

Abstract
Regulation of organellar fusion and fission by Ca2+ has emerged as a central paradigm in intracellular membrane traffic. Originally formulated for Ca2+-driven SNARE-mediated exocytosis in the presynaptic terminals, it was later expanded to explain membrane traffic in other exocytic events within the endo-lysosomal system. The list of processes and conditions that depend on the intracellular membrane traffic includes aging, antigen and lipid processing, growth factor signaling and enzyme secretion. Characterization of the ion channels that regulate intracellular membrane fusion and fission promises novel pharmacological approaches in these processes when their function becomes aberrant. The recent identification of Ca2+ permeability through the intracellular ion channels comprising the mucolipin (TRPMLs) and the two-pore channels (TPCs) families pinpoints the candidates for the Ca2+ channel that drive intracellular membrane traffic. The present review summarizes the recent developments and the current questions relevant to this topic.
Keywords: organelles, TRPMLs, TPCs, calcium, membrane traffic, fusion, fission
Intracellular Membrane Fusion
The exquisite timing of neurotransmitter release is a key determinant of neuronal function. It is an excellent example of a signal-centric paradigm involving interpretation and amplification of an incoming signal. As in all signaling systems, amplification in this model improves fidelity and timing of signaling. The Ca2+-dependence of synaptic vesicle release is a necessary requirement for translating action potentials into a release of neuromediators. The flooding of SNARE docking complexes by Ca2+ entering the cell through voltage-gated Ca2+ channels, followed by conformational changes that ultimately drive membrane fusion, is responsible for the incredibly robust and rapid neurotransmitter release in response to electrical stimulation.1 This signal-interpretation-response paradigm however remains largely unresolved in critical cellular processes involving other intracellular membrane organelles such as endosomes and lysosomes. However, several recent developments, primarily stemming from identification and characterization of new intracellular Ca2+-permeable channels, promise better understanding of these key processes.
The importance of membrane traffic within the endo-lysosomal system cannot be overstated. The endocytic pathway is responsible for lipid and protein uptake,2 membrane remodeling,3 regulation of receptor signaling,4 metal uptake,5 growth factor signaling,6 antigen presentation7,8 and likely a multitude of other functions. The various forms of autophagy, a critical housekeeping function for eliminating dysfunctional organelles and misfolded proteins, are driven by the lysosomal components.9 Melanosome formation is yet another instance of endocytic involvement.10 Such a wide repertoire of roles means that the endo-lysosomal system is likely involved in a plethora of diseases and conditions. Indeed, endo-lysosomal and autophagic dysfunction have been implicated in Parkinson11 and Alzheimer diseases,12,13 over two dozen of lysosomal storage diseases,14,15 cancer16 and metal and drug toxicity.17 It is clear that better understanding of the molecules that regulate the endocytic pathway promises novel interventions into these diseases.
The endocytic pathway begins with the plasma membrane and consists of a series of compartments, each with its own role in the membrane targeting process. The movement of endocytosed material is mediated by fusion/fission and maturation processes, occurring in parallel with acidification of the lumen, in a centripetal direction: from nearly neutral early endosomes to highly acidic lysosomes.18 Acidification controls ionic flow between cytoplasm and the lysosomes, helps activation of lysosomal digestive enzymes and establishes a vector for transport processes driven by cargo receptors, such as those mediating growth factor and LDL processing.19 The specificity of membrane fusion is a function of the membrane recognition system driven by the Rab GTPases, their guanine nucleotide exchange factors (GEFs), their effectors and their associated inositol lipids.20 Each pair of interacting membrane types appears to have a specific Rab and is enriched in a specific inositol lipid. An excellent example of the role of Rabs in endocytic membrane formation and interaction has been recently presented as Rab5 requirement for early endosomal biogenesis. Rab5 deletion appears to completely eliminate early endosomes in rat liver cells.21
In presynaptic terminals, the docking of vesicles at the plasma membrane via SNARE complexes enables extremely fast release of their content in response to Ca2+ influx. SNARE proteins are also found on and appear to be specific to all types of endocytic compartments.22 Their identification is the first clue to Ca2+-dependence of the endocytic fusion events, likely mediated by the lysosomal SNARE complexes.23 Such Ca2+-dependence has been demonstrated directly. First, the buffering of Ca2+ in live cells using membrane-permeable Ca2+ chelators, such as the AM ester of BAPTA, halt the progression of endocytosed material through the endocytic pathway.24 Second, isolated endocytic organelles require Ca2+ for fusion, as shown by the content-mixing assay.25 The Ca2+-dependence of the endocytic function raises three important questions: (1) What are the sources of Ca2+ that drive the fusion within the endo-lysosomal system? (2) What are the ion channels that drive Ca2+ release? And (3) What are the signals that that regulate these elusive calcium release events? The remainder of the present review is focused on these questions.
The Sources of Ca2+ for Intracellular Membrane Fusion
Multiple evidence points to the acidic organelles in the endocytic pathway as the source of Ca2+ that drives their fusion. Ca2+ content of the lysosomes is currently estimated to be ~500 µM, based on direct measurements using endocytosed calcium indicators.26,27 Calcium within lysosomes and other acidic organelles can also be monitored indirectly with cytoplasmic Ca2+ dyes when lysosomal Ca2+ is released into the cytoplasm by the lysosome-lytic agent glycyl-l-phenylalanine 2-naphthylamide (GPN),28 or by the vacuolar H+ pump inhibitor Bafilomycin A129,30 which prevents calcium uptake and initiates lysosomal Ca2+ leak. Accordingly, the lysosomes alkalinize upon release of their Ca2+ 31. Indeed, Bafilomycin A1 is well known to halt membrane traffic in the endocytic pathway, by arresting membrane fusion.32 In another experimental system, heterotypic fusion of isolated endocytic vesicles was facilitated by addition of Ca2+ and halted by addition of a Ca2+ chelator to the reaction buffer.25 The fusion was also halted by depleting lysosomal Ca2+, which was interpreted to support the lysosomal origin of Ca2+ that fuses the endocytic vesicles.25
An independent series of experiments, performed several years before the aforementioned assays, lends an additional support for lysosomes as a significant intracellular source of Ca2+. Two separate pools of intracellular Ca2+ were identified, one filled by a Ca2+ pump that is inhibited by low concentrations of Thapsigargin (TG), but is largely insensitive to tert-Butylhydroquinone (TBHQ), and another, which is insensitive to TG, but very sensitive to TBHQ.33 Subsequently, the TBHQ-sensitive pool was identified as the thapsigargin-insensitive, acidic Ca2+ pool that is likely filled by SERCA3 and accumulates the lysosomal dye lysotracker.34 The high acidification of these organelles suggests that the TBHQ-insensitive pool is lysosomes, or that it comprises lysosomes as its subset.
