulated, at least transiently, after traumatic brain or spinal cord injury(39, 41, 42), pre-conditions that frequently lead to acquired HO(6).
Here, we studied the role of SP in patients with sporadic, post-traumatic, and neurologically-associated HO as well as FOP, and in three independent mouse models of post-traumatic and FOP-like HO(9, 43). We found that SP expression was up-regulated in early pre-osseous sporadic HO and FOP lesions, and that blocking SP secretion or function in the animal models prevented HO. We further determined that mast cells, which robustly express NK1r(30), are required to mediate the downstream events of SP-mediated BMP-dependent HO. These observations identify SP as a critical regulatory factor in the induction of HO, and suggest that blocking SP signaling or the downstream amplification circuit of SP-mediated inflammation could be a novel therapeutic approach to prevent BMP-mediated HO.
Materials and Methods
Patients' tissue samples and processing
Collection of specimens was approved by the Office of Regulatory Affairs and the Institutional Review Board of the University of Pennsylvania (Federal wide Assurance #00004028). Specimens from six FOP patients who underwent biopsy for presumptive neoplasm were obtained from superficial and deep back masses later determined to be early FOP lesions. Tissue samples from ten patients who were diagnosed with post-traumatic or neurologically-associated HO by clinical and radiographic criteria, were divided into the following groups, according to the specific predisposing medical conditions: two from spinal cord injury (SCI), five from traumatic brain injury (TBI), one from non-neurologic trauma (NNT) and two from total hip arthroplasty (THA). Normal muscle tissue was obtained from the backs of four age-matched unaffected males. Tissue samples were fixed in neutral buffered formalin, decalcified, infiltrated, embedded in paraffin, and sectioned at a thickness of five micrometers. Samples were deparaffinized, stained with Harris hematoxylin solution and counterstained with hematoxylin and eosin (H&E) by standard procedures. Prepared specimens were examined under light microscopy for histological characteristics of HO lesion formation. Findings were confirmed by two independent examiners (R.J.P. and F.S.K.).
Transgenic and mouse models
Previously generated Nse-BMP4 and NK1r−/− mice were used in this study(43, 44). Floxed caALK2 transgenic mice were a gift from Dr. Yuji Mishina (University of Michigan). All other lines were from the Jackson Laboratory (Bar Harbor, Maine), unless otherwise specified. All animal experiments were approved by the Animal Care and Use Committee at Northwestern University, or by the Institutional Animal Care and Use Committee at the University of Pennsylvania.
Superficial and deep muscle injury model
The superficial and deep muscle injury was preformed according to previous description(9). Briefly, deep skin incision caused disruption of skin and subcutaneous tissue as well as injury of superficial panniculous carnosus muscle. Deep intramuscular injection of 100μl of 10 μM cardiotoxin (CTX, Calbiochem) caused deep muscle injury(9).
caALK2 mouse model
To induce expression of caALK2, Adenovirus-Cre (Penn Vector Core; 1 × 1011 particles per mouse) together with cardiotoxin (100μl of 10μM solution) was injected into the left hindlimb musculatures of mice at three weeks of age. Tissues were recovered at 4 and 8 days after injections. Tissues were fixed in 4% PFA and frozen in isopentane (2-methylbutane) and cryosections were cut at 10mm for further studies.
BMP4 matrigel injection model
Growth factor-reduced Matrigel (BD Biosciences, Bedford, Massachusetts) was impregnated with recombinant human BMP4 (rhBMP4, R&D) at a concentration of 2.5 μg/50μl and injected intramuscularly into the mid-belly of the tibialis anterior muscle of adult mice, with or without 10 μM cardiotoxin (CTX, Calbiochem). Mice injected with Matrigel only served as controls. Lesional tissue was recovered seven days to three weeks following implantation for histochemical and immunohistochemical analysis.
Colchicine treatment
Control and Nse-BMP4 mice were treated with colchicine (intraperitoneal, 5 mg/kg of body weight) for 24 hours, and then the lumbar dorsal root ganglia (DRG) were fixed and harvested for immunohistochemistry (IHC).
NK1r antagonist treatment
young (1-2 months old) Nse-BMP4 mice were treated with the tachykinin NK1 receptor antagonist, RP-67580 (Tocris Bioscience) at a dose of 2mg/kg body weight, intraperitoneally, twice daily for 4 weeks.(45) Age and sex matched mice treated with PBS were used as controls. RP-67580 treatment was started 4 hours after superficial or deep muscle injury. The same treatment was also administrated to Nse-BMP4 mice that already developed HO to test if these mice could benefit from RP-67580 treatment.
Radiographic Evaluation
Radiographs were taken weekly to monitor the progression of HO after superficial or deep muscle injury. HO formation was assayed by whole body X-ray imaging, using the TruDR Digital Radiography System (Sound Technologies, Carlsbad CA) according to previous description(9). To further quantify the HO after muscle injury and the effect of the SP antagonists, microComputed Tomography (micro-CT) was performed with a MicroCT-40 system (Scanco Medical AG, Bruettisellen, Switzerland). A total of 460 contiguous slices were collected of each tibia (∼17 mm length) at 45 kV, 88 μA, 300 ms integration per projection, and 500 projections of 1024 samples. Reconstruction was with 37 μm isotropic volume elements (voxels) on a 1024 × 1024 grid. The volume of heterotopic ossified tissue was quantified from the micro-CT data sets using the Scanco software suite(46).
Neonatal capsaicin treatment
We permanently ablated SP+ positive neurons in dorsal root ganglia by treating neonatal (1-2 days old) mice with a single subcutaneous injection of capsaicin (50 mg/kg) or vehicle (10% ethanol and 10% Tween 80 in sterile saline) into mouse back, as previously reported(47). Capsaicin and vehicle treated animals were group housed and weaned the same way. These animals were subjected to similar injury at 1-2 months of age. Injury induced SP up-regulation in skin, subcutaneous tissues and muscles were determined by IHC.
Dissociated sensory neuron culture
Lumbar DRGs from 1-2 month old Nse-BMP4 and sex and age matched control mice were harvested, dissociated and cultured according to the published protocol,(48) with or without BMP4/Noggin treatment on 24-well cover slips. Cells were fixed and supernatants were collected three days after culture. The SP concentration in the cell supernatant was determined by Substance P EIA Kit (Cayman Chemical) and the SP positive neurons were determined by counting the SP/NF200 double positive neurons on the cover slips.
Immunohistochemistry
Immunostaining was performed using standard protocols. Briefly, sections were fixed with 4% paraformaldehyde in PBS. Non-specific binding was blocked with 10% normal serum diluted in 1% bovine serum albumin (BSA, Jackson Lab, USA) and 0.25% Triton X-100 for one hour in room temperature. The sections were incubated with primary antibodies diluted with 1% BSA + 0.25% Triton X-100 at 4°C overnight, then incubated with appropriate secondary antibodies (Cy3 or Cy2 conjugated antibodies; Jackson Lab), diluted with 1% BSA + 0.25% Triton X-100 or Alexa Fluor 488, Alexa Fluor 594, and Alexa 647 (1:1000, Invitrogen) in the dark at room temperature for 2 hours, and Counterstained with DAPI (1:5000). Mouse anti-SP antibody (R&D Systems), Rabbit anti-P75 (Promega) and Rabbit Anti-Neurofilament 200 (Sigma), chicken Anti-Neurofilament 200 (Millipore), rabbit anti-GFP (Invitrogen), and rabbit anti-LAMP2 (LifeSpan Biosciences), were used in this study. Fluorescent images were processed by Adobe Photoshop and quantified by ImagJ.
Peptide blocking experiment
SP, NKA and NKB peptides (from AnaSpec) were used in the blocking experiment: SP: RPKPQQFFGLM, NKA: HKTDSFVGLM, and NKB: DMHDFFVGLM were all diluted to 1mM final concentration and incubated with equal amount of antibody (1:1000) in blocking buffer 4°C overnight, and then control (without blocking peptide) and blocked Ab were used to stain the neighboring mouse sections from muscle injury model.
Results
SP is up-regulated in FOP lesions and acquired HO and is neuronal in origin
SP is a potent pro-inflammatory factor(21-23, 49) and has been identified in the most active areas of physiological and pathological postnatal osteogenesis(1, 24, 25). To examine whether SP expression is elevated in HO lesions, immunocytochemistry was used to detect SP protein in both mouse and human pre-osseous lesions. The specificity of the antibody was confirmed by comparing antibody binding of tissues from SP precursor gene (PPT-A) knockout mice(50) with that of WT and Nse-BMP4 mice. We found specific SP staining in the skin, subcutaneous connective tissues, CNS, dorsal root ganglia (DRG) and other tissues from WT and Nse-BMP4 mice. By contrast, this staining was totally absent in the same tissues of SP precursor gene (PPT) knockout mice (Suppl. fig. 1 and data not shown) demonstrating that the antibody is specific and sensitive to probe expression of SP in target tissues. The peptide blocking experiment further confirmed the specificity and excluded the cross-reactivity of this antibody with highly conserved mammalian homologs, such as NKA and NKB (Suppl. fig. 1). Since this antibody is specifically targeted against the mature SP peptide, and the mature peptide is identical across all mammalian species, we also used this antibody to examine SP expression in surgically removed lesional tissues samples from patients with FOP and acquired HO.
