

Abstract
Many cancers shed malignant cells into the circulation. Albeit their rare frequency, these cancer cells serve both diagnostic and therapeutic purposes. However, their purification, quantification and characterization remain challenging. Here, we present a low-cost, rapid microfluidic cell sorter (μFCS) device for the detection and molecular analysis of circulating tumor cells (CTCs). The μFCS employs a weir-shaped microfluidic structure to separate and capture CTCs from unprocessed whole blood cells based on their size difference. The system further allows on-chip culture and molecular profiling of captured cancer cells, and provides easy cell retrieval for subsequent proteomic and genetic analyses. Using a mouse model of cancer metastasis, we show that the μFCS can enrich CTCs from whole blood to unmask their cancer genetic signature. With its rapid processing speed and versatility for downstream analyses, this platform could have a wide range of potential applications in clinical cancer diagnosis.
Keywords: biotechnology, cellular profiling, circulating tumor cells, genetic analysis, microfluidics
Circulating tumor cells (CTCs) can be found in many solid neoplasms. These cells can serve as valuable markers for primary tumor diagnosis;[1-4] both their absolute concentration and their temporal changes in numbers have been used as prognostic and predictive markers, while their molecular and genetic signatures provide significant metrics relevant to tumor stratification, disease progression and therapy management. However, CTCs are rare (as few as one cell per 109 haemotologic cells). The isolation and characterization of rare cancer cells from biofluids therefore pose significant technical challenges, in particular, requiring rapid assessment of large volumes of samples and effective enrichment against a complex biological background.
Different platforms have been developed for the enrichment and detection of CTCs.[5, 6] A prevalent approach is to isolate tumor cells based on their expression of EpCAM (epithelial cell adhesion molecule), an epithelial cell surface marker commonly, though not exclusively, associated with carcinomas; this approach has been adopted to enrich CTCs through immunomagnetic separation (CellSearch) or immunocapture inside microfluidic devices.[7-9] Despite its proven utility in clinical cases where CTCs are abundant, the method is often limited by the presence of EpCAM-negative CTCs and/or fluctuations of EpCAM level in CTCs over tumor growth.[4] Alternatively, CTCs have been separated by exploiting their size differences against blood cells.[5, 10] Obviating the need for antigen-antibody binding steps, the method not only allows for significantly faster sample processing, but also collects putative CTCs regardless of their EpCAM expression. Various microfiltration devices have been implemented, utilizing either membrane or microfabricated filters for CTC collection.[11-14] Closed filter systems, however, can be limited by several factors: 1) as more cells are trapped inside the filters, the required pressure for fluidic flow concomitantly increases, imposing higher shear stress on the trapped cells; 2) flow can eventually stop from filter-clogging; 3) retrieving trapped cells is generally a challenging task with low recovery efficiency. In a bid to address these issues, different types of in-flow filter systems have been developed[15-19] (see Table S1 in Supporting Information for a detailed comparison). Although several devices have demonstrated successful CTC capture from large volumes of samples, they often lack on-chip cellular analytic capabilities; captured cells need to be transferred for further assays, which can cause cell loss and potential phenotypic changes.
We herein report a new type of CTC detection system, a microfluidic cell sorter (μFCS), for the isolation and comprehensive analyses of CTCs in whole blood. The μFCS employs a modified weir-style physical barrier to separate and capture tumor cells based on their size, and subsequently allows on-chip tumor identification through molecular characterization. The operation is performed in a continuous-flow manner, processing large volumes of samples at high flow rates without clogging or pressure buildup. The captured cells can be analyzed in situ for comprehensive and multifaceted evaluation, including single cell enumeration and imaging, molecular and genetic profiling, and drug-treatment responses. They can also be cultured on-chip or retrieved for subsequent off-chip assays. We have implemented the μFCS platform with multiple operational modes and evaluated its capacities. The system not only achieved high enrichment ratios (>104) at high flow rates (up to 20 mL·hr-1) but also enabled versatile cellular analyses at a single cell resolution. The potential clinical application of the technology was demonstrated by capturing CTCs from whole blood of tumor-bearing mice and subsequently performing genetic analyses on the captured cells.
