

Abstract
Voltage-gated sodium selective ion channel NaV1.5 is expressed in the heart and the gastrointestinal tract, which are mechanically active organs. NaV1.5 is mechanosensitive at stimuli that gate other mechanosensitive ion channels. Local anesthetic and antiarrhythmic drugs act upon NaV1.5 to modulate activity by multiple mechanisms. This study examined whether NaV1.5 mechanosensitivity is modulated by local anesthetics. NaV1.5 channels wereexpressed in HEK-293 cells, and mechanosensitivity was tested in cell-attached and excised inside-out configurations. Using a novel protocol with paired voltage ladders and short pressure pulses, negative patch pressure (-30 mmHg) in both configurations produced a hyperpolarizing shift in the half-point of the voltage-dependence of activation (V1/2a) and inactivation (V1/2i) by about -10 mV. Lidocaine (50 µM) inhibited the pressure-induced shift of V1/2a but not V1/2i. Lidocaine inhibited the tonic increase in pressure-induced peak current in a use-dependence protocol, but it did not otherwise affect use-dependent block. The local anesthetic benzocaine, which does not show use-dependent block, also effectively blocked a pressure-induced shift in V1/2a. Lidocaine inhibited mechanosensitivity in NaV1.5 at the local anesthetic binding site mutated (F1760A). However, a membrane impermeable lidocaine analog QX-314 did not affect mechanosensitivity of F1760A NaV1.5 when applied from either side of the membrane. These data suggest that the mechanism of lidocaine inhibition of the pressure-induced shift in the half-point of voltage-dependence of activation is separate from the mechanisms of use-dependent block. Modulation of NaV1.5 mechanosensitivity by the membrane permeable local anesthetics may require hydrophobic access and may involve membrane-protein interactions.
Keywords: voltage-gated, sodium channel, ion channel, stretch, mechanosensitive, pressure, lidocaine, benzocaine, QX-314
Introduction
Voltage-gated sodium selective ion channel NaV1.5 is mechanosensitive
NaV1.5 is found in two mechanically active organs—the heart1 and the gastrointestinal (GI) tract.2 In cardiac myocytes NaV1.5 is responsible for the depolarizing upstroke of the action potential.1 Mutations in NaV1.5 are clearly pathogenic in the heart, as exemplified by disorders such as long QT type 3 (LQT3).3 In the human GI tract NaV1.5 is found in smooth muscle cells and interstitial cells of Cajal.4,5 Sodium currents in the gastrointestinal tract contribute to setting the resting potential and affect the upstroke and frequency of the slow waves,6 which are coordinating electrical events in the GI tract required for cyclical contractions. Mutations in NaV1.5 also result in GI pathologies.7,8 In addition to being voltage-sensitive, NaV1.5 is also mechanically sensitive.6,9,10 Recent evidence suggests that some disease-associated NaV1.5 mutations, both in the heart11 and in the gut,8 have abnormal mechanical sensitivity. Therefore, the mechanism of NaV1.5 mechanosensitivity is of scientific and clinical interest.
NaV1.5 mechanosensitivity is established in both cell-attached patches and whole-cell
NaV1.5 is sensitive to mechanical stimulation in the whole-cell mode to shear-stress6 and in cell-attached patches to pressure.9,10 In both whole cell and cell-attached modes, mechanical stimulation of NaV1.5 results in an increase in peak inward current8 and acceleration of the voltage-dependent activation and inactivation kinetics.9,10 In cell-attached membrane patches in HEK cells, negative pressure additionally hyperpolarizes the half-point of voltage-dependence of activation and inactivation by the same linear constant, ~0.7 mV/mmHg.10 The mechanisms of NaV1.5 mechanosensitivity are unclear. Whole-cell studies demonstrate that shear-sensitivity requires an intact f-actin cytoskeleton as f-actin disassembly or disruption of syntrophin, which likely links NaV1.5 channels with actin result in loss of shear-sensitivity.12,13 Previous studies with other NaV channels (NaV1.4 and NaV1.6) in patches have also demonstrated a requirement of the cytoskeleton for mechanosensitivity.14-16 Patch excision resulted in an irreversible hyperpolarizing shift in the voltage-dependence and kinetic properties.14,16 It is unclear from these studies whether the NaV1.5 α-subunit is mechanosensitive as a stand-alone unit.
