

Abstract
Objectives
Homozygous C1q deficiency is an extremely rare condition and strongly associated with systemic lupus erythematosus. To assess and characterize C1q deficiency in an African-American lupus pedigree, C1q genomic region was evaluated in the lupus cases and family members.
Methods
Genomic DNA from patient was obtained and C1q A, B and C gene cluster was sequenced using next generation sequencing method. The identified mutation was further confirmed by direct Sanger sequencing method in the patient and all blood relatives. C1q levels in serum were measured using sandwich ELISA method.
Results
In an African-American patient with lupus and C1q deficiency, we identified and confirmed a novel homozygote start codon mutation in C1qA gene that changes amino acid Methionine to Arginine at position 1. The Met1Arg mutation prevents protein translation (Met1Arg). Mutation analyses of the patient’s family members also revealed the Met1Arg homozygote mutation in her deceased brother who also had lupus with absence of total complement activity consistent with a recessive pattern of inheritance.
Conclusion
The identification of new mutation in C1qA gene that disrupts the start codon (ATG to AGG (Met1Arg)), has not been reported previously and it expands the knowledge and importance of the C1q gene in the pathogenesis of lupus especially in high risk African-American population.
Introduction
C1q, the first component of the classical pathway of complement system, is a glycoprotein that belongs to the collectin family (lectins containing collagen-like proteins) with molecular mass of 410 kDa. C1q along with the enzymatically active components C1r and C1s form the C1 complex. This process activates the classical pathway of complement. Genetically, three genes in the human genome are responsible for coding C1q protein. This protein is composed of 18 polypeptide chains, 6 each of A, B, and C which are the products of C1qA, C1qB and C1qC genes respectively. These three genes are tandemly arranged 5-prime to 3-prime in the order A-C-B on a 24-kb stretch of DNA and closely linked together on chromosome 1p36 (1).
In the human genome, different coding mutations have been identified that lead to a premature termination codon at different amino acid residues or an amino acid substitution (2). These rare mutations may cause either complete C1q deficiency due to deletion or change of single base that leads to a termination codon, or amino acid exchanges and the formation of an aberrant polypeptide chain. To date, 12 of these coding mutations across the C1qA, B and C loci have been reported; of these, 3 mutations (Gln208X, Gln64X, Glu53fs) were detected in C1qA gene (3-7). These case reports were mostly observed in Caucasian families from Eastern Europe or Turkey (3-7). The incomplete translation of C1q, even when it occurs in only one chain, leads to the failure of synthesis of the C1q triplehelix and secretion of the whole molecule which leads to complete loss of Clq (8). Importantly, while C1q coding mutations are very rare in the general population, they are highly predictive of SLE. Indeed, out of 64 published cases from 38 families with C1q coding mutations, 93% presented with SLE-like symptoms. In contrast to other complement deficiencies, C1q deficiency is usually associated with more severe disease manifestations such as glomerulonephritis, vasculitis or CNS involvement (2). Modeling human lupus with C1q deficiency, C1q knockout mice (C1q -/-) spontaneously develop glomerulonephritis with major immune complex (IC) deposition in the renal glomeruli, and chronic renal damage as well as significant production of autoantibodies (9).
Despite the search for more common alleles that might be relevant to less penetrant but more prevalent forms of C1q-related malfunction, multiple GWAS studies and candidate gene approaches have failed to identify any significant signals in this genomic region, however, suggestive associations have been reported by candidate gene study by us and others (10,11). In our previous results, we showed evidence of association of multiple common SNPs in C1qA gene with disease severity and kidney nephritis in African and Hispanic population samples (10). Subsequently, based on the identified risk haplotype, a subset of patients was selected in order to measure serum C1q protein which might explain the observed allelic associations. Modest correlation with C1q protein and the common risk allele in quantative trait analysis (QTL) analyses was observed (data not shown), Also, we identified several SLE cases with very low level of C1q. In particular, we found one African-American patient with undetectable serum C1q and in this report we describe the results of genomic sequencing and clinical manifestations in this pedigree.