The μFCS is designed to size-selectively enrich target cells from complex biological media (Figure 1a). The device consists of sample inlets, a separation fluidic channel with a weir-style barrier, and collection ports. The barrier, extending from the channel ceiling, defines the height of the physical gap for cell separation. Biological samples and buffer solution are separately introduced onto the input ports, generating two laminar flow streams. Cells smaller than the gap height bypass the barrier, remaining in their original flow stream. Larger cells, on the other hand, move along the barrier and join the buffer stream, thereby are collected at a different outport (Figure 1b). As the sorting operation does not hinder the fluidic flow, the μFCS can sustain a large volume of flow and is free of clogging (Figure S1). Importantly, the system offers versatile multimodal operations for the detection and analysis of CTCs. For example, by embedding cradle-shaped structures along the physical barrier, CTCs can be individually captured (Figure 1c). These cells then can be enumerated and further profiled on-chip by introducing molecular probes (Figure 1d), enabling specific CTC analyses at a single cell resolution. Captured cells can also be retrieved for off-chip assays (e.g., cell culture; Figure 1e), or lysed to elute proteins and nucleic acids (Figure 1f).
Figure 1. Design and modes of the microfluidic cell sorter (μFCS).
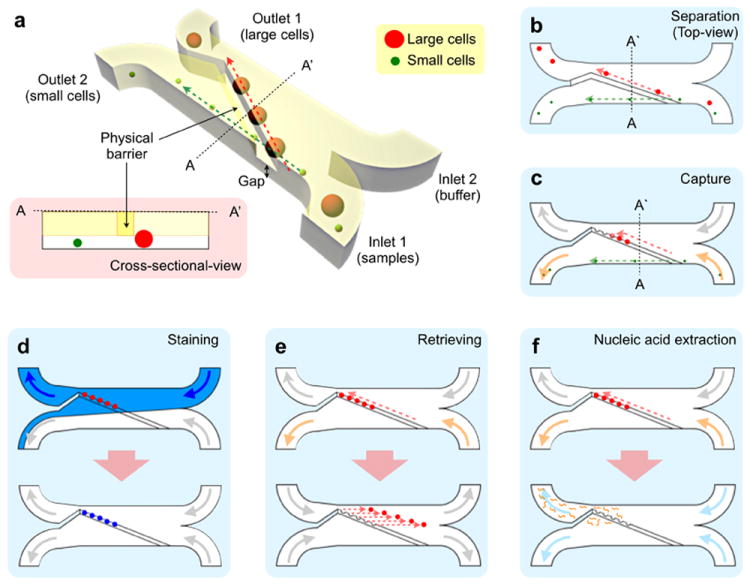
(a) The μFCS consists of a main fluidic channel, two inlets and two outlets. In the main channel, a physical barrier is implemented diagonally to separate cells by their sizes (see the inset for cross-sectional view). Small cells pass through the gap underneath the barrier, and are directed to the collection site (outlet 2) by laminar flow. Large cancer cells move along the barrier, and are collected at a different outlet. The μFCS can be used for different applications. (b) A straight line-shaped barrier is used for continuous, size-dependent cell-sorting and collection. (c) Cell-capture sites are embedded along the barrier to trap individual cells for facile enumeration. (d) By injecting molecular probes into the device, the trapped cells in (c) can be stained and profiled at a single cell resolution. The entire assay procedure (cell capture, targeting, washing) can be performed on the same chip. (e) Captured cells can be released by introducing a reverse flow from the outlet. The released cells can be retrieved for off-chip analyses or can be cultured on-chip. (f) Captured cells are lysed on-chip to extract nucleic acids for genetic analysis.