It should be noted, however, that although the fusion events in the proximal endocytic pathway (formation of endocytic vesicles and early and recycling endosomes) drives the capture and sorting of the endocytosed material, neither the Ca2+ requirement, nor the source of Ca2+ for these events have been established. The TBHQ experiments discussed above suggest existence of a Ca2+ ATPase that pumps Ca2+ into the endocytic organelles. Candidates for such pumps are SERCA333,34 and the secretory pathway Ca2+ pumps (SPCA),35 although their existence has been firmly established largely in the Golgi.35,36 Alternatively, these organelles may receive Ca2+ from the extracellular medium, or from the ER, in the process of organellar maturation.
In summary, these findings establish the functional context and the biochemical identification of the organelles that provide the Ca2+ necessary for the fusion of vesicles in the endocytic pathway. The recent identification of Ca2+ permeable channels residing in the endocytic pathway suggests candidates responsible for Ca2+ release from these organelles that mediate the fusion/fission events.
Endo-Lysosomal Ca2+-Permeable Ion Channels
TRPML and TPCs are two Ca2+-permeable ion channel families that have been localized on the basis of immunestaining and functional evidence to the acidic organelles in the endocytic pathway. This is illustrated in part in Figure 1. TRPML1 (Fig. 1A and ref. 37) and TPC2 are found mostly in late endosomes and lysosomes, TRPML3 is found in multiple intracellular compartments (Fig. 1B and ref. 38), similar to TPC1, in addition to the plasma membrane. TRPML1 and TPC2 show strong colocalization, and accordingly co-immunoprecipitate, while TRPML3 and TPC1 show weak localization with each other and with TRPML1 and TPC1, although some interaction among the channels can be demonstrated by coimmunoprecipitation (Figs. 1C–E and ref. 39). TRPMLs and TPCs are, currently, the best candidates for the role of endocytic Ca2+ release channels, albeit neither one appear to fit that role completely. Another Ca2+ permeable channel that is expressed in and mediates Ca2+ release from intracellular organelles of β cells40 and Macrophages,41,42 is the ADPR-activated TRPM2. Since TRPM2 appears to function as intracellular Ca2+ release channels in specific cells, it will not be discussed here; however, this topic was reviewed recently in references 43–46.

Figure 1. Distribution of the TRPMLs and TPCs in intracellular organelles. Panel (A) shows the localization of TRPML1 relative to the organellar markers LAMP1, MRP and EEA.1. The images were taken from.37 Panel (B) shows the localization of TRPML3 relative to the organellar markers LAMP1, lysotracker and EEA.1. The images were taken from.38 Panel (C) shows the localization of TRPML1 and TRPML3 relative to the localization of TPC1 and TPC2 and panel (D) shows the Co-IP of the TRPMLs and TPCs. The results in (C and D) were taken from.78
TRPMLs
The TRPML family was established with the identification of TRPML1.47,48 TRPML1, the first member of the mucolipins, was cloned in 1999, as a result of genetic analyses of patient tissues with a genetic disease called Mucolipidosis type IV (MLIV).49,50 MLIV is a lysosomal storage disease linked to buildup of poorly characterized material throughout the patient’s tissue in the form of storage bodies.51,52 TRPML1 localizes in lysosomes due to the presence of two N- and a C-terminal lysosomal targeting signals.53 Since then, two more members of this family, TRPML2 and TRPML3, have been found in mammals. A number of excellent review articles on TRPML channels published recently provide comprehensive analysis of TRPML localization, traffic and possible function.47,48,53-55 In the present review, we will focus on their Ca2+ transport function and the evidence for its role in membrane fusion.
TRPML1 permeability was analyzed by patch clamping of lysosomes and by recording the currents through the “activated” mutant form of TRPML, which carries the gain-of-function varitint-waddler mutation of TRPML3.56-58 It is of note that this mutation changes the selectivity of TRPML channels.59 The recent studies with a lysosome-targeted Ca2+ probe showed that TRPML1 is required for Ca2+ release from the lysosomes.60 All TRPML channels function as strong inward rectifiers56,61 which means that, under normal conditions, the predominant direction of ion flow is from the organellar lumen into the cytoplasm. TRPMLs are permeable to monovalent cations and to Ca2+ among other divalent cations.57,58,61,62 Their current is regulated by luminal pH. This was best analyzed for TRPML3, in which the pH regulation is mediated by a series of histidine residues in the loop connecting the 1st and 2nd TM regions that forms a H+ relay pathway.63 Hence, Ca2+ release through TRPML3 is likely controlled by the organellar pH and is facilitated by organellar alkalinization.
The impact of the TRPML permeability on the endocytic Ca2+ content has not been completely settled. Analysis of TRPML1 impact on the lysosomal Ca2+ using the lysomotropic agent GPN yielded no effect of TRPML1 expression or deletion on the lysosomal Ca2+ 37. Thus, expression of channel-dead TRPML164 or knockout of TRPML1 in mice65 did not affect the global cytoplasmic Ca2+ increase evoked by GPN. However, with Ca2+ sensors targeted to the surface of lysosomes, it was possible to detect a small Ca2+ signal by activation of TRPML1.60 This suggests that Ca2+ released from lysosomes mostly regulates Ca2+ in the vicinity of the lysosomes. This is ideal for regulation of local fusion events.