High levels of SP expression were detected in early pre-osseous FOP lesions (fig. 1A). These early lesions were located in muscle tissue, which showed signs of fiber degeneration in phase images (Suppl. fig. 2B) and inflammatory cell infiltration in H&E staining (Suppl. fig. 3). In contrast, minimum SP expression was observed in normal muscle tissue (fig. 1E&2E). Two staining patterns can be recognized in these samples: strong punctuate and weak diffuse staining. We speculated that the diffuse staining could be an artifact of dying/degenerating muscle fibers but further study with specific blocking peptide excluded that possibility (fig. 2A&B). In fact, both punctuate and diffuse staining were blocked by SP peptide. Further double staining and morphologic examination confirmed that majority of the diffuse staining was in muscle fibers (data not shown).
Figure 1. SP is up-regulated in early FOP lesions.

(A) SP up-regulation in an early FOP lesion. Note the widespread, but not universal, SP staining. Two patterns can be recognized: the strong punctuate and semi-diffuse staining. (B) No appreciable level of SP staining was observed in the fibroproliferative (pre-cartilage) stage. (C) shows the SP staining of the mature HO from a FOP patient. (D) Negative control (no 1st antibody). Bar=40 μm. (E) Summary of relative SP expression levels in FOP, acquired-HO patient, and normal controls. Relative SP expressions are graphed based on signal intensity of fluorescence images. fibrodysplasia ossificans progressiva (FOP), Spinal Cord Injury (SCI), Traumatic Brain Injury (TBI), Non-Neurologic Trauma (NNT) and Total Hip Arthroplasty (THA). Note significant SP up-regulation was observed only in early lesions. (* P<0.01 vs control muscles). Error bars represent s.d. early: early lesion, fib: fibrotic lesion, bone: mature bone.
Figure 2. SP staining is specific, and co-localized with a neuronal marker.

(A&B) Peptide blocking experiment indicated that both punctuate and the semi-diffuse staining observed in early lesions are specific. (A) shows a typical image of SP staining in an early lesion from a FOP patient. (B) shows that both punctuated and the semi-diffused staining in a neighboring section are blocked by SP peptide. (C) shows a typical image of SP/NF-200 double staining in an early lesion from a traumatic brain injury (TBI) patient. Note that there is extensive co-localization of SP and NF-200 (white arrows in C). Also note that there are some NF-200− cells also express high level of SP (white arrow heads in C). (C′ and C″) shows the split channel of NF-200 and SP, respectively. (D) shows the typical image of SP stained in mature HO from a sample from a TBI patient. (E) shows the typical image of SP staining from a sample of normal control muscle. Bar=40 μm.
We also tested SP expression in samples of heterotopic bone from patients with four types of acquired HO: Spinal Cord Injury (SCI), Traumatic Brain Injury (TBI), Non-Neurologic Trauma (NNT) and Total Hip Arthroplasty (THA). Due to the maturity of lesions at the time of collection, early lesional stages were found only in small portions of these samples (Fig. 1E & Suppl. fig. 4). Consistently, SP was up-regulated in early lesions in acquired HO, but less dramatically compared to FOP early lesions. however, no appreciable SP expression was observed in later stage lesions from acquired HO samples (fig. 2D).
To help clarify whether neurons or non-neuronal cells contributed to the observed SP up-regulation, we double-stained sections with SP and NF-200, a heavy neurofilament protein that is commonly used as a biomarker of neurons(51), and we found extensive co-localization of SP and NF-200 both in the early FOP lesions (Suppl. fig. 2A) and in early lesions of acquired HO (fig. 2C). However, some NF-200− cells that express high levels of SP were also found in the early lesions of acquired HO (fig. 2C), interestingly, these NF-200− and SP+ cells were not closely associated with or surrounded by diffused SP staining as NF-200+ cells do, suggesting that they do not contribute significantly to the SP up-regulation in the lesion. In contrast, SP signals rarely co-localized with NF-200 in mature stages of FOP HO (Suppl. fig. 2C). These data suggest that the predominant source of SP in the early pre-osseous lesions is neuronal (NF-200+). Overall, our data support that SP dysregulation may play key roles in the pathophysiology of common human HO and in response to dysregulated BMP signaling in patients with FOP.
SP up-regulation in target tissues of animal models is BMP dependent and injury induced
In order to better understand the functional implications of SP up-regulation in HO, we studied the Nse-BMP4 transgenic mouse model that closely recapitulates FOP and also displays the histological hallmarks of acquired HO(9). We first examined the expression of SP in subcutaneous connective tissue, and muscles of the hind limbs, target tissues where HO formation occurs in response to injury(9). As a control, we also compared SP expression in other neuronal and non-neuronal tissues that could potentially be indirectly involved in the HO process, including the secondary immune system (spleen and lymph nodes), primary immune system (thymus), DRG and central nervous system (CNS) (Suppl. fig. 5, and data not shown). There were no obvious transgene dependent changes in SP expression in any tissues from postnatal or young uninjured Nse-BMP4 mice.
To determine whether the SP up-regulation in target tissues is triggered by injury, we performed superficial and deep muscle injury in young (1 month old) Nse-BMP4 mice and examined SP expression in the injured and uninjured limbs of the same mice. No transgene dependent SP up-regulation is detected in naïve animals (data not shown). However, in response to injury, the limbs of Nse-BMP4 mice showed substantially increased SP expression compared to the uninjured limbs as early as 1.5 hours after injury (Fig. 3 A-C). In contrast, the increase in SP was minimal in WT mice under the same conditions (data not shown). At one day after injury, dramatically increased SP expression was observed in injured Nse-BMP4 mice compared to WT mice (Fig. 3 D-F). More importantly, Similar SP up-regulation was also observed in CTX induced deep muscle injury model (Fig. 3 G-I), which further strengthened our conclusion.
Figure 3. Injuries induced transgene dependent SP up-regulation in connective tissue and muscles.
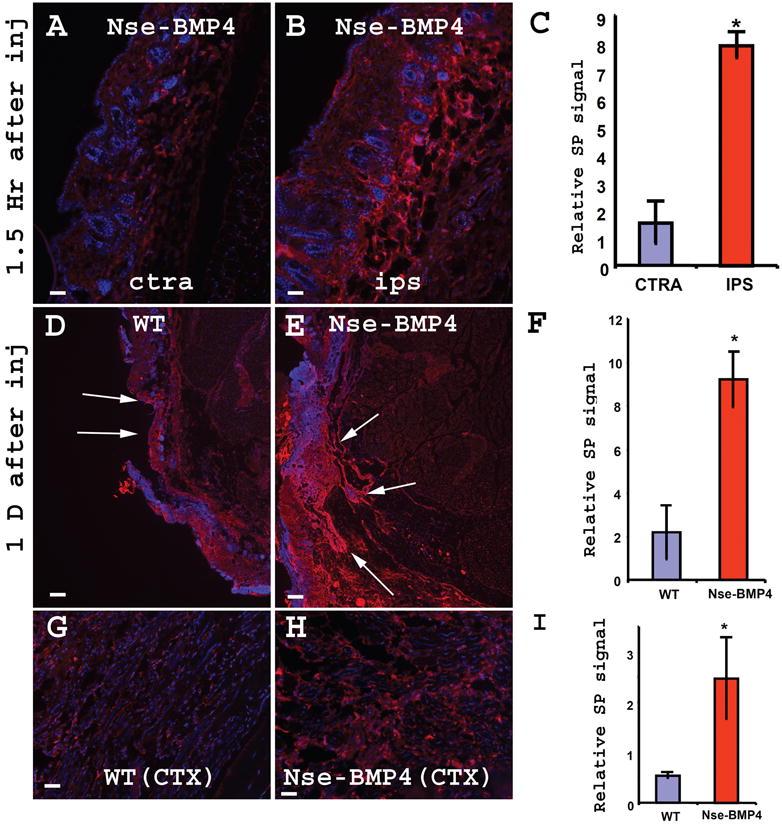
Both superficial muscles injury (A-F) and CTX induced deep muscles injury (G-I) induced SP up-regulation. (A&B) show typical images of SP immunostained cross sections of contralateral uninjured (A) and injured (B) limbs 1.5 hr after superficial muscles injury. Note the dramatic SP up-regulation and edema in the injured site (B). (C) quantified the relative SP signal in (A&B). (* P<0.01 vs control). Error bars represent s.d. (D&E) show typical images of SP immunostained cross sections of injured WT (D) and Nse-BMP4 (E) limbs one day after superficial muscles injury. Note the dramatic SP up-regulation and edema in Nse-BMP4 mice (E). (F) quantified the relative SP signal in (D&E). (* P<0.01 vs control). Error bars represent s.d. (G&H) show typical images of SP immunostained cross sections of injured WT (G) and Nse-BMP4 (H) limbs after CTX induced injury. (I) quantified the relative SP signal in (G&H). (* P<0.01 vs control). Error bars represent s.d. Bar=40 μm in A, B, G&H, Bar=200 μm in D&E.