A prototype μFCS was fabricated by replicating a SU-8 photoresist (Microchem) mold with polydimethylsiloxane (PDMS, Dow Corning) as the structural material (see Materials and Methods in Supporting Information for details). The device was built to include a wide and tall fluidic channel (3 mm × 50 μm in cross-section) to have low hydraulic resistance for operation at high flow rates (Figure S2a; see Materials and Methods in Supporting Information for design details). To reduce the resistance further, the barrier was designed to have a small footprint (80 μm in width) and to cross the main channel at a small angle (10°). Small pillars were embedded across the channel, including the underside of the physical barrier, to prevent channel deformation and to thus maintain a uniform barrier-channel gap at high flow rates (Figure S2b). A rigid support (a 150-μm thick glass cover slip) was also embedded on the ceiling of the PDMS channel during the polymer cure. With such structural improvements, we were able to enhance the maximum flow rate for stable cell separation by ~10-fold (from 2 mL·hr-1 to 20 mL·hr-1). Whilst minor distortion of flow occurred around each post, both the sample and the buffer streams maintained their laminar nature, a feature that is crucial for the sorting operation. The optimal gap height for the passage of human blood cells (erythrocytes, 6-8 μm; leukocytes, 8-10 μm) was experimentally determined; at a height of 10 μm, most blood cells (>99%) were found to pass under the barrier (Figure S3). Note that samples can be pre-labeled with a cocktail of cancer-specific microbeads (Figure S4) to extrinsically enlarge the overall CTC size (>10 μm). This strategy incorporates cancer molecular selectivity and helps to ensure effective CTC sorting with broad size ranges.
We first assessed the sorting capability of the μFCS, utilizing a device with a straight line-shaped barrier (Figure 1b). Samples containing two differently-sized bead populations (7 and 15 μm in diameter, respectively) were prepared to mimic a biological mixture of human leukocytes and cancer cells. The bead mixture and a buffer solution were simultaneously introduced to the chip at the same flow rate, and the beads were sorted in a continuous-flow manner (see Supplementary Movies 1 and 2 for bead and cell separation, respectively). The collected bead populations were then analyzed by flow cytometry. Even a very low concentration of large beads (~0.23%) in the initial mixture could be enriched to a highly purified fraction (>98%) after the μFCS processing (Figure 2a).
Figure 2. System characterization.
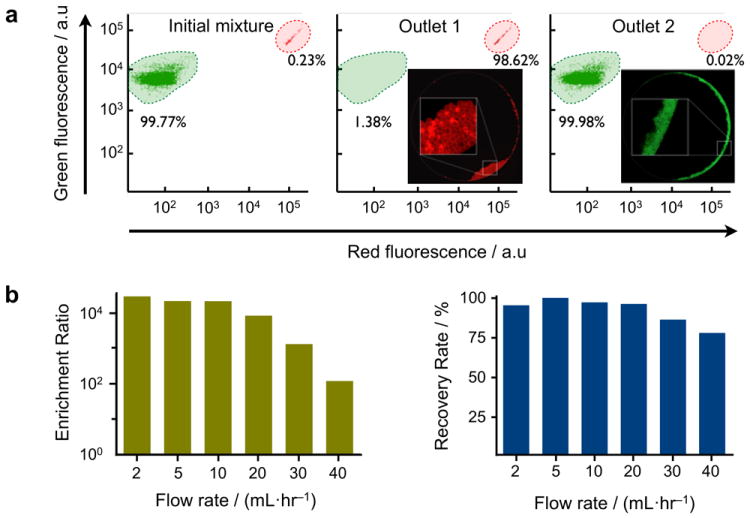
(a) The size-sorting capacity of the μFCS was evaluated using microbeads. A mixture of two differently-sized microbeads (green and red fluorescent beads with diameters of 7 and 15 μm, respectively) was introduced to the device. The initial mixture was highly enriched with small beads (99.77%). After sorting, large beads were recovered at outlet 1 with >30,000-fold enrichment. Negligible amount of large beads was collected at outlet 2. The composition of bead population was determined by flow cytometry. The insets show fluorescence images of the collected beads in outlet 1 and 2, respectively. (b) Enrichment ratios (left) and recovery rates (right) of size separation were measured at varying flow rates. Considerably high enrichment ratios (>2×104) and recovery rates (>95%) could be achieved even at a high flow rate of 20 mL·hr-1. Note that the maximum flow rate could be further increased by implementing devices with rigid materials (e.g., glass, plastic).