Although a role for organellar Ca2+ in membrane trafficking is firmly established, the exact role and mechanism by which the TRPML impact on membrane traffic remains unsettled. The effect of TRPML1 on membrane traffic has been analyzed using human fibroblasts from MLIV patients. Limitations of these approaches have been discussed before.66,67 siRNA-driven TRPML1 knockdown (KD) models24,68 yielded pre-lysosomal membrane traffic delays in one model68 and post-lysosomal delays with no pre-lysosomal delays in another study.24 Localization of TRPML3 is broader than that of TRPML1 and it is expressed in all compartments of the endocytic pathway38 (see also Figure 1). Knockdown of TRPML3 led to an increase in the fusion of vesicles in the proximal portion of the endocytic pathway, which is consistent with TRPML3 localization.38 Conversely, overexpression of TRPML3 caused autophagy and TRPML3 is massively recruited to autophagosomes when autophagy was induced by several stressors.38,69
TPCs
TPCs were cloned from a rat kidney library long before their function was revealed.70 In a search for the intracellular channels activated by the potent second messenger NAADP,71 two recent independent studies70,72 identified the TPCs as the channels activated by NAADP to mediate Ca2+ release from acidic organelles. Subsequent work showed that overexpression of TPC172 and TPC270,73,74 potentiates NAADP-mediated Ca2+ release whereas knockdown of TPCs70,72 or expression of the channel-dead pore mutants TPC1(L273P)72 and TPC2(L265P)75,76 inhibits NAADP-mediated Ca2+ release. NAADP-dependent Ca2+ release is ubiquitous and in a subset of cells is a prerequisite for secretion, most prominently in sea urchin eggs, the original model for studying NAADP signaling.71 In other cells, such as pancreatic acini,77 it serves as a trigger to facilitate Ca2+ release from the ER when stimulated with agonists such as CCK. The TPCs are coded by three genes in most animals; however the gene coding for TPC3 is degenerate in certain mammals including humans.74 To the extent examined, the level of TPC1 transcripts is about 10 times higher than TPC2.70,78,79 TPCs have a unique structure comprising two repeated domains each containing six transmembrane (TM) spans where the 5th and 6th TM spans of each repeat likely form the pore, and TM4 and TM10 act as voltage sensors.80 This contrasts to TRPMLs, whose structure largely reproduces the blueprint for voltage-gated K+ channels comprising a single 6 TM domain.
Ca2+ mobilization by NAADP, which activates the TPCs, has been linked to several key physiological functions that include regulated exocytosis, fertilization, glucose sensing and neuronal growth.81 Fusion and fission events of intracellular organelles mediate many aspects of these functions, suggesting a role for the TPCs in membrane trafficking. This suggestion is supported by a lysosomal storage disease-like phenotype observed in cells overexpressing TPC2 and swelling of the endocytic pathway loaded with fluid phase markers in cells opverexpressing TPC1.82 Although knockout of TPC2 eliminates the NAADP responses in pancreatic β cells,70 no trafficking phenotype has been reported in these mice yet.
Regulation of TRPMLs and TPCs
The real puzzle of Ca2+ signaling in endocytic membrane fusion is the nature of the cue(s) initiating Ca2+ release. Although biochemical and electrophysiological studies have clearly identified TRPML and TPC activation mechanisms, their significance in the context of membrane fusion is not known. Moreover, although present in the same organelles (Fig. 1), the TRPML and TPC appear to function independently of each other. This is most clearly illustrated for TRPML1 and TPC2, which show almost perfect co-localizations and strong co-precipitation (Fig. 1). However, as shown in Figure 2, TRPML1 is not activated by NAADP (Fig. 2A) and the dominant negative channel-dead TRPML1 does not affect NAADP-mediated Ca2+ release (Fig. 2B and C). Furthermore, the response to NAADP is unaltered in cells deficient with TRPML1 (Fig. 2D and E). Returning to the presynaptic fusion paradigm, it involves the arrival of a defined signal (depolarization), which is then immediately translated into membrane fusion. Are there corresponding signals for membrane fusion within the endo-lysosomal system or is fusion truly constitutive? Perhaps by understanding regulation of the Ca2+ release channels we can obtain information to address these questions, at least in part. The main regulatory modalities of the TRPMLs and TPCs and the principals of synaptic and endo/lysosomal fusion/fission are illustrated in Figure 3.

Figure 2. Independent function of TRPML1 and the TPCs. Panels (A-C) show cytosolic Ca2+ responses of individual Fura-2 loaded SKBR3 cells stimulated with NAADP. In (A) the cells were transfected with TRPML1 and injected with 10 nM NAADP. The insert shows typical response of cells transfected with TPC2 as a control. In (B and C) the native TPCs were activated by injection of 10 µM NAADP in control cells and cell transfected with the dominant negative TRPML1(D471K). In panels (D and E), acinar cells isolated from the pancreas of wild type (D) or TRPML1−/− mice (E) were used to record the Ca2+-activated Cl- current in response to infusion with 50 nM NAADP through the patch pipette. As controls, the cells were stimulated with 100 µM carbachol. Note that deletion of TRPML1 has no effect on the response to NAADP. The results were taken from.39

Figure 3. A schematic representation of the role of TPC and TRPML channels in membrane fusion. Left panel: The classic scheme of membrane fusion in presynaptic terminals: Ca2+ influx through the voltage-regulated L-type CaV channels (orange) triggers conformational change in SNARE complexes (not shown) and membrane fusion (green). This influx it triggered by a change in the membrane potential (red). Right panel: Membrane fusion in the endocytic pathway. Ca2+ efflux out of the H+- and Ca2+- rich endo/lysosomes (orange) through TPC and TRPML channels triggers conformational change in SNARE complexes (not shown) and membrane fusion (green). Ca2+ efflux through TPC channels is triggered by NAADP and modulated by membrane potential (red) and Ca2+. TRPML Ca2+ fluxes are activated by PI(3,5)P2 and TRPML1 activity is terminated by cleavage.
An interesting and likely important regulator of TRPML1, and perhaps other organellar Ca2+ release channels, is Phosphatidylinositol 3,5-bisphosphate [PI(3,5)P2].60,83 PI(3,5)P2 is the only physiological signal that has been shown to date to activate TRPML1. PI(3,5)P2 localization is restricted to the late endocytic pathway, limiting the functional range of TRPML1. Perhaps this is also true for TPCs. This notion is supported by the recent finding that PI(4,5)P2 and sphingomyelin (SM), which are predominant in the plasma membrane, inhibit TRPML1,60,83 possibly ensuring that this channel is not active in the plasma membrane. Inhibition by SM may be even more significant for TRPML3 that traffics between the plasma membrane and intracellular organelles.38 PI(3,5)P2 is not known to undergo dramatic changes in response to cell signaling or during cell stress. Models of the functional consequences of its effect on TRPML1 and membrane traffic have been discussed before.47 In brief, the PI(3,5)P2-TRPML1 interaction may be a priming mechanism for newly delivered TRPML1 into lysosomes, or it may serve as a proximity sensor for organelles in the endocytic pathway. According to this model, the interacting membranes are matched by Rab and SNARE proteins, followed by PI(3,5)P2 binding to the C-termini of TRPML1, Ca2+ release and conformational changes in SNARE proteins which drive membrane fusion.