Mechanism and pharmacology of NaV1.5 mechanosensitivity is unclear
The mechanism of voltage-gated ion channel mechanosensitivity independent of the cytoskeleton is unclear. Previous studies suggest that amphiphiles alter mechanosensitivity of voltage-gated sodium channels,17 but their specificity is unclear.18 Further, no pharmacologic tools exist that specifically address mechanosensitivity of voltage-gated ion channels, and there is little knowledge on the impact of voltage-gated ion channel drugs on their mechanosensitivity. Our recent work showed that lidocaine and a piperazine derivative ranolazine, a molecule similar to lidocaine and with similar mechanism of NaV1.5 block,19 both inhibit NaV1.5 mechanosensitivity.20 We showed in cardiac myocytes and HEK cells heterologously expressing NaV1.5 that ranolazine and lidocaine inhibited mechanosensitivity to shear stress in whole cells and patch pressure in cell-attached patches. Initial evaluation suggested that block of mechanosensitivity by ranolazine was similar to lidocaine, and if the molecules were charged they were ineffective blockers of NaV1.5 mechanosensitivity when applied from the outside. This work is limited by the availability of the extracellular face of the membrane to pharmacologic manipulation. Our aim here was to thoroughly test the extracellular and intracellular facing membrane responses to modulation by local anesthetics.
Reversible NaV1.5 mechanosensitivity in excised patches has not been shown
Direct examination of ion channel mechanosensitivity requires fast (millisecond), reversible and reproducible responses that are independent of other factors.21 The duration of mechanical stimulation is important in directly assessing mechanosensitivity. Previous work on NaV1.5 mechanosensitivity used pressure pulses spanning at the shortest hundreds of milliseconds to seconds,9,10 which results in significant cell membrane restructuring22 and allows the possibility of other mechanosensitive factors contributing to response. Shorter duration pulses are therefore needed. Further, excised patches allow testing of mechanosensitivity in an environment where cytoskeleton and other cytoplasmic factors are less important. However, of the voltage-gated ion channels studied to date only the Shaker KV channel has been shown to be reversibly mechanosensitive in excised patches.23
In this work we demonstrate a robust method for studying reversible mechanosensitivity of NaV1.5 in excised patches. We test NaV1.5 mechanosensitivity of inside-out patches and compare results to the established behavior in cell-attached patches. We then demonstrate lidocaine modulation of NaV1.5 mechanosensitivity in excised patches and that the mechanism of lidocaine inhibition of the pressure-induced shift in the voltage-dependence of activation is separate from the mechanisms of use-dependent block.
Results
Reversible pressure-dependent response in membrane patches
In this study we aimed to minimize membrane restructuring by using the shortest possible pressure pulses. In preliminary experiments for this study we determined that 25 msec pressure steps resulted in a sufficient stimulation and fully reversible behavior. We used a protocol with paired voltage ladders to test voltage-dependence of activation (steps from -140 mV to +10 mV) and voltage-dependence of inactivation (step to -10 mV) (Fig. 1A). By using a 10 msec pre-pulse to -200 mV we were able to accelerate recovery from inactivation which allowed us to obtain a 5-fold average of 16 voltage steps for both control and test without using inter-pulse delays. We tested this protocol extensively for fidelity. In both cell-attached and inside-out patch configurations when no pressure was applied, the voltage-dependent currents were identical for the two ladders, meaning that complete recovery from fast-inactivation had taken place between steps (Fig. S1).