The sorting performance of the μFCS was quantitatively evaluated by measuring two parameters: enrichment ratio (COL·COS-1)·(CIL·CIS-1)−1 and recovery rate COL·CIL-1, where CIS and COS are the concentrations of small cells before and after sorting respectively, and CIL and COL are the concentrations of large cells before and after sorting respectively. Figure 2b shows the measured enrichment ratios and recovery rates at different volumetric flow rates. High enrichment (>2×104) could be achieved even up to 10 mL·hr-1, while the recovery rate remained consistently high (>95%). The μFCS thus can serve as a platform to concentrate very sparse entities in a large volume of complex biological sample. The maximum flow rate for the current PDMS prototype was set by changes in the gap height, as induced from the pressure buildup; we expect further enhancement in the achievable flow rate by switching to rigid materials (e.g., glass, poly(methyl methacrylate), polycarbonate) for the fluidic structure.
We next tested the capacity of the μFCS for on-chip capture, enumeration and molecular screening. We implemented the μFCS in its capturing mode, by embedding cell capture sites along the physical barrier (Figure 3a). The capture sites have narrow bypassing channels, which help to retain trapped cells by exploiting differences in hydrodynamic resistances.[20] The capture efficiency was first evaluated using whole blood spiked with fluorescently-labeled cancer cells (human A431 cancer cells, ~15 μm diameter in suspension, see Materials and Methods in Supporting Information for experimental details). Samples with varying cancer cell concentrations were prepared in anti-coagulated whole blood and introduced to the μFCS at a flow rate of 5 mL·hr-1. Indeed, the target cancer cells were individually captured at the pre-defined sites, which served to facilitate the enumeration process. The measured cell counts showed excellent agreement with expected cell counts, even at very low initial cancer cell concentrations (Figure 3b).
Figure 3. On-chip capture, culture and molecular profiling of cancer cells from whole blood.
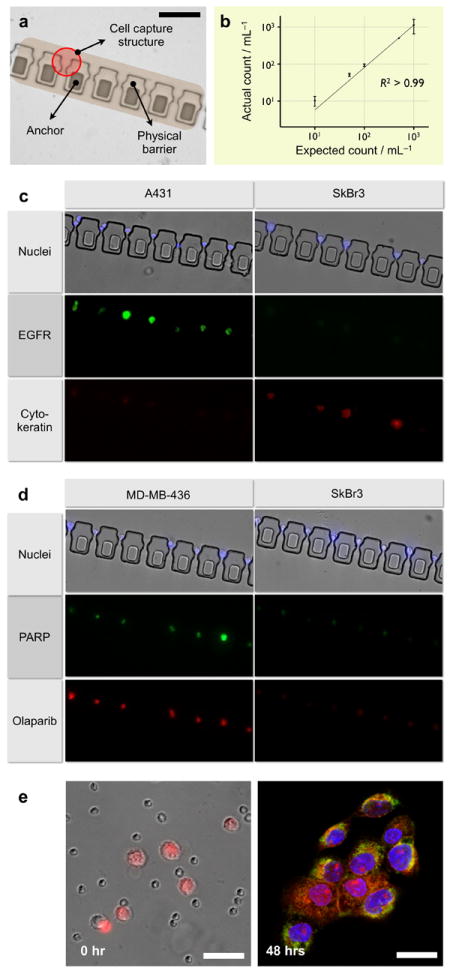
(a) Cell capturing sites were embedded in the physical barrier to trap individual cancer cells from whole blood. The captured cells can be enumerated and molecularly characterized at a single cell resolution. Scale bar, 100 μm. (b) The system allowed high precision CTC capture and quantification. Varying amounts of cancer cells (A431) were spiked into whole blood and sorted through the device without any off-chip purification. Captured cells were counted via optical microscopy. Even at a low cell concentration (10 cells mL-1 whole blood), the device successfully captured and recovered all spiked cancer cells. The measured counts showed excellent agreement with the expected numbers. (c) Captured cells were molecularly profiled for multiple biomarkers. The entire process including membrane-permeablization, targeting, incubation and washing could be performed on-chip. Cells were simultaneously stained for extracellular (EGFR) and intracellular (cytokeratin) markers along with nucleus (Hoechst). (d) The μFCS was used to evaluate the efficacy of drug binding on CTCs. A molecular inhibitor (Olaparib) of poly ADP ribose polymerase (PARP) was introduced to the captured cells. Cells with high PARP expression (MDA-MB-436), which was independently confirmed by on-chip immunostaining (middle), showed a correspondingly efficient drug binding. (e) Captured CTCs (A431, pre-stained red with live cell tracker) were released from the physical barrier by reversing the flow direction (left), and were cultured in the collection outlet. Although a small number of leukocytes were initially carried over due to their high abundance, only adherent cancer cells remained after culture. The cancer cells were on-chip fixed and stained for EGFR (green) and nucleus (blue). Scale bar, 50 μm.