Another very important form of TRPML1 regulation is proteolytic cleavage. The majority of native and expressed TRPML1 is cleaved at R202 in the large extracellular loop between TM regions 1 and 2. The cleavage is mediated by Cathepsin B and results in inactivation of the channel.37 It appears that the TRPML1 channels remain active for a short time and inactivate shortly after arriving at the lysosomes by cleavage. How this cleavage is regulated or whether the cleavage is regulated by PI(3,5)P2 is not known at present. It seems to be important to follow this mode of regulation of TRPML1 that is likely to have a prominent cellular role. Unlike TRPML1, neither TRPML3 nor the TPCs appear to be cleaved.
The kinetic and regulatory properties of the TPCs and how they are activated by NAADP is still in its infancy. NAADP is a second messenger that rises in the cytoplasm in response to cell activation albeit through a poorly defined pathway and enzyme(s). Until recently, studying TPC activation by NAADP was limited to recording Ca2+ signals induced by NAADP delivery into the cytoplasm. The adoption of bilayer technique to TPC research and targeting the TPCs to the plasma membrane enabled testing the kinetics and regulation of TPC activation by NAADP through biophysical means. Additional information that is relevant in part to the mammalian TPCs, are the properties of the plant TPC1 homolog AtTPC1 that has been more extensively studied although likely NAADP-insensitive.80
What we have learned over the last few years is that when activated by NAADP, human TPC2 behaved as a channel with a conductance of 100–120 pS in symmetrical Cs+ 75, or a 300 pS in symmetrical K+ and with 1:3 K+:Ca2+ selectivity.84 Both, TPC1 and TPC2 are regulated by lysosomal lumen pH and Ca2+.79,84 The Arabidopsis AtTPC1 that mediates the slow vacuolar current,85 is also regulated by luminal Ca2+.86 Ca2+ and pH affect the apparent affinity of TPC2 to NAADP with higher Ca2+ increasing the apparent affinity for NAADP and lower pH reducing channel open probability.84 However, this topic is still not settled as TPC2 analysis in a planar patch clamp system suggested that TPC2 is active only at acidic luminal pH.84 Recently, we characterized several features of human TPC1 and reported that it is regulated by luminal Ca2+ and pH, with both high luminal H+ and Ca2+ activating the channel.79 The molecular basis for this regulation is not known at present but in the Arabidopsis channel, a putative regulatory calcium binding site has been identified between the first and second TM region of domain II (S7 and S8).87 Another interesting feature of TPC1 is that it exists in two strictly coupled conductances of 50 and 200 pS, both of which are activated by NAADP. The large conductance has short openings but once open it markedly increases the open time of the small conductance.79 This unique mode of regulation may serve as a guard to control the duration of endo-lysosomal Ca2+ permeability.
An interesting mode of regulation of TPC1 is by the membrane potential. Shifting the membrane potential from 0 to -60 mV increased linearly both the small and the large conductances and the mean open probability.79 Notably, the mammalian TPCs have two putative voltage sensors in fourth TM regions of each domain.80 The voltage sensors are likely to mediate the voltage dependence of TPC1 as they do in AtTPC1.80 But perhaps the most intriguing aspect of regulation of TPC1 by voltage is the crosstalk between the lysosomal lumen, membrane potential and NAADP binding on the cytoplasmic side of TPC1. We have shown recently,79 (illustrated in Fig. 4), that the membrane potential regulates the apparent affinity for NAADP activation of TPC1 by 10 nM/mV. Dynamic affinity for NAADP that is regulated by membrane potential provides a plausible mechanism for Ca2+ oscillations generated by intracellular organelles. NAADP is known to induce oscillatory changes in cytoplasmic Ca2+ involving both acidic organelles and the ER (Fig. 2 and refs. 78, 88, 89). The lysosomal and endosomal membrane potentials are about 20 mV lumen-positive,90,91 at which the apparent affinity for activation of TPC1 by NAADP is relatively low such that the channel is expected to be in a closed state. Generation of NAADP in response to receptor stimulation activates TPC1 to cause Ca2+ release and perhaps membrane depolarization by counter ion flow of Cl- through ClC792 and/or K+ influx.93 The depolarization reduces the apparent affinity for NAADP, while the reduction in organellar Ca2+ reduces channel NPo, to terminate Ca2+ release. Ca2+ reloading of the organelles and repolarization of the membrane potential restore responsiveness of TPC1 to NAADP to evoke the next Ca2+ spike.