The observed SP up-regulation in the Nse-BMP4 mouse model could arise from neuronal tissue, non-neuronal tissue, or a combination of both. However, data from double staining of human samples suggested that neuronal SP is the predominant source, at least in early lesions. Double staining of the mouse sections also supports this conclusion (Suppl. Fig. 6).
To explore the underling mechanism and further confirm the injury induced and BMP signaling dependent SP up-regulation in other in vivo systems, we took the advantage of two other well established mouse models: the caALK2 mouse model (a mouse model of FOP)(52), and BMP4 matrigel injection model. Adenovirus-Cre was mixed with CTX and injected into hindlimb muscles of caALK2 transgenic mice to induce muscle injury and local caALK2 expressing cells. We repeated the injury-induced, caALK2 dependent, SP up-regulation in this model (Suppl. Fig. 7). Co-localization study with NF200 further suggested the neuronal contribution to SP up-regulation (Suppl. fig. 8). Detailed study suggested a paracrine, rather than an autocrine mediated mechanism of action, because robust SP did not co-localize with GFP (caALK2+) cells (Suppl. fig. 8). To further test whether injury and exogenous BMP4 signaling function synergistically in SP up-regulation and HO induction, we mixed BMP4 with matrigel, with or without CTX, followed by intramuscular injection to induce HO. Indeed, we also confirmed the BMP4 dependent SP up-regulation in this model (Suppl. fig. 9). Interestingly, even though CTX alone does not appreciably up-regulate SP, local SP up-regulation was more dramatic in the mice that were treated with CTX+BMP4 matrigel than that of BMP4 matrigel alone (Suppl. fig. 9).
Since hind limb musculature is richly innervated by SP+ sensory nerve fibers, we examined lumbar dorsal root ganglia (DRG) in Nse-BMP4 mice to determine the possible contribution of SP+ sensory neurons to the injury induced increase in SP expression. In young (3-4 week old), uninjured Nse-BMP4 mice, the number and pattern of SP expression by DRG neurons did not differ from WT mice (Suppl. fig. 10). However, in injured, or adult Nse-BMP4 mice with HO, we observed an unusual mesh-like pattern of SP expression in which cellular staining of SP was not prominent whereas staining in the tissue surrounding neurons was dramatically increased (Fig. 4C). The p75 low affinity neurotrophin receptor (p75) is expressed by almost all sensory neurons in the adult DRG(53). Quantitative analysis found that the number of SP+ p75+ cells in the DRG of old Nse-BMP4 mice that had HO was lower, even though the number of total p75+ neurons was similar (Fig. 4E). We reasoned that over-release of SP from peripheral sensory neurons could lead to this staining pattern by depleting the cytoplasmic SP in the cell bodies of the DRG. To directly test this hypothesis, we pretreated Nse-BMP4 old mice that had HO with colchicine, which disrupts and blocks the axoplasmic transport and release of SP(54). We then compared the SP expression pattern of treated and untreated lumbar DRG, and found that the normal expression pattern of SP was largely restored in colchicine treated Nse-BMP4 mice (Fig. 4D). Further, the number of SP+ neurons was dramatically increased after colchicine treatment in Nse-BMP4 mice but not in age-matched WT mice (Fig. 4E). These observations exclude the possibility that the low number of SP+ neurons in DRG of older Nse-BMP4 mice was due to reduced survival and strongly support the hypothesis of injury-induced over-release of the peptide.
Figure 4. Injury induced a unique immunohistochemical phenotype (mesh-like SP staining pattern) of lumbar DRGs of Nse-BMP4 mice.

Sections of lumbar DRGs from age matched 6 month old WT (A&B) and Nse-BMP4 with HO (C&D) mice were immunostained for SP. A&C, untreated, and B&D, pretreated with colchcine (COL). Note that the unique mesh-like SP staining pattern was found only in Nse-BMP4 (C), and that the normal pattern was restored by colchcine treatment in Nse-BMP4 mice (D). (E) depicts the SP+ cell densities of different subpopulations of neurons in 6 month old Nse-BMP4 and age matched WT lumbar DRGs. Note that colchcine treatment increased the density of SP+ neurons in Nse-BMP4 DRGs without significantly changing the total number of P75+ neurons. (F&G) The mesh-like staining pattern is induced by superficial muscles injury (G, 1.5 hours after injury) in younger (one month old) Nse-BMP4 mice. (F) SP immunostained sections of lumbar DRGs from 1 month old naïve Nse-BMP4. Note that cytoplasmic staining of SP is still observed at this time point. White arrows point to the SP+ cell bodies in (G). NK1r antagonist (RP-67580) treatment had variable efficiency in restoring the normal staining pattern of SP in old Nse-BMP4 mice (H&I). SP stained sections of lumbar DRGs from 6 month old Nse-BMP4 treated with PBS (H), and 6 months old Nse-BMP4 pre-treated with NK1r antagonist for 2 weeks (I). Note that the normal staining pattern of SP in old Nse-BMP4 mice was only partially restored with NK1r antagonist treatment. White arrows point to the SP+ cells. Bar=40 μm.
Since the observed peripheral SP up-regulation was induced by injury, we speculated that SP over-release/depletion in DRG neurons could also be induced by injury. To directly test the hypothesis, superficial muscles were injured in young (one month old) Nse-BMP4 mice which do not display the mesh-like pattern of SP immunostaining. The mesh-like pattern was reproduced as early as 1.5 hours after injury (Fig. 4F&G), coincident with the up-regulation of SP in the injured skin (Fig. 3). These observations suggest that SP+ DRG neurons contribute to the injury induced increase in SP levels.
Since the specific SP receptor, NK1r is expressed by DRG neurons(55), it is possible that the SP release acts through paracrine and/or autocrine mechanisms to regulate SP expression. To test this hypothesis, SP levels were examined in DRG of NK1r antagonist treated old Nse-BMP4 mice. Unlike colchicine, RP-67580 treatment of old Nse-BMP4 mice restored only a small and variable degree of normal immunostaining (Fig. 4H&I) suggesting that paracrine/autocrine SP signaling is not the predominate mechanism underlying the increased release of SP. However the mesh-like pattern of SP immunostaining was less prominent after RP-67580 treatment suggesting at least some role for paracrine/autocrine signaling.
To further test the role of neuronal SP, we selectively ablated SP+ neurons in DRG of neonatal (1-2 days old) Nse-BMP4 mice by capsaicin treatment(47) and waited until the treated animals were 1-2 months old to test injury-induced short-term and long-term effects. Neonatal capsaicin treatment is known to selectively and irreversibly destroy small diameter DRG sensory neurons without obvious effects on non-neuronal cells(56). The effectiveness of ablation was further confirmed by comparing SP stained lumbar DRG sections from the capsaicin and vehicle pretreated young WT and Nse-BMP4 mice (1 month old). We found that the small diameter SP+ neurons were almost completely absent in lumbar DRG of capsaicin treated mice (Suppl. fig. 11). Once the effectiveness was confirmed, capsaicin and vehicle pretreated adult WT and Nse-BMP4 mice were subjected to standard injury. Injured and control hind limbs and DRG were harvested at 1 hour and 1 day after injury and examined for SP expression. We found that in DRG, the injury-induced mesh-like pattern was absent in capsaicin pretreated Nse-BMP4 mice (fig. 5B). In hind limbs, injury-induced SP up-regulation in target tissues of the capsaicin treatment group was also minimal, compared to vehicle treated mice (fig. 5E, E&I). Consistently, H&E staining demonstrated the luck of massive inflammatory response in capsaicin pretreated Nse-BMP4 mice, compared to vehicle pretreated ones (fig 5G,H&J). These data further support the hypothesis that the mesh-like pattern is caused by over release of SP, that neuronal SP is necessary for the injury induced SP up-regulation in target tissues, and that SP is necessary to amplify the inflammatory response. Importantly, the efficiency of HO formation (4 weeks after injury) was greatly reduced in capsaicin pretreated adult Nse-BMP4 mice, a finding that further supports that the abnormal inflammatory response induced by neuronal SP secretion mediates HO formation (fig. 6).
Figure 5. Ablation of SP+ neurons inhibits injury induced SP up-regulation and inflammation.

Small diameter SP+ sensory neurons were virtually absent in capsaicin pretreated Nse-BMP4 DRG (A) & (B), compared to vehicle treated ones (C)&(D). DRG depicted in (A&C) are from uninjured side, while (B & D) show the DRG from injured side. Note that injury did not lead to injury induced mesh-like pattern in capsaicin pretreated Nse-BMP4 mice (B), compared to vehicle treated one (D). Ablation of SP+ neurons inhibits injury-induced SP up-regulation in target tissues of Nse-BMP4 mice (E &F). Cross sections of hind limbs from the injury site are shown here. Vehicle (E) and capsaicin pretreated (F) mice were subjected to similar superficial muscles injury, hind limbs were harvested 1 hour after injury (shown) or 1 day after injury (not shown). Reduced SP up-regulation was observed in lesions from capsaicin pretreated Nse-BMP4 mice (F), compared to vehicle pretreated mice (E). The strong SP staining was found closely associated with muscle fibers. Infiltrating cells are indicated by dense DAPI+ regions. (G & H) H&E staining clearly indicated that ablation of SP+ neurons inhibited injury induced inflammatory response. (G) Image from injured site (1 hour after injury) of vehicle pretreated Nse-BMP4 mice demonstrated massive infiltration of inflammatory cells. In contrast, much less inflammatory cells infiltration was observed in injured site of capsaicin pretreated Nse-BMP4 mice (H). (I) quantified the relative SP levels observed in (E&F). * P<0.01 vs vehicle control. (J) Quantified the numbers of inflammatory cells observed in (G&H). * P<0.01 vs vehicle control.