The μFCS was further applied to perform multiplexed molecular analyses on captured cells, a crucial task not only to correctly identifying cancer cells, but also to investigating heterogeneity in cancer population and drug responsiveness. Different types of cancer cells (A431 and SkBr3) were separately spiked into anti-coagulated whole blood and processed by the μFCS. Captured cells were labeled for nucleus and semi-permeablized by sequentially flowing fluorescent dyes (Hoechst nuclear dye) and saponin. Subsequently, a mixture of antibodies was injected to probe for extracellular (EGFR: epidermal growth factor receptor) as well as intracellular (cytokeratin) biomarkers. Briefly, 5 μg·mL-1 of EGFR and cytokeratin antibodies were introduced as a antibody cocktail for simultaneous staining (see Materials and Methods in Supporting Information for experimental details). Fluorescence micrographs (Figure 3c) confirmed efficient cell-staining without any cell loss. In addition, multiplexed screening using different biomarkers helps to differentiate tumor types: A431 cells show high EGFR but low cytokeratin expression while SKBR3 cells indicate the reverse, demonstrating robust CTC detection and identification on-chip.
Captured cells can be monitored to evaluate drug treatment efficacy. As a model system, we probed the cells for their cellular affinity for Olaparib™, a small molecule inhibitor of the nuclear DNA repair enzyme poly ADP ribose polymerase (PARP). Two different types of breast cancer cells (MDA-MB-436, PARPhigh; SkBr3, PARPlow) were separately spiked into whole blood, and captured by the μFCS. We next introduced a fluorescent version of PARP-inhibitor[21] to the captured cells. For comparative analysis, the cells were also immunostained for PARP expression by flowing fluorescent antibodies to PARP. Even within the same tumor type, there was significant cellular heterogeneity in protein expression and drug binding. This operation allowed direct and simultaneous measurements of an intracellular drug target and relevant drug concentration/affinity, as well as the correlation (Figure 3d).
The μFCS offers a facile way to release and retrieve captured cells for comprehensive off-chip analyses (e.g., proteomic, genomic assays), a key feature often missing in most CTC detection platforms. By reversing the flow direction, the captured cells can be dislodged and channeled to a sample inlet by laminar flow (Supplementary Movie 3). Figure 3e shows collected cancer cells at an inlet, where cells can adhere and be cultured on-chip. Due to the large abundance of haemotologic cells in whole blood, the reverse flow carried over residual leukocytes and red blood cells inside the fluidic channel; however, only the cancer cells adhered to the device surface while residual blood cells were easily removed by a buffer flush. The purity of the on-chip cultured cancer cells was confirmed by their membrane expression of EGFR (Figure 3e).
The utility of the μFCS platform was further demonstrated by investigating CTCs in tumor-bearing animals. A tumor metastasis model was obtained by intravenously injecting murine melanoma cells (B16-F10 expressing red fluorescent protein) into mice.[22, 23] Two weeks post injection, lung metastases were apparent as confirmed by computerized tomography (CT) scanning (Figure 4a). Whole blood was obtained from mice and treated with sodium citrate to prevent coagulation. The samples were then aliquoted for μFCS detection and flow cytometry. As shown in Figure 4b, CTCs in whole blood could be effectively captured and enumerated by the μFCS. The presence of CTCs was also independently confirmed and quantified by fluorescence flow cytometry analysis on red blood cell-lysed samples. Note that CTC counts by the μFCS were significantly higher (153 %) than those by flow cytometry, mainly due to the minimal sample processing requirements by the μFCS.