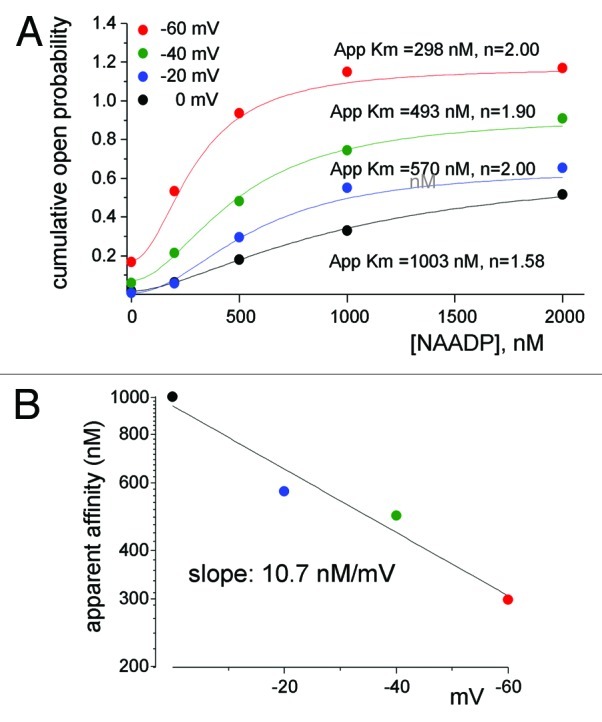
Figure 4. The apparent affinity for activation of TPC1 by NAADP is determined by the membrane potential. Shown in (A) is the NAADP dependence of TPC1 open probability (NPo) at holding potentials of 0, −20, −40 and −60 mV. Fitting parameters for Apparent Km and the Hill coefficient are shown next to the traces. (B) Plots the voltage dependence of the apparent affinity for NAADP obtained for panel (A). Results were taken from.79
Outlook
It is clear the endo-lysosomal system undergoes Ca2+-dependent membrane trafficking and that it is endowed with at least two distinct families of Ca2+-permeable channels that are capable of mediating Ca2+ release from acidic organelles. It is puzzling why lysosomes and endosomes express multiple Ca2+ release channels (Fig. 1) that appear to function independently78 (Fig. 2). One possibility is that they respond to different stimuli to mediate selective organellar functions. A recent study provided direct evidence for Ca2+ release from lysosomes evoked by pharmacological activation of TRPML1.60 But how the TRPMLs are activated physiologically is not known. We know that TPCs are physiologically activated by NAADP to generate local signals that are subsequently amplified by the ER to generate global Ca2+ signals. What we don’t know is whether local signals can be generated independently to regulate trafficking. Finding the mechanisms that activate endo-lysosomal Ca2+ channels is thus paramount. Addressing this puzzle and the possible specific roles of the TRPMLs and TPCs in organellar functions deserve further investigation, and is a central challenge in this field. This is necessary, if the role of Ca2+ in membrane trafficking and organellar fusion/fission is to be fully understood.
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/21723
References
- 1.Pang ZP, Südhof TC. Cell biology of Ca2+-triggered exocytosis. Curr Opin Cell Biol. 2010;22:496–505. doi: 10.1016/j.ceb.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Groves E, Dart AE, Covarelli V, Caron E. Molecular mechanisms of phagocytic uptake in mammalian cells. Cell Mol Life Sci. 2008;65:1957–76. doi: 10.1007/s00018-008-7578-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwarz LA, Patrick GN. Ubiquitin-dependent endocytosis, trafficking and turnover of neuronal membrane proteins. Mol Cell Neurosci. 2012;49:387–93. doi: 10.1016/j.mcn.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Platta HW, Stenmark H. Endocytosis and signaling. Curr Opin Cell Biol. 2011;23:393–403. doi: 10.1016/j.ceb.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 5.Valerio LG. Mammalian iron metabolism. Toxicol Mech Methods. 2007;17:497–517. doi: 10.1080/15376510701556690. [DOI] [PubMed] [Google Scholar]
- 6.Haglund K, Dikic I. The role of ubiquitylation in receptor endocytosis and endosomal sorting. J Cell Sci. 2012;125:265–75. doi: 10.1242/jcs.091280. [DOI] [PubMed] [Google Scholar]
- 7.Raposo G, Fevrier B, Stoorvogel W, Marks MS. Lysosome-related organelles: a view from immunity and pigmentation. Cell Struct Funct. 2002;27:443–56. doi: 10.1247/csf.27.443. [DOI] [PubMed] [Google Scholar]
- 8.Watts C. The endosome-lysosome pathway and information generation in the immune system. Biochim Biophys Acta. 2012;1824:14–21. doi: 10.1016/j.bbapap.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009;1793:664–73. doi: 10.1016/j.bbamcr.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 10.Raposo G, Marks MS. The dark side of lysosome-related organelles: specialization of the endocytic pathway for melanosome biogenesis. Traffic. 2002;3:237–48. doi: 10.1034/j.1600-0854.2002.030401.x. [DOI] [PubMed] [Google Scholar]
- 11.Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain. 2008;131:1969–78. doi: 10.1093/brain/awm318. [DOI] [PubMed] [Google Scholar]
- 12.Barnett A, Brewer GJ. Autophagy in aging and Alzheimer’s disease: pathologic or protective? J Alzheimers Dis. 2011;25:385–94. doi: 10.3233/JAD-2011-101989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horan MP, Pichaud N, Ballard JW. Review: Quantifying Mitochondrial Dysfunction in Complex Diseases of Aging. J Gerontol A Biol Sci Med Sci. 2012 doi: 10.1093/gerona/glr263. In Press. [DOI] [PubMed] [Google Scholar]
- 14.Kiselyov K, Jennigs JJ, Jr., Rbaibi Y, Chu CT. Autophagy, mitochondria and cell death in lysosomal storage diseases. Autophagy. 2007;3:259–62. doi: 10.4161/auto.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiselyov K, Yamaguchi S, Lyons CW, Muallem S. Aberrant Ca2+ handling in lysosomal storage disorders. Cell Calcium. 2010;47:103–11. doi: 10.1016/j.ceca.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–34. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 17.Schneider P, Korolenko TA, Busch U. A review of drug-induced lysosomal disorders of the liver in man and laboratory animals. Microsc Res Tech. 1997;36:253–75. doi: 10.1002/(SICI)1097-0029(19970215)36:4<253::AID-JEMT4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 18.Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol. 2010;11:50–61. doi: 10.1038/nrm2820. [DOI] [PubMed] [Google Scholar]
- 19.Weisz OA. Organelle acidification and disease. Traffic. 2003;4:57–64. doi: 10.1034/j.1600-0854.2003.40201.x. [DOI] [PubMed] [Google Scholar]
- 20.Jean S, Kiger AA. Coordination between RAB GTPase and phosphoinositide regulation and functions. Nat Rev Mol Cell Biol. 2012;13:463–70. doi: 10.1038/nrm3379. [DOI] [PubMed] [Google Scholar]
- 21.Zeigerer A, Gilleron J, Bogorad RL, Marsico G, Nonaka H, Seifert S, et al. Rab5 is necessary for the biogenesis of the endolysosomal system in vivo. Nature. 2012;485:465–70. doi: 10.1038/nature11133. [DOI] [PubMed] [Google Scholar]