Figure 6. Inhibition of SP signaling blocks injury induced inflammation and HO.

(A) summarizes superficial muscles injury-induced edema and HO in different conditions. Nse-BMP4 mice without endogenous SP signaling (PPT-A−/−, Nk1r−/−, or ablation of SP+ neurons), or mast cells (c-kitw-sh/w-sh) did not develop injury induced HO efficiently. (B) Effect of SP receptor (NK1r) inhibition (RP-67580) in response to superficial muscles injury. Frequency (percent of mice) of HO formation over time (1-6 weeks) with RP-67580 or PBS treatment is shown. (C-G) RP-67580 inhibited HO in CTX induced muscle injury model. (C) shows the CTX induced HO (3D reconstruction) from a PBS treated Nse-BMP4 mouse. (D) shows that CTX induced HO was inhibited by RP-67580 treatment. (E) depicts the total HO volumes one month after muscles injury in RP-67580 and PBS treated groups. (F) shows gross images from the same groups. (G) depicts the total wet weights of affected hindlimbs in the same groups. * P<0.01 vs control. Note that the total HO volumes, total wet weights and gross images consistently indicated the inhibition effect of RP-67580. (H-K) RP-67580 specifically inhibited injury induced edema and inflammation. (H) shows gross images seven days after injury from RP-67580 and PBS treated limbs. Note that the edema in PBS treated limbs are much obvious than the limbs from RP-67580 group (reflected by circumferences). (I) depicts the total wet weights of affected hindlimbs from the same groups. * P<0.01 vs control. (J&K) show the typical images of H&E staining from RP-67580 and PBS treated group, respectively. Note that the inflammatory response is dramatically reduced in RP-67580 treated lesion (J), comparing to PBS (K) treated lesion.
Taken together, these in vivo studies suggest that SP up-regulation is dependent on increased BMP signaling, and that the neuronal source is the major contributor. To directly test whether BMP4 could influence the expression and release of SP in a dissociated neuronal population, lumbar DRGs were harvested, dissociated and cultured with or without BMP4 treatment. We found that SP release and expression was up-regulated by BMP4 treatment in WT sensory neurons. Interestingly, both the expression and release of SP from cultured Nse-BMP4 neurons were up-regulated, and this effect was blocked by Noggin treatment indicating a ligand-dependent paracrine effect (suppl fig 12 and data not shown). The caveat of this in vitro study is that behavior of dissociated and cultured sensory neurons likely reflect the in vivo function of injured instead of naïve sensory neurons, and the observed plateaued response of Nse-BMP4 neurons to BMP4 treatment likely reflects the saturation of BMP signaling in this condition.
NK1r antagonist inhibits injury induced inflammation and HO
These observations suggested that inhibition of the SP receptor, NK1r, might therefore be a novel treatment for preventing the early events that lead to HO. To test this hypothesis, we treated Nse-BMP4 mice (2 months old, without pre-existing HO) with the specific NK1r antagonist RP-67580(45, 57, 58) or PBS after superficial or deep muscle injury. The percentage of mice that developed HO after superficial muscle injury in RP-67580 treated groups (40%) was significantly lower than in the control group (80%) four weeks after injury (Fig. 6), indicating that RP-67580 inhibited HO formation. Similar result was also observed by micro-CT analysis from CTX induced deep muscle injury model (fig. 6). Overall these findings suggest that an NK1r antagonist can effectively prevent HO.
To directly test if NK1r antagonists prevent HO by inhibiting the early inflammatory response, two additional experiments were preformed. We found the edema and inflammation were markedly reduced in RP-67580 treated Nse-BMP4 mice both grossly and histologically (fig. 6). Conversely, administering the same treatment to older Nse-BMP4 mice that had already developed HO provided no observable beneficial effect in RP-67580 treated group (data not shown). Both experiments suggested that the Nk1r antagonist (RP-67580) worked mainly through blocking the initial inflammatory response.
Endogenous SP signaling is essential for HO
To directly test the requirement for SP signaling for HO, we mated Nse-BMP4 mice with SP precursor null mutant (PPT-A−/−) mice to generate Nse-BMP4; PPT-A−/− double mutant mice. These mice survive without any gross phenotype. However, unlike the Nse-BMP4 mice, the double mutant mice failed to form HO in response to injury (Fig. 6) indicating that endogenous SP signaling is essential for HO formation. To further determine if the observed phenotype is SP signaling specific, we mated Nse-BMP4 mice with SP receptor NK1r−/− mice(44) and subjected them to similar injury. We found that Nse-BMP4;NK1r−/− mice form HO with reduced efficacy (Fig. 6), which essentially mimicked the phenotype of Nse-BMP4; PPT-A−/− double mutant mice, and further supporting the conclusion that SP signaling plays a central role in HO formation.
Mast cells are required for the SP-mediated induction of HO
SP induces release of other inflammatory mediators through Nk1r mast cell-dependent pathways(23, 59), which stimulates further leukocyte recruitment, thereby amplifying the inflammatory response. We reasoned that this amplification might be crucial for the transgene dependent abnormal injury responses. More importantly, recent work further suggested that degranulation of mast cells required direct interaction between mast cells and sensory nerve terminals(60, 61) and specific NK1r expression was observed on cell surface of mast cells(30).
Not surprisingly, we observed massive mast cells infiltration after muscle injury in Nse-BMP4 mice, compared to that of WT controls. More interestingly, mast cells were highly enriched in areas surrounding early inflammation, often immediately adjacent to, or contacted directly with fibers of neurons (NF200+) (data not shown), and also in proximity to small-medium sized blood vessels. This pattern is consistent with the proposed early proinflammatory function of mast cells (Suppl. Fig. 13 &14).
To directly test whether mast cells are necessary for HO, we utilized the mast cell deficient mouse line, c-kitw-sh/w-sh (62). We first generated Nse-BMP4; c-kitw-sh/w-sh double transgenic mice and confirmed that mast cells are indeed deficient in these double transgenic mice by counting the toluidine blue-positive cells (mast cells) in the skin of Nse-BMP4;c-kitw-sh/w-sh double mutant and c-kitw-sh/w-sh mice. We found that mast cells were virtually absent in these double mutants (Suppl. Fig. 15). Previous reports have indicated that mast cells mediate downstream effects of SP(59). Consistently, we found that both Nse-BMP4 single transgenic mice and Nse-BMP4; c-kitw-sh/w-sh double mutant mice had similar levels of injury-induced SP up-regulation (data not shown). However, the efficiency of HO formation was dramatically reduced in Nse-BMP4; c-kitw-sh/w-sh double mutant mice in response to injury, compared to Nse-BMP4 single transgenic mice (Fig. 6), indicating that mast cells (or factors from mast cells) are required to mediate the downstream injury response to produce HO. Since mast cells are not known to produce any chondrogenic or osteogenic factors directly, it is likely that mast cells contribute to HO indirectly by amplifying the injury response, at least in part through the SP receptor (NK1r).
Discussion
An elusive neuro-inflammatory connection to HO has long been suspected. We show here that neuro-inflammatory signaling through Substance P (SP) induces and mediates BMP-dependent heterotopic ossification. Our study shows that a critical inductive event in HO is the neural release of BMP-dependent SP. Dramatic up-regulation of SP was observed not only in patients with FOP and acquired HO, but also in three independent animal models of HO. Importantly, blocking neuron-specific SP signaling through the NK1r receptor abrogates HO formation. Since the Nk1r inhibitor RP-67580 crosses the blood-brain barrier poorly, this drug likely acted on peripheral tissues rather than the central nervous system.(63, 64) This conclusion is supported by the observations that ablation of SP+ neurons in DRG or ablation of the mast cell-dependent local amplifying circuitry dramatically reduced injury-induced HO. Null mutation of the PPT gene completely blocked HO formation, further implicating SP signaling in the pathogenesis of the disorder. Taken together, these data implicate neuron-specific SP up-regulation as a common neuro-inflammatory inductive factor for hereditary and sporadic HO and provide a molecular target for therapeutic intervention.
Our data also show that anterograde stimulation of DRG likely induces the retrograde transport of SP not only to the skin, but also to skeletal muscle and other connective tissues where it acts on NK1r receptors on tissue mast cells to amplify the inflammatory response and stimulate HO. Taken together, our studies in three independent animal models suggest that elevated BMP signaling indirectly influences the expression and release of SP in injured site through a paracrine mediated mechanism.