Materials and methods
Samples
The African-American lupus pedigree (2 SLE cases and 3 blood relatives and Spouse) included in this study, was enrolled in the Lupus Genetics Studies at OMRF. All protocols were approved by the Institutional Review Boards at the institution. All patients met the revised 1997 ACR criteria for the classification of SLE (12). All available medical records were reviewed to obtain serological and clinical manifestations and to confirm the diagnosis. Blood samples were collected from each participant, and genomic DNA was isolated and stored using standard methods. CH50 titers for complement classic pathway activity are routinely measured at the time of patients’ participation into the study in OMRF lab. C1q levels in serum and anti-C1q were measured using a sandwich ELISA method as previously described (13).
Deep sequencing
Genomic DNA from a set of 46 African-American SLE cases were sheared and prepared for sequencing using an Illumina Paired-End Genomic DNA Sample Prep Kit. Targeted regions of interest from each sample were then enriched with a SureSelect Target Enrichment System. Resequencing was undertaken on an Illumina GAIIx platform using standard procedures. Post sequence data was processed with Illumina Pipeline software v.1.7. All samples were sequenced to minimum average fold coverage of 25X. Variants were identified by a custom analysis pipeline utilizing the BWA alignment program and the Genome Analysis Tool Kit (GATK).
Sanger sequencing
Briefly, A 302 bp fragment covering C1qA exon 1 was amplified using Advantage 2 GC polymerase (Clontech) from genomic DNA from a set of 6 DNA samples of interest and 6 controls. PCR and sequencing primers used were TTGTGTGCATGGGACTCAAG – forward and GGCCAAGTCAGGCCAAG – reverse. This product was sequenced from each pcr primer using BigDye terminator v1.1 (Life Technologies) and injected on a 3730xl Genetic Analyzer (ABI). All sequencing was done in both directions. Sequence alignment and mutation analysis was performed with Mutation Surveyor v4.0 (SoftGenetics).
Results
We found one African-American lupus patient with undetectable serum C1q. In fact, no total complement hemolytic activity (CH50) was present in this patient. In addition, anti-C1q antibodies, which are commonly seen in SLE patients were not detectable in the serum of this patient and, thus, were not the likely cause for the absence of C1q.
To follow-up our initial observation of absent C1q in this patient, C1q deep sequencing data for this patient was obtained from ongoing next generation sequencing studies of lupus related loci. Evaluation of this patient revealed a novel homozygote mutation at start codon of C1qA gene. The mutation was a T-to-G nucleotide transition in start codon of C1qA at position (chr1:22,836,698) according to NCBI36/hg18 genomic map, which changed the codon from ATG (methionine) to AGG (arginine) (ATG >AGG, 1M>R). No further alterations in the coding or non-coding sequences of the three C1q genes were detected in this patient. Since the start codon mutation could lead to the absence of protein translation in the ribosome and therefore lack of C1q synthesis, we confirmed this primary sequencing result in all available DNA samples from this patient and family members using Sanger sequencing. Analysis of her family members established a classic homozygous recessive mechanism for this mutation and explanation for the absent C1q and absent CH50 activity. The mutation analyses using Sanger sequencing confirmed the original finding, as the mutation was detected at the same codon and with high quality score. The plot of the electropherogram at the mutation site from the patient and one control sample is shown in Fig. 1. The mutation was only detected in the patient and her relatives but not in her spouse or control samples.
Figure 1.
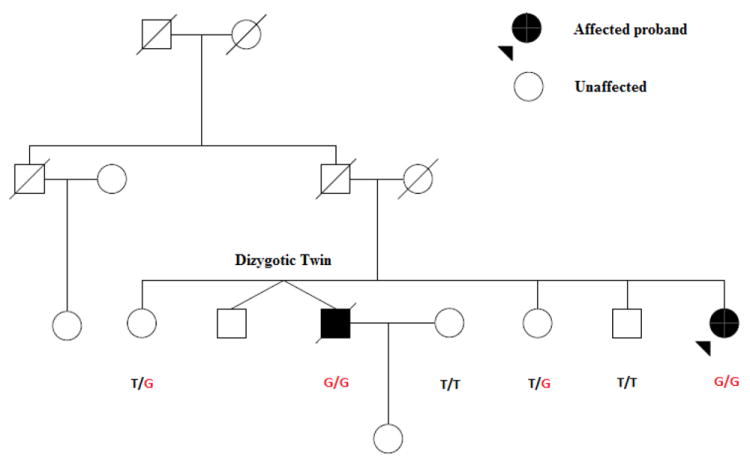
The family structure of an African-American pedigree with Novel C1qA mutation (T>G, MET1ARG).