- 22.Luzio JP, Pryor PR, Gray SR, Gratian MJ, Piper RC, Bright NA. Membrane traffic to and from lysosomes. Biochem Soc Symp. 2005;•••:77–86. doi: 10.1042/bss0720077. [DOI] [PubMed] [Google Scholar]
- 23.Lang T, Jahn R. Core proteins of the secretory machinery. Handb Exp Pharmacol. 2008:107–27. doi: 10.1007/978-3-540-74805-2_5. [DOI] [PubMed] [Google Scholar]
- 24.Miedel MT, Rbaibi Y, Guerriero CJ, Colletti G, Weixel KM, Weisz OA, et al. Membrane traffic and turnover in TRP-ML1-deficient cells: a revised model for mucolipidosis type IV pathogenesis. J Exp Med. 2008;205:1477–90. doi: 10.1084/jem.20072194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pryor PR, Mullock BM, Bright NA, Gray SR, Luzio JP. The role of intraorganellar Ca(2+) in late endosome-lysosome heterotypic fusion and in the reformation of lysosomes from hybrid organelles. J Cell Biol. 2000;149:1053–62. doi: 10.1083/jcb.149.5.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christensen KA, Myers JT, Swanson JA. pH-dependent regulation of lysosomal calcium in macrophages. J Cell Sci. 2002;115:599–607. doi: 10.1242/jcs.115.3.599. [DOI] [PubMed] [Google Scholar]
- 27.Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, et al. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14:1247–55. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- 28.Churchill GC, Okada Y, Thomas JM, Genazzani AA, Patel S, Galione A. NAADP mobilizes Ca(2+) from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell. 2002;111:703–8. doi: 10.1016/S0092-8674(02)01082-6. [DOI] [PubMed] [Google Scholar]
- 29.Tapper H, Sundler R. Bafilomycin A1 inhibits lysosomal, phagosomal, and plasma membrane H(+)-ATPase and induces lysosomal enzyme secretion in macrophages. J Cell Physiol. 1995;163:137–44. doi: 10.1002/jcp.1041630116. [DOI] [PubMed] [Google Scholar]
- 30.Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem. 1991;266:17707–12. [PubMed] [Google Scholar]
- 31.Morgan AJ, Galione A. NAADP induces pH changes in the lumen of acidic Ca2+ stores. Biochem J. 2007;402:301–10. doi: 10.1042/BJ20060759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- 33.Cavallini L, Coassin M, Alexandre A. Two classes of agonist-sensitive Ca2+ stores in platelets, as identified by their differential sensitivity to 2,5-di-(tert-butyl)-1,4-benzohydroquinone and thapsigargin. Biochem J. 1995;310:449–52. doi: 10.1042/bj3100449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosado JA. Acidic Ca(2+) stores in platelets. Cell Calcium. 2011;50:168–74. doi: 10.1016/j.ceca.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 35.Vandecaetsbeek I, Vangheluwe P, Raeymaekers L, Wuytack F, Vanoevelen J. The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a004184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baron S, Vangheluwe P, Sepúlveda MR, Wuytack F, Raeymaekers L, Vanoevelen J. The secretory pathway Ca(2+)-ATPase 1 is associated with cholesterol-rich microdomains of human colon adenocarcinoma cells. Biochim Biophys Acta. 2010;1798:1512–21. doi: 10.1016/j.bbamem.2010.03.023. [DOI] [PubMed] [Google Scholar]
- 37.Kiselyov K, Chen J, Rbaibi Y, Oberdick D, Tjon-Kon-Sang S, Shcheynikov N, et al. TRP-ML1 is a lysosomal monovalent cation channel that undergoes proteolytic cleavage. J Biol Chem. 2005;280:43218–23. doi: 10.1074/jbc.M508210200. [DOI] [PubMed] [Google Scholar]
- 38.Kim HJ, Soyombo AA, Tjon-Kon-Sang S, So I, Muallem S. The Ca(2+) channel TRPML3 regulates membrane trafficking and autophagy. Traffic. 2009;10:1157–67. doi: 10.1111/j.1600-0854.2009.00924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zbidi H, Jardin I, Bartegi A, Salido GM, Rosado JA. Ca2+ leakage rate from agonist-sensitive intracellular pools is altered in platelets from patients with type 2 diabetes. Platelets. 2011;22:284–93. doi: 10.3109/09537104.2010.528813. [DOI] [PubMed] [Google Scholar]
- 40.Lange I, Yamamoto S, Partida-Sanchez S, Mori Y, Fleig A, Penner R. TRPM2 functions as a lysosomal Ca2+-release channel in beta cells. Sci Signal. 2009;2:ra23. doi: 10.1126/scisignal.2000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Starkus JG, Fleig A, Penner R. The calcium-permeable non-selective cation channel TRPM2 is modulated by cellular acidification. J Physiol. 2010;588:1227–40. doi: 10.1113/jphysiol.2010.187476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sumoza-Toledo A, Lange I, Cortado H, Bhagat H, Mori Y, Fleig A, et al. Dendritic cell maturation and chemotaxis is regulated by TRPM2-mediated lysosomal Ca2+ release. FASEB J. 2011;25:3529–42. doi: 10.1096/fj.10-178483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sumoza-Toledo A, Penner R. TRPM2: a multifunctional ion channel for calcium signalling. J Physiol. 2011;589:1515–25. doi: 10.1113/jphysiol.2010.201855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heiner I, Eisfeld J, Lückhoff A. Role and regulation of TRP channels in neutrophil granulocytes. Cell Calcium. 2003;33:533–40. doi: 10.1016/S0143-4160(03)00058-7. [DOI] [PubMed] [Google Scholar]
- 45.Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47:122–9. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 46.Takahashi N, Kozai D, Kobayashi R, Ebert M, Mori Y. Roles of TRPM2 in oxidative stress. Cell Calcium. 2011;50:279–87. doi: 10.1016/j.ceca.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 47.Colletti GA, Kiselyov K. Trpml1. Adv Exp Med Biol. 2011;704:209–19. doi: 10.1007/978-94-007-0265-3_11. [DOI] [PubMed] [Google Scholar]