Although it is generally recognized that SP expression is increased in many inflammatory conditions, there is currently no consensus on how SP is regulated at the mRNA and protein levels in DRG in response to injury(65) (66). Measurement of total levels of SP or its transcripts did not reveal the critical pattern changes that were found in this study suggesting that these measures are not sufficient to gauge changes in the regulation of SP in DRG. Rather, our data suggest that the unique staining pattern in DRG reflect increased release of SP in response to injury. DRG neurons are pseudo-unipolar with axons that bifurcate into two branches, a distal process that innervates peripheral tissues (e.g., skin and muscles) and a central process that innervates spinal cord. The velocity or efficiency of axoplasmic transportation along the two branches is asymmetrical, even though the diameters of two branches are similar(67), and 80% of SP is transported peripherally and only 20% centrally(68). This may explain why the dramatic SP up-regulation was observed in the periphery, but not in the spinal cord. SP is also known to be expressed by some non-neuronal cells, and release of the peptide from these cells could be up-regulated in some pathological conditions(69). However, the role of the non-neuronal SP in HO is still unclear.
Our demonstration that mast cells are a downstream target of SP in this neuro-inflammatory process has important therapeutic implications and is consistent with prior studies.(70-72) Mast cells are present in all HO lesions and are particularly abundant at all stages of FOP lesions and in BMP4-induced HO(72). Our findings are also consistent with previous findings showing that sprouting peripheral nerve fibers is a common observation in new bone formation(73) and fracture healing(74). Interestingly, it was also reported that, in context of other lesions, SP-positive nerves had more contacts with mast cells compared to VIP- or CGRP-containing fibers, and tryptase, a mast cell specific proteinase, could degrade VIP and CGRP, but not SP(75). These, together with our data, may explain why only SP is specifically up-regulated.
Our current working hypothesis is that following injury, BMP signaling up-regulates SP release in DRG neurons which, in turn, activates mast cells that produce factors that amplify local inflammation. Little is known about the post-inflammatory pre-osseous events in HO, but SP may also recruit local progenitors/stem cells to form a fibroproiferative lesion that is common to all forms of sporadic and hereditary heterotopic endochondral ossification(40). A recent study indicates that vascular endothelial cells can be transformed into multipotent stem-like cells in an ACVR1 or BMP4-dependent manner through an endothelial-to-mesenchymal transition, consistent with both the molecular genetics and pathology of FOP(76). Based on the study, it is reasonable to postulate that dysregulated BMP signaling plays dual roles in the chronicle phase of HO induction: first, in the induction of stem-like cells, and then in their endochondral transformation to heterotopic bone. Our data show that SP acts downstream of BMP signaling to orchestrate this complex process.
Overall, the current study has profound therapeutic implications. Our study identifies SP as a potent inductive neuro-inflammatory factor for HO and places it in a neuro-inflammatory amplification circuit that provides novel cellular and molecular targets for therapeutic intervention. Our findings specifically suggest that NK1r antagonists or mast cell inhibitors might provide specific chronic targets for preventing bone formation, in both hereditary and acquired forms of HO.
Supplementary Material
Tissue sections (adult tail sections shown) from PPT-A knockout (A), and Nse-BMP4 (B) mice were stained with SP antibody according to standard protocol. Specific signal was found in skin and connective tissues in Nse-BMP4 (shown, B) and WT (not shown) mice, but no detectable signal was found in the same tissue of PPT-A−/− animals (A). (C-F) peptide blocking experiment further confirmed that this antibody doesn't crossreact with NKA and NKB peptides. (C) shows a typical image of SP staining in CTX injured lesion from Nse-BMP4 mice. (D) shows that SP peptide completely blocked the staining, while peptide NKA (E) or NKB (F) didn't block the specific staining.
(A) shows a typical image of SP/NF-200 double staining in an early lesion from sample of FOP. Note that there is extensive co-localization of SP and NF-200. (B) shows the phase image of (A). White * indicated degenerating/degenerated muscle fibers. (C) shows a typical image of SP/NF-200 double staining in mature HO from the same patient. Note that there is only one instance of co-localization of SP and NF-200 (white arrow). (D) is the phase image of (C).
Note that 5/6 samples have well defined early lesions, and only 2/6 samples have typical mature bone.
i.e., Spinal Cord Injury (SCI), Traumatic Brain Injury (TBI), Non-Neurologic Trauma (NNT) and Total Hip Arthroplasty (THA).
Adult tissue sections (lymph node, A-D, spleen, E&F, thymus, G&H and brain, I-M) were immunostained for SP (A-D), or SP/CD11b (E&F), or SP/NPY (G-N). Subtle SP up-regulation with a more diffuse staining pattern was observed in lymph node, spleen, and thymus of Nse-BMP4 mice (compare A&B, C&D, E&F and G&H). There were no appreciable changes in levels or patterns of expression in the CNS (compare I&J, K&L and M&N) except for very subtle SP up-regulation in the CAN(central amygdalar nucleus) (M&N). GC (in E&F), germinal center, ARH (in J&K), arcuate hypothalamic nucleus, DMHa (in L&M), dorsomedial nucleus of the hypothalamus, anterior part.
(A & B) show the SP/NF-200 double staining in subcutaneous connective tissue (A) and muscles (B) of Nse-BMP4 mice. White arrows point to the co-localization of SP and NF-200 in (A&B).
(A & B) show the typical images of GFP/SP double staining from mice that 4 days after injection of adeno cre(A) or empty vector (B). Note that in the induced lesion (A), SP was markedly up-regulated, comparing to the unaffected region (A) or induced lesion in control (B). (C & D) show the similar caALK2 dependent SP up-regulation 8 days after injection. Broken white lines in (A-D) represent the boundaries between the unaffected tissues and induced lesions.
(A) shows the merged image of SP/GFP/NF-200 triple staining, and (A′, A″ and A‴) show the split channel of NF-200, SP and GFP, respectively. Note that robust SP co-localized with NF-200 extensively (thin white arrows), but not co-localized with GFP (caALK2+) cells. Bold white arrow points to a GFP/NF-200 double staining.
Typical images of SP staining from the injection site of a mouse that injected with BMP4 matrigel (A), or BMP4 matrigel+ CTX (B), or matrigel only (C), or matrigel +CTX (D). Note that even though CTX alone (D) has unappreciable ability to up-regulate SP, local SP up-regulation was more obvious in the mice that were treated with CTX+BMP4 matrigel (B) than that of BMP4 matrigel alone(A). (E) quantified the relative SP levels in different conditions. * P<0.05 vs gel control or BMP4+CTX.
(A) lumbar DRG from 3 week old WT (A) and (B) Nse-BMP4 mice. (C) quantitation of single SP+ and double SP+p75+ neurons in lumbar DRG of WT and Nse-BMP4 mice (3 weeks old).
Small diameter SP+ sensory neurons were virtually absent in capsaicin pretreated DRG (A) & (B), compared to vehicle treated ones (C)&(D). DRG depicted in (A&C) are from uninjured side (ctr), while (B & D) show the DRG from injured side. Note that superficial muscles injury did not lead to the phenotypic change in WT DRG that was observed in Nse-BMP4 mice.
(A-D) show the images of SP/NF-200 double stained cultured sensory neurons from Nse-BMP4 (A&B) and WT(C&D) under control (A&C) or BMP4 treated (B&D) conditions. (E) depicts SP concentration in the supernents. * P<0.05 vs control. ** P<0.01 vs WT control, NS, none significant. (F) depicts the percent of SP+ neurons in each condition. * P<0.05 vs WT control Note that neurons from WT mice release more SP in response to BMP4 treatment, and neurons from Nse-BMP4 mice have up-regulated SP expression and release more SP, conversely, the expression and release of SP were inhibited by Noggin treatment (data not shown).
Images of Toluidine blue staining from early (day 4 after injury, A&B) and later (day 8 after injury, C&D) show dramatic mast cell infiltration in Nse-BMP4 (A&C), comparing to WT (B&D) mice response to the similar injury. Arrows in (A-C) point to metachromatic stained (red-purple) mast cells. Note that mast cells enriched in surrounding early inflammatory areas not more mature regions, often in proximity to small-medium sized blood vessels. BV, blood vessels. (E) (E) quantifies the Toluidine blue+ cells/area in (A-D). * P<0.01 vs WT control,
Massive mast cells infiltration was found in injured muscle of Nse-BMP4 mice (A), while the mast cells infiltration in similarly injured WT mice was much milder (B). No appreciable staining was found in uninjured muscles, either from Nse-BMP4 (C) or WT (D) mice. (E) quantifies the LAMP2+ cells/area in (A-D). * P<0.01 vs WT control,
Typical toluidine blue stained skin sections from (A) c-kitw-sh/w-sh, (B) Nse-BMP4;c-kitw-sh/w-sh and (C) WT mice. Black arrows in (C) point to metachromatic stained (red-purple) mast cells. (D) quantitative summary of (A-C).
Acknowledgments
We appreciate the help from many members of the Kessler lab, the assistance provided by CCM Animal Health Technicians in the acquisition of digital radiographs, and the professional help provided by Dr. Xiao-Qi Wang of Department of Dermatology, Northwestern University. We thank Dr. Mary Ann Keenan and Dr. John Esterhai in the Department of Orthopaedic Surgery, University of Pennsylvania, for samples of acquired HO. LK was supported in part by grants to from The Center for Research in FOP and Related Disorders of The University of Pennsylvania School of Medicine. JAK was supported by NIH grants NS20013 and NS20778. This work was also supported in part by the Center for Research in FOP and Related Disorders at The Perelman School of Medicine of The University of Pennsylvania, the International FOP Association, the Ian Cali Endowment, the Weldon Family Endowment, the Penn Center for Musculoskeletal Disorders, The Isaac & Rose Nassau Professorship of Orthopaedic Molecular Medicine and by grants from the Rita Allen Foundation and the NIH (R01-AR40196) to FSK and EMS.