The pedigree diagram of this family is shown in Fig. 2. In addition to confirming the mutation in the proband case, we also found the same homozygote mutation in another family member (male sibling). This sibling is deceased, but was affected with lupus and with absent total complements hemolytic activity. Importantly, other siblings who were either heterozygote or homozygote for the wild type allele were healthy and have a negative or low titer of anti-nuclear antibodies.
Figure 2.
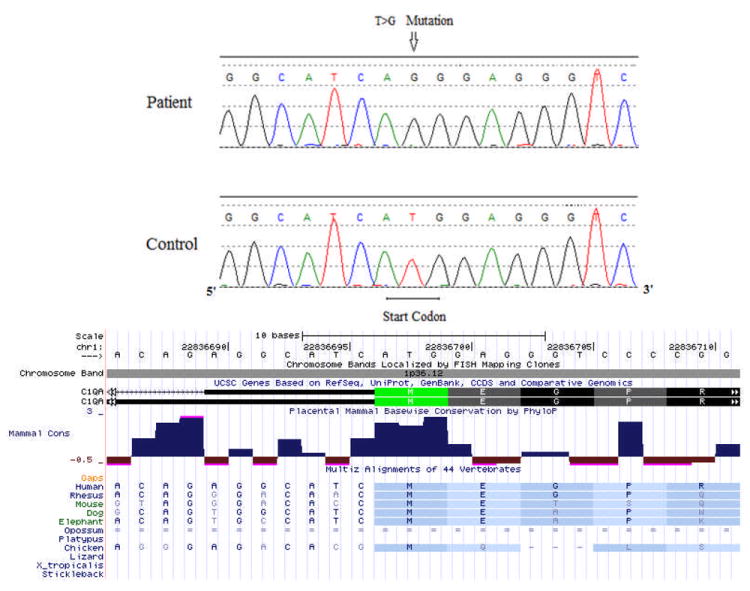
The electropherogram from Sanger sequencing shows mutation site (T>G) at the start codon of C1qA gene. The corresponding NCBI 36 human genome and the position of the first amino acid (M, methionine) is also shown (from USCS genome browser). Methionine is conserved among different mammalian species.
The ACR (American College of Rheumatology) criteria and clinical manifestations of the patient and her sibling are shown in Table 1, and there are some important shared clinical features consistent with severe lupus phenotype observed in C1q deficient patients. Both patients who were diagnosed with lupus in the 1980’s, had history of nephritis, arthritis, serositis (pericarditis), pancytopenia including (leukopenia, lymphopenia and thrombocytopenia) and Raynaud’s. In addition, both had high titers of anti-nuclear antibodies and were positive for anti-RNP, anti-Sm and anti-Ro antibodies. In addition both had elevated liver enzymes and Creatine phosphokinase (Table 1). Other available complement component in medical records include C3 which were near normal to normal in both cases (proband C3=60 (70-150 mg/dl), deceased sibling C3= reported as Normal)). The deceased male sibling also had a history of pulmonary hypertension, pancreatitis, and diabetes. Unfortunately, the serum of the deceased sibling patient was not available. Both the allelic segregation and phenotype distribution in this pedigree are consistent with a recessive pattern of inheritance. Although the parents are deceased and their DNA was not available, the genotype information in siblings suggests that both parents were obligate heterozygotes (Fig.2).
Table 1.