- 48.Zeevi DA, Frumkin A, Bach G. TRPML and lysosomal function. Biochim Biophys Acta. 2007;1772:851–8. doi: 10.1016/j.bbadis.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 49.Bargal R, Avidan N, Ben-Asher E, Olender Z, Zeigler M, Frumkin A, et al. Identification of the gene causing mucolipidosis type IV. Nat Genet. 2000;26:118–23. doi: 10.1038/79095. [DOI] [PubMed] [Google Scholar]
- 50.Acierno JS, Jr., Kennedy JC, Falardeau JL, Leyne M, Bromley MC, Colman MW, et al. A physical and transcript map of the MCOLN1 gene region on human chromosome 19p13.3-p13.2. Genomics. 2001;73:203–10. doi: 10.1006/geno.2001.6526. [DOI] [PubMed] [Google Scholar]
- 51.Bach G, Cohen MM, Kohn G. Abnormal ganglioside accumulation in cultured fibroblasts from patients with mucolipidosis IV. Biochem Biophys Res Commun. 1975;66:1483–90. doi: 10.1016/0006-291X(75)90526-4. [DOI] [PubMed] [Google Scholar]
- 52.Tellez-Nagel I, Rapin I, Iwamoto T, Johnson AB, Norton WT, Nitowsky H. Mucolipidosis IV. Clinical, ultrastructural, histochemical, and chemical studies of a case, including a brain biopsy. Arch Neurol. 1976;33:828–35. doi: 10.1001/archneur.1976.00500120032005. [DOI] [PubMed] [Google Scholar]
- 53.Puertollano R, Kiselyov K. TRPMLs: in sickness and in health. Am J Physiol Renal Physiol. 2009;296:F1245–54. doi: 10.1152/ajprenal.90522.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng X, Shen D, Samie M, Xu H. Mucolipins: Intracellular TRPML1-3 channels. FEBS Lett. 2010;584:2013–21. doi: 10.1016/j.febslet.2009.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kiselyov K, Colletti GA, Terwilliger A, Ketchum K, Lyons CW, Quinn J, et al. TRPML: transporters of metals in lysosomes essential for cell survival? Cell Calcium. 2011;50:288–94. doi: 10.1016/j.ceca.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim HJ, Li Q, Tjon-Kon-Sang S, So I, Kiselyov K, Muallem S. Gain-of-function mutation in TRPML3 causes the mouse Varitint-Waddler phenotype. J Biol Chem. 2007;282:36138–42. doi: 10.1074/jbc.C700190200. [DOI] [PubMed] [Google Scholar]
- 57.Nagata K, Zheng L, Madathany T, Castiglioni AJ, Bartles JR, García-Añoveros J. The varitint-waddler (Va) deafness mutation in TRPML3 generates constitutive, inward rectifying currents and causes cell degeneration. Proc Natl Acad Sci U S A. 2008;105:353–8. doi: 10.1073/pnas.0707963105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grimm C, Cuajungco MP, van Aken AF, Schnee M, Jörs S, Kros CJ, et al. A helix-breaking mutation in TRPML3 leads to constitutive activity underlying deafness in the varitint-waddler mouse. Proc Natl Acad Sci U S A. 2007;104:19583–8. doi: 10.1073/pnas.0709846104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim HJ, Yamaguchi S, Li Q, So I, Muallem S. Properties of the TRPML3 channel pore and its stable expansion by the Varitint-Waddler-causing mutation. J Biol Chem. 2010;285:16513–20. doi: 10.1074/jbc.M109.078204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shen D, Wang X, Li X, Zhang X, Yao Z, Dibble S, et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun. 2012;3:731. doi: 10.1038/ncomms1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu H, Delling M, Li L, Dong X, Clapham DE. Activating mutation in a mucolipin transient receptor potential channel leads to melanocyte loss in varitint-waddler mice. Proc Natl Acad Sci U S A. 2007;104:18321–6. doi: 10.1073/pnas.0709096104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dong XP, Cheng X, Mills E, Delling M, Wang F, Kurz T, et al. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature. 2008;455:992–6. doi: 10.1038/nature07311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim HJ, Li Q, Tjon-Kon-Sang S, So I, Kiselyov K, Soyombo AA, et al. A novel mode of TRPML3 regulation by extracytosolic pH absent in the varitint-waddler phenotype. EMBO J. 2008;27:1197–205. doi: 10.1038/emboj.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soyombo AA, Tjon-Kon-Sang S, Rbaibi Y, Bashllari E, Bisceglia J, Muallem S, et al. TRP-ML1 regulates lysosomal pH and acidic lysosomal lipid hydrolytic activity. J Biol Chem. 2006;281:7294–301. doi: 10.1074/jbc.M508211200. [DOI] [PubMed] [Google Scholar]
- 65.Chandra M, Zhou H, Li Q, Muallem S, Hofmann SL, Soyombo AA. A role for the Ca2+ channel TRPML1 in gastric acid secretion, based on analysis of knockout mice. Gastroenterology. 2011;140:857–67. doi: 10.1053/j.gastro.2010.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kiselyov K, Soyombo A, Muallem S. TRPpathies. J Physiol. 2007;578:641–53. doi: 10.1113/jphysiol.2006.119024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miedel MT, Rbaibi Y, Guerriero CJ, Colletti G, Weixel KM, Weisz OA, et al. Membrane traffic and turnover in TRP-ML1-deficient cells: a revised model for mucolipidosis type IV pathogenesis. J Exp Med. 2008;205:1477–90. doi: 10.1084/jem.20072194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thompson EG, Schaheen L, Dang H, Fares H. Lysosomal trafficking functions of mucolipin-1 in murine macrophages. BMC Cell Biol. 2007;8:54. doi: 10.1186/1471-2121-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martina JA, Lelouvier B, Puertollano R. The calcium channel mucolipin-3 is a novel regulator of trafficking along the endosomal pathway. Traffic. 2009;10:1143–56. doi: 10.1111/j.1600-0854.2009.00935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Calcraft PJ, Ruas M, Pan Z, Cheng X, Arredouani A, Hao X, et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature. 2009;459:596–600. doi: 10.1074/jbc.M110.190694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee HC, Aarhus R. A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J Biol Chem. 1995;270:2152–7. doi: 10.1074/jbc.270.5.2152. [DOI] [PubMed] [Google Scholar]