Footnotes
Author contributions: L.K.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; S. R. S., R. J. P., E. M. S. and F. S. K.: provision of study material, technical advice, data analysis and manuscript editing, V. Y. L., L. D., Y.L. and T.M.: collection and/or assembly of data; B.L. and N.P.G.: provision of study material; J.A.K.: data analysis, manuscript editing, approval of manuscript.
References
- 1.Salisbury E, Sonnet C, Heggeness M, Davis AR, Olmsted-Davis E. Heterotopic ossification has some nerve. Crit Rev Eukaryot Gene Expr. 2010;20:313–324. doi: 10.1615/critreveukargeneexpr.v20.i4.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Forsberg JA, Potter BK. Heterotopic ossification in wartime wounds. J Surg Orthop Adv. 2010;19:54–61. [PubMed] [Google Scholar]
- 3.Cullen N, Perera J. Heterotopic ossification: pharmacologic options. J Head Trauma Rehabil. 2009;24:69–71. doi: 10.1097/HTR.0b013e31819a8fcc. [DOI] [PubMed] [Google Scholar]
- 4.Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan FS, Glaser DL, Shore EM, Pignolo RJ, Xu M, Zhang Y, Senitzer D, Forman SJ, Emerson SG. Hematopoietic stem-cell contribution to ectopic skeletogenesis. J Bone Joint Surg Am. 2007;89:347–357. doi: 10.2106/JBJS.F.00472. [DOI] [PubMed] [Google Scholar]
- 6.Sawyer JR, Myers MA, Rosier RN, Puzas JE. Heterotopic ossification: clinical and cellular aspects. Calcif Tissue Int. 1991;49:208–215. doi: 10.1007/BF02556120. [DOI] [PubMed] [Google Scholar]
- 7.Kaplan FS, Shen Q, Lounev V, Seemann P, Groppe J, Katagiri T, Pignolo RJ, Shore EM. Skeletal metamorphosis in fibrodysplasia ossificans progressiva (FOP) J Bone Miner Metab. 2008;26:521–530. doi: 10.1007/s00774-008-0879-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forss-Petter S, Danielson PE, Catsicas S, Battenberg E, Price J, Nerenberg M, Sutcliffe JG. Transgenic mice expressing beta-galactosidase in mature neurons under neuron-specific enolase promoter control. Neuron. 1990;5:187–197. doi: 10.1016/0896-6273(90)90308-3. [DOI] [PubMed] [Google Scholar]
- 9.Kan L, Liu Y, McGuire TL, Berger DM, Awatramani RB, Dymecki SM, Kessler JA. Dysregulation of local stem/progenitor cells as a common cellular mechanism for heterotopic ossification. Stem Cells. 2009;27:150–156. doi: 10.1634/stemcells.2008-0576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mukhopadhyay A, McGuire T, Peng CY, Kessler JA. Differential effects of BMP signaling on parvalbumin and somatostatin interneuron differentiation. Development. 2009;136:2633–2642. doi: 10.1242/dev.034439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kan L, Kessler JA. Animal models of typical heterotopic ossification. J Biomed Biotechnol. 2011;2011:309287. doi: 10.1155/2011/309287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 13.Czura CJ, Tracey KJ. Autonomic neural regulation of immunity. J Intern Med. 2005;257:156–166. doi: 10.1111/j.1365-2796.2004.01442.x. [DOI] [PubMed] [Google Scholar]
- 14.Marques-Deak A, Cizza G, Sternberg E. Brain-immune interactions and disease susceptibility. Mol Psychiatry. 2005;10:239–250. doi: 10.1038/sj.mp.4001643. [DOI] [PubMed] [Google Scholar]
- 15.Savastano S, Tommaselli AP, Valentino R, Scarpitta MT, D'Amore G, Luciano A, Covelli V, Lombardi G. Hypothalamic-pituitary-adrenal axis and immune system. Acta Neurol (Napoli) 1994;16:206–213. [PubMed] [Google Scholar]
- 16.Rowley DA, Kohler H, Cowan JD. An immunologic network. Contemp Top Immunobiol. 1980;9:205–230. doi: 10.1007/978-1-4615-9131-3_8. [DOI] [PubMed] [Google Scholar]
- 17.Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–859. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- 18.Lerner UH, Persson E. Osteotropic effects by the neuropeptides calcitonin gene-related peptide, substance P and vasoactive intestinal peptide. J Musculoskelet Neuronal Interact. 2008;8:154–165. [PubMed] [Google Scholar]
- 19.Bergstrom J, Ahmed M, Li J, Ahmad T, Kreicbergs A, Spetea M. Opioid peptides and receptors in joint tissues: study in the rat. J Orthop Res. 2006;24:1193–1199. doi: 10.1002/jor.20132. [DOI] [PubMed] [Google Scholar]
- 20.Allison SJ, Baldock PA, Enriquez RF, Lin E, During M, Gardiner EM, Eisman JA, Sainsbury A, Herzog H. Critical interplay between neuropeptide Y and sex steroid pathways in bone and adipose tissue homeostasis. J Bone Miner Res. 2009;24:294–304. doi: 10.1359/jbmr.081013. [DOI] [PubMed] [Google Scholar]
- 21.McGillis JP, Organist ML, Payan DG. Substance P and immunoregulation. Fed Proc. 1987;46:196–199. [PubMed] [Google Scholar]
- 22.Mantyh PW. Substance P and the inflammatory and immune response. Ann N Y Acad Sci. 1991;632:263–271. doi: 10.1111/j.1749-6632.1991.tb33114.x. [DOI] [PubMed] [Google Scholar]
- 23.Yano H, Wershil BK, Arizono N, Galli SJ. Substance P-induced augmentation of cutaneous vascular permeability and granulocyte infiltration in mice is mast cell dependent. J Clin Invest. 1989;84:1276–1286. doi: 10.1172/JCI114295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edoff K, Hellman J, Persliden J, Hildebrand C. The developmental skeletal growth in the rat foot is reduced after denervation. Anat Embryol (Berl) 1997;195:531–538. doi: 10.1007/s004290050073. [DOI] [PubMed] [Google Scholar]
- 25.Li J, Ahmed M, Bergstrom J, Ackermann P, Stark A, Kreicbergs A. Occurrence of substance P in bone repair under different load comparison of straight and angulated fracture in rat tibia. J Orthop Res. 2010;28:1643–1650. doi: 10.1002/jor.21169. [DOI] [PubMed] [Google Scholar]
- 26.Millward-Sadler SJ, Mackenzie A, Wright MO, Lee HS, Elliot K, Gerrard L, Fiskerstrand CE, Salter DM, Quinn JP. Tachykinin expression in cartilage and function in human articular chondrocyte mechanotransduction. Arthritis Rheum. 2003;48:146–156. doi: 10.1002/art.10711. [DOI] [PubMed] [Google Scholar]
- 27.Goto T, Yamaza T, Kido MA, Tanaka T. Light- and electron-microscopic study of the distribution of axons containing substance P and the localization of neurokinin-1 receptor in bone. Cell Tissue Res. 1998;293:87–93. doi: 10.1007/s004410051100. [DOI] [PubMed] [Google Scholar]
- 28.Goto T, Kido MA, Yamaza T, Tanaka T. Substance P and substance P receptors in bone and gingival tissues. Med Electron Microsc. 2001;34:77–85. doi: 10.1007/s007950170001. [DOI] [PubMed] [Google Scholar]
- 29.Wang L, Zhao R, Shi X, Wei T, Halloran BP, Clark DJ, Jacobs CR, Kingery WS. Substance P stimulates bone marrow stromal cell osteogenic activity, osteoclast differentiation, and resorption activity in vitro. Bone. 2009;45:309–320. doi: 10.1016/j.bone.2009.04.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okada T, Hirayama Y, Kishi S, Miyayasu K, Hiroi J, Fujii T. Functional neurokinin NK-1 receptor expression in rat peritoneal mast cells. Inflamm Res. 1999;48:274–279. doi: 10.1007/s000110050459. [DOI] [PubMed] [Google Scholar]
- 31.Barbut D, Polak JM, Wall PD. Substance P in spinal cord dorsal horn decreases following peripheral nerve injury. Brain Res. 1981;205:289–298. doi: 10.1016/0006-8993(81)90340-1. [DOI] [PubMed] [Google Scholar]
- 32.Datar P, Srivastava S, Coutinho E, Govil G. Substance P: structure, function, and therapeutics. Curr Top Med Chem. 2004;4:75–103. doi: 10.2174/1568026043451636. [DOI] [PubMed] [Google Scholar]
- 33.Zubrzycka M, Janecka A. Substance P: transmitter of nociception (Minireview) Endocr Regul. 2000;34:195–201. [PubMed] [Google Scholar]
- 34.Pinto FM, Almeida TA, Hernandez M, Devillier P, Advenier C, Candenas ML. mRNA expression of tachykinins and tachykinin receptors in different human tissues. Eur J Pharmacol. 2004;494:233–239. doi: 10.1016/j.ejphar.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 35.Nelson DA, Bost KL. Non-neuronal mammalian tachykinin expression. Front Biosci. 2004;9:2166–2176. doi: 10.2741/1372. [DOI] [PubMed] [Google Scholar]
- 36.Quartara L, Maggi CA. The tachykinin NK1 receptor. Part II: Distribution and pathophysiological roles. Neuropeptides. 1998;32:1–49. doi: 10.1016/s0143-4179(98)90015-4. [DOI] [PubMed] [Google Scholar]