Figure 4. Tumor molecular signature of chip-captured CTCs from mouse metastasis model.
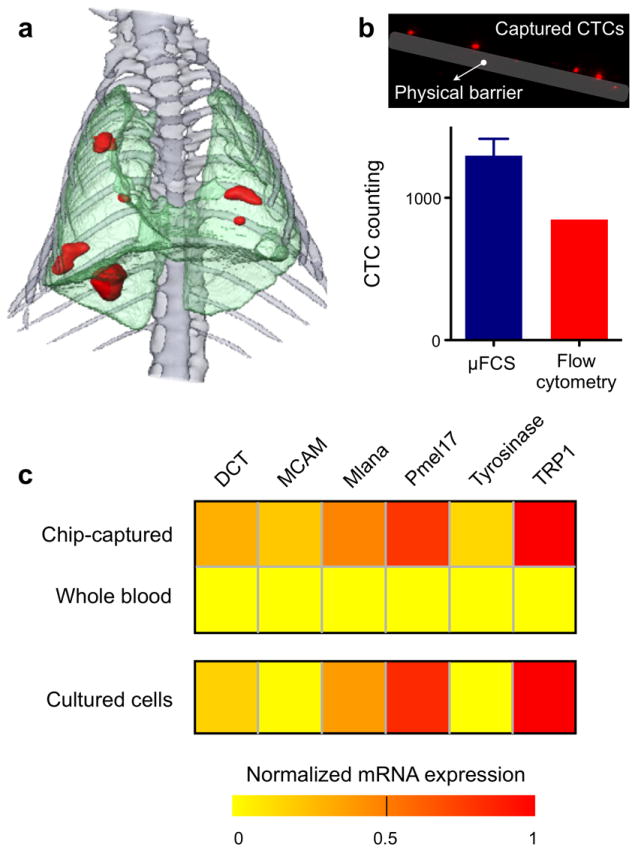
(a) 3D CT of mouse thorax shows melanoma metastasis (red), two weeks after intravenous injection of melanoma cell line (B16-F10 with RFP). (b) CTCs from whole blood were effectively captured by the μFCS and enumerated. Fluorescence imaging showed CTCs in the blood samples. The μFCS showed many more CTCs compared to that by flow cytometry analysis. Data is shown as mean ± s.e.m. (c) Total cellular RNA was extracted on-chip from the captured CTCs and used for quantitative RT-PCR analyses of tumor-specific molecular markers. As compared to RNA extracted from an equal volume of unsorted whole blood, RNA from the chip-purified sample showed significant enrichment of tumor-specific mRNA, and displayed a similar signature to that of parental tumor cells (p > 0.1).
We next performed genetic analyses on the captured CTCs. Cell-lysis buffer was injected through the device to rupture cells, and total cellular RNA was extracted. As a control, RNA was extracted from an equal volume of unsorted whole blood from the same animal. The collected nucleic acid was then analyzed for tumor-specific markers by quantitative reverse transcription polymerase chain reaction (qRT-PCR). The transcriptional activities of the following melanoma-associated markers were measured: dopachrome tautomerase (DCT), melanoma cell adhesion molecule (MCAM), Melan-A (Mlana), melanocyte protein 17 (Pmel17), tyrosinase and tyrosinase related protein 1 (TRP1).[24-27] Whole blood-derived RNA was unable to generate a discernible tumor signature (Figure S5); the low abundance of tumor RNA was entirely masked by the predominance of RNA from host cells (e.g., leukocytes). Such interference was absent in the μFCS-purified samples. Moreover, the transcriptional signature was statistically similar (p > 0.1) to that of parental melanoma tumor cells in culture (Figure 4c).