The ACR criteria and clinical manifestation of the lupus siblings.
| SLE Sibling Cases | ||
|---|---|---|
| 1 | 2 | |
| Gender | F | M |
| Race | African-American | |
| Age of SLE onset | 20 | 32 |
| Malar Rash | N | N |
| Discoid Rash | P | N |
| Photosensitivity | P | N |
| Oral ulcers | N | N |
| Arthritis | P | P |
| Serositis | P | P |
| Pleuritis | N | P |
| Pericarditis | P | P |
| Nephritis | P | P |
| Neurologic | N | N |
| Hematologic | P | P |
| Hemolytic Anemia | N | N |
| Leukopenia | P | P |
| Lymphopenia | P | P |
| Thrombocytopenia | P | P |
| Immunologic | P | P |
| Anti-dsDNA | P | N |
| Anti-Sm/RNP | P/P | P/P |
| Anti-Ro/La | P/N | P/N |
| aPL* | N | P |
| ANA† | P | P |
| Other clinical/serological manifestations | Raynauds/ Elevate Liver enzymes/Elevated CPK‡ | Raynauds/Elevated liver enzymes/elevated CPK‡/Pancreatitis/Pulmonary Hypertension/Systolic Hypertension/Diabetes |
aPL=antiphopholipid antibodies
Anti-nuclear antibodies with highest titer of 1/2560 for proband and 1/3240 for the sibling.
Liver enzymes: (AST=115, NL=5-50),(ALT=19, NL= 0-50), Creatin phosphokinase (CPK=351, NL=20-190)
Liver enzymes:(AST=397, NL=0-47), (ALT=200, NL=3-36), CPK=441 NL=0-225)
Discussion
The mutation affecting the ATG start codon has not been reported previously in C1q genes and in fact, these kinds of mutations are rare. In mammals, ATG (AUG in RNA) start codon is typical for ribosomal translation and it is recognized more efficiently when it is located in a favorable nucleotide context known as the Kozak consensus sequence (in particular a purine at position -3 and G at position +4, RccAUGG (R=purine)) (14). The augmenting effect of G at position +4 however is diminished when it is combined with U at position (+5) (14). Very rarely in higher organisms non-AUG codons may also initiate translation but this does not include AGG or AAG (15). As mentioned above, in the family studied, the mutation produced an AGG codon, which is not competent to initiate translation and therefore explains the functional consequence of this mutation. In addition, in theory, it is rarely possible that when the consensus is weak or disrupted, the ribosomal translation starts from secondary downstream AUG codon, a process called leaky scanning. However, in the C1qA gene, the downstream AUG is more than 50 nucleotides away with a suboptimal consensus sequence TctAUGGU (14). Even if translation was initiated from this codon, the translated protein would result in an incomplete or truncated protein that would lack the N-terminal leading signal peptide. In any case, the complete absence of C1q protein in serum of the patient suggests that macromolecule triple helix cannot be built and secreted. Indeed, we have not been able to detect C1q subunits by western blot in any of the C1q deficient samples we have tested, which supports that the entire C1q protein is missing in serum. This is consistent with the previous report that when a defect occurs in only one chain of C1q, the entire synthesis and secretion of the C1q triplehelix fails (8).
As mentioned above no other novel coding mutation in C1q gene cluster or in other selected lupus candidate genes was identified in the proband of this family using deep sequencing. In addition, based on available genome wide data from index case (unpublished data), the proband was not homozygote for the more common known risk alleles of lupus such as STAT4 (rs7574865), PTPN22 (rs2476601), IRF5 (rs3807306) and ITGAM (rs1143679). Moreover, in HLA region, the patient mainly showed an enrichment of protective haplotypes. For example, one of the strongest signals in African-American lupus population that has been recently reported was near to HLA-DRB1 (rs9271366), in which our index case was homozygote for protective alleles (16). All of these observations provide further support that the detected mutation in this pedigree most likely operates as a single gene effect and it is the solely causative mutation of lupus by inducing a complete C1q deficiency.
In summary, the identification of the new C1qA mutation that disrupts the start codon of the gene (ATG to AGG (Met1Arg)), in an African-American patient with lupus and a C1q deficiency, expands the knowledge and importance of the C1q gene in the pathogenesis of lupus in different populations. It is likely that further mutations in the C1q gene cluster will be identified through additional sequencing especially in those with complete C1q deficiency.