- 37.Goto T, Tanaka T. Tachykinins and tachykinin receptors in bone. Microsc Res Tech. 2002;58:91–97. doi: 10.1002/jemt.10123. [DOI] [PubMed] [Google Scholar]
- 38.Nakaya Y, Kaneko T, Shigemoto R, Nakanishi S, Mizuno N. Immunohistochemical localization of substance P receptor in the central nervous system of the adult rat. J Comp Neurol. 1994;347:249–274. doi: 10.1002/cne.903470208. [DOI] [PubMed] [Google Scholar]
- 39.Donkin JJ, Nimmo AJ, Cernak I, Blumbergs PC, Vink R. Substance P is associated with the development of brain edema and functional deficits after traumatic brain injury. J Cereb Blood Flow Metab. 2009;29:1388–1398. doi: 10.1038/jcbfm.2009.63. [DOI] [PubMed] [Google Scholar]
- 40.Hong HS, Lee J, Lee E, Kwon YS, Ahn W, Jiang MH, Kim JC, Son Y. A new role of substance P as an injury-inducible messenger for mobilization of CD29(+) stromal-like cells. Nat Med. 2009;15:425–435. doi: 10.1038/nm.1909. [DOI] [PubMed] [Google Scholar]
- 41.Sharma HS, Nyberg F, Olsson Y, Dey PK. Alteration of substance P after trauma to the spinal cord: an experimental study in the rat. Neuroscience. 1990;38:205–212. doi: 10.1016/0306-4522(90)90386-i. [DOI] [PubMed] [Google Scholar]
- 42.Donkin JJ, Turner RJ, Hassan I, Vink R. Substance P in traumatic brain injury. Prog Brain Res. 2007;161:97–109. doi: 10.1016/S0079-6123(06)61007-8. [DOI] [PubMed] [Google Scholar]
- 43.Kan L, Hu M, Gomes WA, Kessler JA. Transgenic mice overexpressing BMP4 develop a fibrodysplasia ossificans progressiva (FOP)-like phenotype. Am J Pathol. 2004;165:1107–1115. doi: 10.1016/S0002-9440(10)63372-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bozic CR, Lu B, Hopken UE, Gerard C, Gerard NP. Neurogenic amplification of immune complex inflammation. Science. 1996;273:1722–1725. doi: 10.1126/science.273.5282.1722. [DOI] [PubMed] [Google Scholar]
- 45.Kennedy PG, Rodgers J, Jennings FW, Murray M, Leeman SE, Burke JM. A substance P antagonist, RP-67,580, ameliorates a mouse meningoencephalitic response to Trypanosoma brucei brucei. Proc Natl Acad Sci U S A. 1997;94:4167–4170. doi: 10.1073/pnas.94.8.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stock SR, Ignatiev KI, Foster SA, Forman LA, Stern PH. MicroCT quantification of in vitro bone resorption of neonatal murine calvaria exposed to IL-1 or PTH. J Struct Biol. 2004;147:185–199. doi: 10.1016/j.jsb.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 47.Sugimoto T, Xiao C, Ichikawa H. Neonatal primary neuronal death induced by capsaicin and axotomy involves an apoptotic mechanism. Brain Res. 1998;807:147–154. doi: 10.1016/s0006-8993(98)00788-4. [DOI] [PubMed] [Google Scholar]
- 48.Malin SA, Davis BM, Molliver DC. Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nat Protoc. 2007;2:152–160. doi: 10.1038/nprot.2006.461. [DOI] [PubMed] [Google Scholar]
- 49.Thornton E, Ziebell JM, Leonard AV, Vink R. Kinin receptor antagonists as potential neuroprotective agents in central nervous system injury. Molecules. 2010;15:6598–6618. doi: 10.3390/molecules15096598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao YQ, Mantyh PW, Carlson EJ, Gillespie AM, Epstein CJ, Basbaum AI. Primary afferent tachykinins are required to experience moderate to intense pain. Nature. 1998;392:390–394. doi: 10.1038/32897. [DOI] [PubMed] [Google Scholar]
- 51.Kaku Y, Yonekawa Y, Tsukahara T, Ogata N, Kimura T, Taniguchi T. Alterations of a 200 kDa neurofilament in the rat hippocampus after forebrain ischemia. J Cereb Blood Flow Metab. 1993;13:402–408. doi: 10.1038/jcbfm.1993.54. [DOI] [PubMed] [Google Scholar]
- 52.Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML, Hong DW, McManus PM, Katagiri T, Sachidanandan C, Kamiya N, Fukuda T, Mishina Y, Peterson RT, Bloch KD. BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat Med. 2008;14:1363–1369. doi: 10.1038/nm.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV, Jaenisch R. Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell. 1992;69:737–749. doi: 10.1016/0092-8674(92)90286-l. [DOI] [PubMed] [Google Scholar]
- 54.Paulson JC, McClure WO. Inhibition of axoplasmic transport by colchicine, podophyllotoxin, and vinblastine: an effect on microtubules. Ann N Y Acad Sci. 1975;253:517–527. doi: 10.1111/j.1749-6632.1975.tb19225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andoh T, Nagasawa T, Kuraishi Y. Expression of tachykinin NK1 receptor mRNA in dorsal root ganglia of the mouse. Brain Res Mol Brain Res. 1996;35:329–332. doi: 10.1016/0169-328x(95)00244-m. [DOI] [PubMed] [Google Scholar]
- 56.Nagy JI, Hunt SP, Iversen LL, Emson PC. Biochemical and anatomical observations on the degeneration of peptide-containing primary afferent neurons after neonatal capsaicin. Neuroscience. 1981;6:1923–1934. doi: 10.1016/0306-4522(81)90032-4. [DOI] [PubMed] [Google Scholar]
- 57.Shepheard SL, Williamson DJ, Hill RG, Hargreaves RJ. The non-peptide neurokinin1 receptor antagonist, RP 67580, blocks neurogenic plasma extravasation in the dura mater of rats. Br J Pharmacol. 1993;108:11–12. doi: 10.1111/j.1476-5381.1993.tb13432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moussaoui SM, Montier F, Carruette A, Blanchard JC, Laduron PM, Garret C. A non-peptide NK1-receptor antagonist, RP 67580, inhibits neurogenic inflammation postsynaptically. Br J Pharmacol. 1993;109:259–264. doi: 10.1111/j.1476-5381.1993.tb13562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saban R, Gerard NP, Saban MR, Nguyen NB, DeBoer DJ, Wershil BK. Mast cells mediate substance P-induced bladder inflammation through an NK(1) receptor-independent mechanism. Am J Physiol Renal Physiol. 2002;283:F616–629. doi: 10.1152/ajprenal.00096.2002. [DOI] [PubMed] [Google Scholar]
- 60.Folgueras AR, Valdes-Sanchez T, Llano E, Menendez L, Baamonde A, Denlinger BL, Belmonte C, Juarez L, Lastra A, Garcia-Suarez O, Astudillo A, Kirstein M, Pendas AM, Farinas I, Lopez-Otin C. Metalloproteinase MT5-MMP is an essential modulator of neuro-immune interactions in thermal pain stimulation. Proc Natl Acad Sci U S A. 2009;106:16451–16456. doi: 10.1073/pnas.0908507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suzuki A, Suzuki R, Furuno T, Teshima R, Nakanishi M. N-cadherin plays a role in the synapse-like structures between mast cells and neurites. Biol Pharm Bull. 2004;27:1891–1894. doi: 10.1248/bpb.27.1891. [DOI] [PubMed] [Google Scholar]
- 62.Duttlinger R, Manova K, Chu TY, Gyssler C, Zelenetz AD, Bachvarova RF, Besmer P. W-sash affects positive and negative elements controlling c-kit expression: ectopic c-kit expression at sites of kit-ligand expression affects melanogenesis. Development. 1993;118:705–717. doi: 10.1242/dev.118.3.705. [DOI] [PubMed] [Google Scholar]
- 63.Holzer-Petsche U, Rordorf-Nikolic T. Central versus peripheral site of action of the tachykinin NK1-antagonist RP 67580 in inhibiting chemonociception. Br J Pharmacol. 1995;115:486–490. doi: 10.1111/j.1476-5381.1995.tb16359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barr AJ, Watson SP. Non-peptide antagonists, CP-96,345 and RP 67580, distinguish species variants in tachykinin NK1 receptors. Br J Pharmacol. 1993;108:223–227. doi: 10.1111/j.1476-5381.1993.tb13466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Castagliuolo I, Riegler M, Pasha A, Nikulasson S, Lu B, Gerard C, Gerard NP, Pothoulakis C. Neurokinin-1 (NK-1) receptor is required in Clostridium difficile- induced enteritis. J Clin Invest. 1998;101:1547–1550. doi: 10.1172/JCI2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reinshagen M, Patel A, Sottili M, Nast C, Davis W, Mueller K, Eysselein V. Protective function of extrinsic sensory neurons in acute rabbit experimental colitis. Gastroenterology. 1994;106:1208–1214. doi: 10.1016/0016-5085(94)90011-6. [DOI] [PubMed] [Google Scholar]
- 67.Ochs S, Erdman J, Jersild RA, Jr, McAdoo V. Routing of transported materials in the dorsal root and nerve fiber branches of the dorsal root ganglion. J Neurobiol. 1978;9:465–481. doi: 10.1002/neu.480090606. [DOI] [PubMed] [Google Scholar]