In summary, we have developed a new diagnostic μFCS platform for accurate and rapid CTC detection in whole blood, based on size-separating technique. The system provides versatile, on-chip characterization on CTCs, including tumor cell capture and counting, molecular profiling on multiple biomarkers, and monitoring of drug treatment, all at a single-cell resolution. Importantly, the μFCS permits facile retrieval of the captured cells for extensive off-chip studies (e.g., culture, genomic studies). Through extrinsic manipulation of cell size with immuno-microbeads, the μFCS platform could be extended to enrich CTCs of broader size range as well as other types of rare cells in circulation (e.g., nucleated red blood cells,[28] stem cells, immune cells). We envision further advancing the technology 1) by cascading devices with different barrier height to perform multiple-size separation at finer resolution, and 2) through integration with other functional modalities (e.g., different types of cell separators, detectors, on-chip PCR). Such systems would enable comprehensive, point-of-care analyses on rare cells, finding significant diagnostic applications.
Supplementary Material
Acknowledgments
The authors thank Gregory Wojtkiewicz for his assistance in CT imaging; Y. Fisher-Jeffes for critical reading of the manuscript. H. Shao acknowledges financial support from the B.S.-Ph.D. National Science Scholarship awarded by the Agency for Science, Technology and Research, Singapore. This work was supported in part by National Institute of Health Grants R01-EB0044626, R01-EB010011, P50 grant P50CA86355, CCNE contract U54CA151884, and TPEN contract HHSN268201000044C.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Dr. Jaehoon Chung, Center for Systems Biology, Massachusetts General Hospital, 185 Cambridge St, CPZN 5206, Boston, MA 02114, USA.
Dr. Huilin Shao, Center for Systems Biology, Massachusetts General Hospital, 185 Cambridge St, CPZN 5206, Boston, MA 02114, USA; Harvard Biophysics Program, Harvard Medical School Boston, MA 02115, USA.
Dr. Thomas Reiner, Center for Systems Biology, Massachusetts General Hospital, 185 Cambridge St, CPZN 5206, Boston, MA 02114, USA
Dr. David Issadore, Center for Systems Biology, Massachusetts General Hospital, 185 Cambridge St, CPZN 5206, Boston, MA 02114, USA
Prof. Ralph Weissleder, Center for Systems Biology, Massachusetts General Hospital, 185 Cambridge St, CPZN 5206, Boston, MA 02114, USA, rweissleder@mgh.harvard.edu Department of Systems Biology, Harvard Medical School Boston, MA 02115, USA.
Prof. Hakho Lee, Center for Systems Biology, Massachusetts General Hospital, 185 Cambridge St, CPZN 5206, Boston, MA 02114, USA, hlee@mgh.harvard.edu
References
- 1.Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH, Norton L, Massague J. Cell. 2009;139:1315. doi: 10.1016/j.cell.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maheswaran S, Haber DA. Curr Opin Genet Dev. 2010;20:96. doi: 10.1016/j.gde.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haber DA, Gray NS, Baselga J. Cell. 2011;145:19. doi: 10.1016/j.cell.2011.03.026. [DOI] [PubMed] [Google Scholar]
- 4.Chaffer CL, Weinberg RA. Science. 2011;331:1559. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 5.Pantel K, Brakenhoff RH, Brandt B. Nat Rev Cancer. 2008;8:329. doi: 10.1038/nrc2375. [DOI] [PubMed] [Google Scholar]
- 6.Gerges N, Rak J, Jabado N. Br Med Bull. 2010;94:49. doi: 10.1093/bmb/ldq011. [DOI] [PubMed] [Google Scholar]