Acknowledgments
The cooperation of the patient and family members involved in this study is gratefully acknowledged. This project was supported by the NIH (R37 AI024717, P01 AI083194,), Lupus Foundation of America, the Alliance for Lupus Research, the U.S. Department of Veterans Affairs, and the U.S. Department of Defense (PR094002). We would also like to thank the sequencing facilities from Oklahoma and Texas in which the work was supported by NIH grant 1RC2AR058959-01.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Sellar GC, Blake DJ, Reid KB. Characterization and organization of the genes encoding the A-, B- and C-chains of human complement subcomponent C1q. The complete derived amino acid sequence of human C1q. Biochem J. 1991;274:481–90. doi: 10.1042/bj2740481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walport MJ, Davies KA, Botto M. C1q and systemic lupus erythematosus. Immunobiology. 1998;199:265–85. doi: 10.1016/S0171-2985(98)80032-6. [DOI] [PubMed] [Google Scholar]
- 3.Hoppenreijs EP, van Dijken PJ, Kabel PJ, Th Draaisma JM. Hereditary C1q deficiency and secondary Sjögren’s syndrome. Ann Rheum Dis. 2004;63:1524–5. doi: 10.1136/ard.2003.016592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petry F, Berkel AI, Loos M. Multiple identification of a particular type of hereditary C1q deficiency in the Turkish population: review of the cases and additional genetic and functional analysis. Hum Genet. 1997;100:51–6. doi: 10.1007/s004390050464. [DOI] [PubMed] [Google Scholar]
- 5.Topaloglu R, Bakkaloglu A, Slingsby JH, Mihatsch MJ, Pascual M, Norsworthy P, et al. Molecular basis of hereditary C1q deficiency associated with SLE and IgA nephropathy in a Turkish family. Kidney Int. 1996;50:635–42. doi: 10.1038/ki.1996.359. [DOI] [PubMed] [Google Scholar]
- 6.Sun-Tan C, Ozgür TT, Kilinç G, Topaloğlu R, Gököz O, Ersoy-Evans S, et al. Hereditary C1q deficiency: a new family with C1qA deficiency. Turk J Pediatr. 2010;52:184–6. [PubMed] [Google Scholar]
- 7.Schejbel L, Skattum L, Hagelberg S, Ahlin A, Schiller B, Berg S, et al. Molecular basis of hereditary C1q deficiency-revisited: identification of several novel disease-causing mutations. Genes Immun. 2011;9:1038–1039. doi: 10.1038/gene.2011.39. [DOI] [PubMed] [Google Scholar]
- 8.Petry F, Le DT, Kirschfink M, Loos M. Non-sense and missense mutations in the structural genes of complement component C1q A and C chains are linked with two different types of complete selective C1q deficiencies. J Immunol. 1995;155:4734–8. [PubMed] [Google Scholar]
- 9.Botto M, Dell’Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–9. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- 10.Namjou B, Gray-McGuire C, Sestak AL, Gilkeson GS, Jacob CO, Merrill JT, et al. Evaluation of C1q genomic region in minority racial groups of lupus. Genes Immun. 2009;10:517–24. doi: 10.1038/gene.2009.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martens HA, Zuurman MW, de Lange AH, Nolte IM, van der Steege G, Navis GJ, et al. Analysis of C1q polymorphisms suggests association with systemic lupus erythematosus, serum C1q and CH50 levels and disease severity. Ann Rheum Dis. 2009;68:715–20. doi: 10.1136/ard.2007.085688. [DOI] [PubMed] [Google Scholar]
- 12.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 13.Dillon SP, D’Souza A, Kurien BT, Scofield RH. Systemic lupus erythematosus and C1q: A quantitative ELISA for determining C1q levels in serum. Biotechnol J. 2009;4:1210–4. doi: 10.1002/biot.200800273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kozak M. Recognition of AUG and alternative initiator codons is augmented by G in position +4 but is not generally affected by the nucleotides in positions +5 and +6. EMBO J. 1997;16:2482–92. doi: 10.1093/emboj/16.9.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peabody DS. Translation initiation at non-AUG triplets in mammalian cells. J Biol Chem. 1989;264:5031–5. [PubMed] [Google Scholar]
- 16.Ruiz-Narvaez EA, Fraser PA, Palmer JR, Cupples LA, Reich D, Wang YA, et al. MHC region and risk of systemic lupus erythematosus in African American women. Hum Genet. 2011;130:807–15. doi: 10.1007/s00439-011-1045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]