- 72.Brailoiu E, Churamani D, Cai X, Schrlau MG, Brailoiu GC, Gao X, et al. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J Cell Biol. 2009;186:201–9. doi: 10.1083/jcb.200904073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zong X, Schieder M, Cuny H, Fenske S, Gruner C, Rötzer K, et al. The two-pore channel TPCN2 mediates NAADP-dependent Ca(2+)-release from lysosomal stores. Pflugers Arch. 2009;458:891–9. doi: 10.1007/s00424-009-0690-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brailoiu E, Hooper R, Cai X, Brailoiu GC, Keebler MV, Dun NJ, et al. An ancestral deuterostome family of two-pore channels mediates nicotinic acid adenine dinucleotide phosphate-dependent calcium release from acidic organelles. J Biol Chem. 2010;285:2897–901. doi: 10.1074/jbc.C109.081943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brailoiu E, Rahman T, Churamani D, Prole DL, Brailoiu GC, Hooper R, et al. An NAADP-gated two-pore channel targeted to the plasma membrane uncouples triggering from amplifying Ca2+ signals. J Biol Chem. 2010;285:38511–6. doi: 10.1074/jbc.M110.162073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pereira GJ, Hirata H, Fimia GM, do Carmo LG, Bincoletto C, Han SW, et al. Nicotinic acid adenine dinucleotide phosphate (NAADP) regulates autophagy in cultured astrocytes. J Biol Chem. 2011;286:27875–81. doi: 10.1074/jbc.C110.216580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Petersen OH, Tepikin AV. Polarized calcium signaling in exocrine gland cells. Annu Rev Physiol. 2008;70:273–99. doi: 10.1146/annurev.physiol.70.113006.100618. [DOI] [PubMed] [Google Scholar]
- 78.Yamaguchi S, Jha A, Li Q, Soyombo AA, Dickinson GD, Churamani D, et al. Transient receptor potential mucolipin 1 (TRPML1) and two-pore channels are functionally independent organellar ion channels. J Biol Chem. 2011;286:22934–42. doi: 10.1074/jbc.M110.210930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rybalchenko V, Ahuja M, Coblentz J, Churamani D, Patel S, Kiselyov K, et al. Membrane potential regulates nicotinic acid adenine dinucleotide phosphate (NAADP) dependence of the pH- and Ca2+-sensitive organellar two-pore channel TPC1. J Biol Chem. 2012;287:20407–16. doi: 10.1074/jbc.M112.359612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hedrich R, Marten I. TPC1-SV channels gain shape. Mol Plant. 2011;4:428–41. doi: 10.1093/mp/ssr017. [DOI] [PubMed] [Google Scholar]
- 81.Guse AH, Lee HC. NAADP: a universal Ca2+ trigger. Sci Signal. 2008;1:re10. doi: 10.1126/scisignal.144re10. [DOI] [PubMed] [Google Scholar]
- 82.Ruas M, Rietdorf K, Arredouani A, Davis LC, Lloyd-Evans E, Koegel H, et al. Purified TPC isoforms form NAADP receptors with distinct roles for Ca(2+) signaling and endolysosomal trafficking. Curr Biol. 2010;20:703–9. doi: 10.1016/j.cub.2010.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang X, Li X, Xu H. Phosphoinositide isoforms determine compartment-specific ion channel activity. Proceedings of the National Academy of Sciences of the United States of America (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pitt SJ, Funnell TM, Sitsapesan M, Venturi E, Rietdorf K, Ruas M, et al. TPC2 is a novel NAADP-sensitive Ca2+ release channel, operating as a dual sensor of luminal pH and Ca2+ J Biol Chem. 2010;285:35039–46. doi: 10.1074/jbc.M110.156927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Peiter E, Maathuis FJ, Mills LN, Knight H, Pelloux J, Hetherington AM, et al. The vacuolar Ca2+-activated channel TPC1 regulates germination and stomatal movement. Nature. 2005;434:404–8. doi: 10.1038/nature03381. [DOI] [PubMed] [Google Scholar]
- 86.Pottosin II, Martínez-Estévez M, Dobrovinskaya OR, Muñiz J, Schönknecht G. Mechanism of luminal Ca2+ and Mg2+ action on the vacuolar slowly activating channels. Planta. 2004;219:1057–70. doi: 10.1007/s00425-004-1293-7. [DOI] [PubMed] [Google Scholar]
- 87.Dadacz-Narloch B, Beyhl D, Larisch C, López-Sanjurjo EJ, Reski R, Kuchitsu K, et al. A novel calcium binding site in the slow vacuolar cation channel TPC1 senses luminal calcium levels. Plant Cell. 2011;23:2696–707. doi: 10.1105/tpc.111.086751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cancela JM, Gerasimenko OV, Gerasimenko JV, Tepikin AV, Petersen OH. Two different but converging messenger pathways to intracellular Ca(2+) release: the roles of nicotinic acid adenine dinucleotide phosphate, cyclic ADP-ribose and inositol trisphosphate. EMBO J. 2000;19:2549–57. doi: 10.1093/emboj/19.11.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Churchill GC, Galione A. NAADP induces Ca2+ oscillations via a two-pool mechanism by priming IP3- and cADPR-sensitive Ca2+ stores. EMBO J. 2001;20:2666–71. doi: 10.1093/emboj/20.11.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Koivusalo M, Steinberg BE, Mason D, Grinstein S. In situ measurement of the electrical potential across the lysosomal membrane using FRET. Traffic. 2011;12:972–82. doi: 10.1111/j.1600-0854.2011.01215.x. [DOI] [PubMed] [Google Scholar]
- 91.Sonawane ND, Thiagarajah JR, Verkman AS. Chloride concentration in endosomes measured using a ratioable fluorescent Cl- indicator: evidence for chloride accumulation during acidification. J Biol Chem. 2002;277:5506–13. doi: 10.1074/jbc.M110818200. [DOI] [PubMed] [Google Scholar]
- 92.Weinert S, Jabs S, Supanchart C, Schweizer M, Gimber N, Richter M, et al. Lysosomal pathology and osteopetrosis upon loss of H+-driven lysosomal Cl- accumulation. Science. 2010;328:1401–3. doi: 10.1126/science.1188072. [DOI] [PubMed] [Google Scholar]
- 93.Steinberg BE, Huynh KK, Brodovitch A, Jabs S, Stauber T, Jentsch TJ, et al. A cation counterflux supports lysosomal acidification. J Cell Biol. 2010;189:1171–86. doi: 10.1083/jcb.200911083. [DOI] [PMC free article] [PubMed] [Google Scholar]