- 7.Gleghorn JP, Pratt ED, Denning D, Liu H, Bander NH, Tagawa ST, Nanus DM, Giannakakou PA, Kirby BJ. Lab Chip. 2010;10:27. doi: 10.1039/b917959c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M. Nature. 2007;450:1235. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, Rothenberg SM, Shah AM, Smas ME, Korir GK, Floyd FPJ, Gilman AJ, Lord JB, Winokur D, Springer S, Irimia D, Nagrath S, Sequist LV, Lee RJ, Isselbacher KJ, Maheswaran S, Haber DA, Toner M. Proc Natl Acad Sci U S A. 2010;107:18392. doi: 10.1073/pnas.1012539107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paterlini-Brechot P, Benali NL. Cancer Lett. 2007;253:180. doi: 10.1016/j.canlet.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 11.De Giorgi V, Pinzani P, Salvianti F, Panelos J, Paglierani M, Janowska A, Grazzini M, Wechsler J, Orlando C, Santucci M, Lotti T, Pazzagli M, Massi D. J Invest Dermatol. 2010;130:2440. doi: 10.1038/jid.2010.141. [DOI] [PubMed] [Google Scholar]
- 12.Hosokawa M, Hayata T, Fukuda Y, Arakaki A, Yoshino T, Tanaka T, Matsunaga T. Anal Chem. 2010;82:6629. doi: 10.1021/ac101222x. [DOI] [PubMed] [Google Scholar]
- 13.Lin HK, Zheng S, Williams AJ, Balic M, Groshen S, Scher HI, Fleisher M, Stadler W, Datar RH, Tai YC, Cote RJ. Clin Cancer Res. 2010;16:5011. doi: 10.1158/1078-0432.CCR-10-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng S, Lin H, Liu JQ, Balic M, Datar R, Cote RJ, Tai YC. J Chromatogr A. 2007;1162:154. doi: 10.1016/j.chroma.2007.05.064. [DOI] [PubMed] [Google Scholar]
- 15.Chen Z, Zhang S, Tang Z, Xiao P, Guo X, Lu Z. Surf Interface Anal. 2006;38:996. [Google Scholar]
- 16.Tan SJ, Yobas L, Lee GYH, Ong CN, Lim CT. Biomed Microdevices. 2009;11:883. doi: 10.1007/s10544-009-9305-9. [DOI] [PubMed] [Google Scholar]
- 17.Jain A, Munn LL. Lab Chip. 2011;11:2941. doi: 10.1039/c1lc20401g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhagat AAS, Hou HW, Li LD, Lim CT, Han J. Lab Chip. 2011;11:1870. doi: 10.1039/c0lc00633e. [DOI] [PubMed] [Google Scholar]
- 19.Hur SC, Mach AJ, Di Carlo D. Biomicrofluidics. 2011;5:022206. doi: 10.1063/1.3576780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung J, Kim YJ, Yoon E. Appl Phys Lett. 2011;98:123701. doi: 10.1063/1.3565236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reiner T, Earley S, Turetsky A, Weissleder R. ChemBioChem. 2010;11:2374. doi: 10.1002/cbic.201000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanna N, Fidler IJ. Cancer Res. 1981;41:438. [PubMed] [Google Scholar]
- 23.Koop S, MacDonald IC, Luzzi K, Schmidt EE, Morris VL, Grattan M, Khokha R, Chambers AF, Groom AC. Cancer Res. 1995;55:2520. [PubMed] [Google Scholar]
- 24.Medic S, Pearce RL, Heenan PJ, Ziman M. Pigment Cell Res. 2007;20:80. doi: 10.1111/j.1600-0749.2006.00356.x. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt H, Sorensen BS, Fode K, Nexo E, von der Maase H. Melanoma Res. 2005;15:409. doi: 10.1097/00008390-200510000-00009. [DOI] [PubMed] [Google Scholar]
- 26.Visus C, Andres R, Mayordomo JI, Martinez-Lorenzo MJ, Murillo L, Saez-Gutierrez B, Diestre C, Marcos I, Astier P, Godino J, Carapeto-Marquez de Prado FJ, Larrad L, Tres A. Melanoma Res. 2007;17:83. doi: 10.1097/CMR.0b013e3280a60878. [DOI] [PubMed] [Google Scholar]
- 27.Xi L, Nicastri DG, El-Hefnawy T, Hughes SJ, Luketich JD, Godfrey TE. Clin Chem. 2007;53:1206. doi: 10.1373/clinchem.2006.081828. [DOI] [PubMed] [Google Scholar]
- 28.Huang R, Barber TA, Schmidt MA, Tompkins RG, Toner M, Bianchi DW, Kapur R, Flejter WL. Prenat Diagn. 2008;28:892. doi: 10.1002/pd.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.