- 68.Harmar A, Keen P. Synthesis, and central and peripheral axonal transport of substance P in a dorsal root ganglion-nerve preparation in vitro. Brain Res. 1982;231:379–385. doi: 10.1016/0006-8993(82)90374-2. [DOI] [PubMed] [Google Scholar]
- 69.Castagliuolo I, Keates AC, Qiu B, Kelly CP, Nikulasson S, Leeman SE, Pothoulakis C. Increased substance P responses in dorsal root ganglia and intestinal macrophages during Clostridium difficile toxin A enteritis in rats. Proc Natl Acad Sci U S A. 1997;94:4788–4793. doi: 10.1073/pnas.94.9.4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kulka M, Sheen CH, Tancowny BP, Grammer LC, Schleimer RP. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology. 2008;123:398–410. doi: 10.1111/j.1365-2567.2007.02705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Freeman TA, Parvizi J, Dela Valle CJ, Steinbeck MJ. Mast cells and hypoxia drive tissue metaplasia and heterotopic ossification in idiopathic arthrofibrosis after total knee arthroplasty. Fibrogenesis Tissue Repair. 2010;3:17. doi: 10.1186/1755-1536-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gannon FH, Glaser D, Caron R, Thompson LD, Shore EM, Kaplan FS. Mast cell involvement in fibrodysplasia ossificans progressiva. Hum Pathol. 2001;32:842–848. doi: 10.1053/hupa.2001.26464. [DOI] [PubMed] [Google Scholar]
- 73.Hedberg A, Messner K, Persliden J, Hildebrand C. Transient local presence of nerve fibers at onset of secondary ossification in the rat knee joint. Anat Embryol (Berl) 1995;192:247–255. doi: 10.1007/BF00184749. [DOI] [PubMed] [Google Scholar]
- 74.Madsen JE, Hukkanen M, Aune AK, Basran I, Moller JF, Polak JM, Nordsletten L. Fracture healing and callus innervation after peripheral nerve resection in rats. Clin Orthop Relat Res. 1998:230–240. [PubMed] [Google Scholar]
- 75.Naukkarinen A, Harvima IT, Aalto ML, Horsmanheimo M. Mast cell tryptase and chymase are potential regulators of neurogenic inflammation in psoriatic skin. Int J Dermatol. 1994;33:361–366. doi: 10.1111/j.1365-4362.1994.tb01069.x. [DOI] [PubMed] [Google Scholar]
- 76.Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med. 2010;16:1400–1406. doi: 10.1038/nm.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tissue sections (adult tail sections shown) from PPT-A knockout (A), and Nse-BMP4 (B) mice were stained with SP antibody according to standard protocol. Specific signal was found in skin and connective tissues in Nse-BMP4 (shown, B) and WT (not shown) mice, but no detectable signal was found in the same tissue of PPT-A−/− animals (A). (C-F) peptide blocking experiment further confirmed that this antibody doesn't crossreact with NKA and NKB peptides. (C) shows a typical image of SP staining in CTX injured lesion from Nse-BMP4 mice. (D) shows that SP peptide completely blocked the staining, while peptide NKA (E) or NKB (F) didn't block the specific staining.
(A) shows a typical image of SP/NF-200 double staining in an early lesion from sample of FOP. Note that there is extensive co-localization of SP and NF-200. (B) shows the phase image of (A). White * indicated degenerating/degenerated muscle fibers. (C) shows a typical image of SP/NF-200 double staining in mature HO from the same patient. Note that there is only one instance of co-localization of SP and NF-200 (white arrow). (D) is the phase image of (C).
Note that 5/6 samples have well defined early lesions, and only 2/6 samples have typical mature bone.
i.e., Spinal Cord Injury (SCI), Traumatic Brain Injury (TBI), Non-Neurologic Trauma (NNT) and Total Hip Arthroplasty (THA).
Adult tissue sections (lymph node, A-D, spleen, E&F, thymus, G&H and brain, I-M) were immunostained for SP (A-D), or SP/CD11b (E&F), or SP/NPY (G-N). Subtle SP up-regulation with a more diffuse staining pattern was observed in lymph node, spleen, and thymus of Nse-BMP4 mice (compare A&B, C&D, E&F and G&H). There were no appreciable changes in levels or patterns of expression in the CNS (compare I&J, K&L and M&N) except for very subtle SP up-regulation in the CAN(central amygdalar nucleus) (M&N). GC (in E&F), germinal center, ARH (in J&K), arcuate hypothalamic nucleus, DMHa (in L&M), dorsomedial nucleus of the hypothalamus, anterior part.
(A & B) show the SP/NF-200 double staining in subcutaneous connective tissue (A) and muscles (B) of Nse-BMP4 mice. White arrows point to the co-localization of SP and NF-200 in (A&B).
(A & B) show the typical images of GFP/SP double staining from mice that 4 days after injection of adeno cre(A) or empty vector (B). Note that in the induced lesion (A), SP was markedly up-regulated, comparing to the unaffected region (A) or induced lesion in control (B). (C & D) show the similar caALK2 dependent SP up-regulation 8 days after injection. Broken white lines in (A-D) represent the boundaries between the unaffected tissues and induced lesions.
(A) shows the merged image of SP/GFP/NF-200 triple staining, and (A′, A″ and A‴) show the split channel of NF-200, SP and GFP, respectively. Note that robust SP co-localized with NF-200 extensively (thin white arrows), but not co-localized with GFP (caALK2+) cells. Bold white arrow points to a GFP/NF-200 double staining.
Typical images of SP staining from the injection site of a mouse that injected with BMP4 matrigel (A), or BMP4 matrigel+ CTX (B), or matrigel only (C), or matrigel +CTX (D). Note that even though CTX alone (D) has unappreciable ability to up-regulate SP, local SP up-regulation was more obvious in the mice that were treated with CTX+BMP4 matrigel (B) than that of BMP4 matrigel alone(A). (E) quantified the relative SP levels in different conditions. * P<0.05 vs gel control or BMP4+CTX.
(A) lumbar DRG from 3 week old WT (A) and (B) Nse-BMP4 mice. (C) quantitation of single SP+ and double SP+p75+ neurons in lumbar DRG of WT and Nse-BMP4 mice (3 weeks old).
Small diameter SP+ sensory neurons were virtually absent in capsaicin pretreated DRG (A) & (B), compared to vehicle treated ones (C)&(D). DRG depicted in (A&C) are from uninjured side (ctr), while (B & D) show the DRG from injured side. Note that superficial muscles injury did not lead to the phenotypic change in WT DRG that was observed in Nse-BMP4 mice.
(A-D) show the images of SP/NF-200 double stained cultured sensory neurons from Nse-BMP4 (A&B) and WT(C&D) under control (A&C) or BMP4 treated (B&D) conditions. (E) depicts SP concentration in the supernents. * P<0.05 vs control. ** P<0.01 vs WT control, NS, none significant. (F) depicts the percent of SP+ neurons in each condition. * P<0.05 vs WT control Note that neurons from WT mice release more SP in response to BMP4 treatment, and neurons from Nse-BMP4 mice have up-regulated SP expression and release more SP, conversely, the expression and release of SP were inhibited by Noggin treatment (data not shown).
Images of Toluidine blue staining from early (day 4 after injury, A&B) and later (day 8 after injury, C&D) show dramatic mast cell infiltration in Nse-BMP4 (A&C), comparing to WT (B&D) mice response to the similar injury. Arrows in (A-C) point to metachromatic stained (red-purple) mast cells. Note that mast cells enriched in surrounding early inflammatory areas not more mature regions, often in proximity to small-medium sized blood vessels. BV, blood vessels. (E) (E) quantifies the Toluidine blue+ cells/area in (A-D). * P<0.01 vs WT control,
Massive mast cells infiltration was found in injured muscle of Nse-BMP4 mice (A), while the mast cells infiltration in similarly injured WT mice was much milder (B). No appreciable staining was found in uninjured muscles, either from Nse-BMP4 (C) or WT (D) mice. (E) quantifies the LAMP2+ cells/area in (A-D). * P<0.01 vs WT control,
Typical toluidine blue stained skin sections from (A) c-kitw-sh/w-sh, (B) Nse-BMP4;c-kitw-sh/w-sh and (C) WT mice. Black arrows in (C) point to metachromatic stained (red-purple) mast cells. (D) quantitative summary of (A-C).