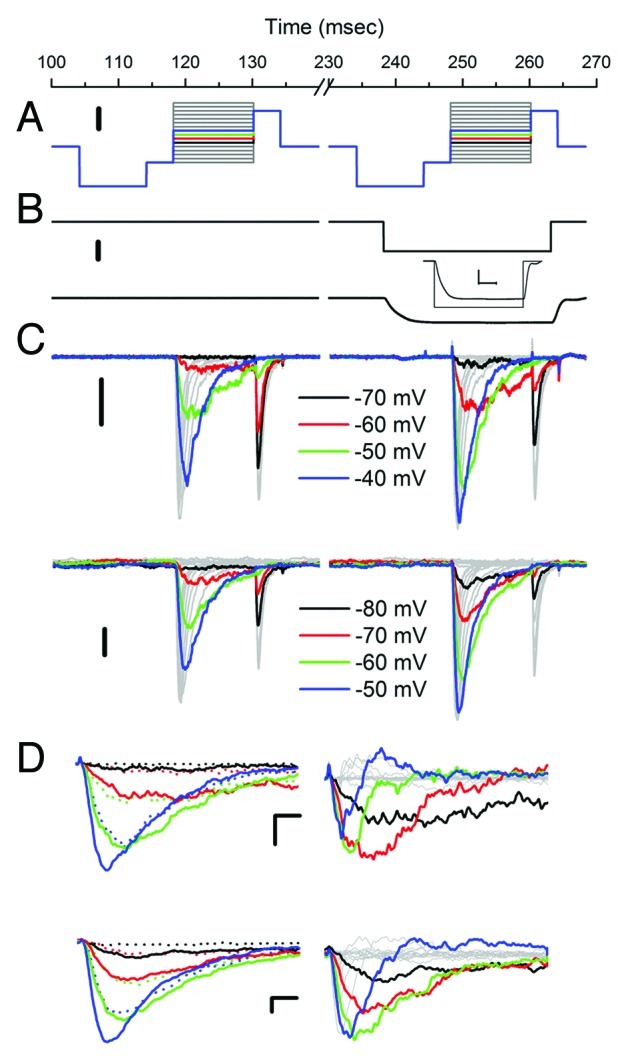
Figure 1. NaV1.5 is reversibly pressure-sensitive in cell-attached and inside-out patches. (A) Voltage-ladder protocol includes a pre-pulse to -200 mV for 10 msec to accelerate recovery from inactivation. Patches are stepped from -140 to +10 mV in 10 mV increments to test activation and then a step to -10 mV to test inactivation. Scale bar is 50 mV. (B) Pressure step to -35 mmHg was applied during the second voltage ladder. Scale bar is 20 mmHg. Lower trace shows pressure at the patch in response to a -30 mmHg step. Inset shows that a -35 mmHg applied pressure results in a -30 mmHg pressure at the patch after a 5 msec risetime. (C) Voltage-dependent currents in a cell-attached (upper) and inside-out patches (lower) at rest (0 mmHg, first ladder) and during pressure stimulation (-30 mmHg, second ladder). Color highlighted steps demonstrate typical differences (larger currents, faster kinetics, greater inactivation) between 0 mmHg and -30 mmHg at multiple voltages spanning the steep voltage-dependent activation. Scale bar is 20 pA for upper trace and 50 pA for lower trace. (D) On the left voltage-dependent currents highlighted in (C) overlaid for 0 mmHg (dotted traces) and -30 mmHg (solid traces) for cell-attached (top) and inside-out (bottom) patches. These traces show larger currents and faster kinetics with pressure. On the right are difference currents Idiff = I-30mmHg – I0mmHg for cell-attached (top) and inside-out (bottom) patches. Pressure-induced changes in the voltage-dependent currents for cell-attached and inside-out patches are very similar. Scale bars 20 pA and 1 msec for cell-attached (upper) traces and 50 pA and 1 msec for inside-out (lower) traces.
To test mechanosensitivity we used -30 mmHg patch pressure, and each voltage step was obtained first at 0 mmHg (rest) and then -30 mmHg stimulus (pressure) (Fig. 1A and B). In our system -35 mmHg of applied pressure translated to -30 mmHg pressure in the patch (Fig. 1B) with a rise-time of ~5 msec (Fig. 1B, inset). Thus, voltage was stepped 10 msec after the start of the pressure pulse. A reverse protocol in which the ladder at -30 mmHg preceded the one at 0 mmHg yielded consistent results with those we present below (Fig. S2).
Negative pressure altered the voltage-dependent currents as illustrated by records obtained from a representative patch in Figure 1C (upper cell-attached, lower inside-out). Qualitatively, no significant differences were noted between the currents in the cell-attached and inside-out configurations. In both sets negative pressure resulted in activation (first voltage step) at more negative voltages (black traces), larger peak currents (blue traces) and faster kinetics (red, green and blue traces) (Fig. 1C and D). Following activation, channel availability (second voltage step) was diminished more significantly with pressure (black, red and green traces) suggesting that the increase in peak current is not due to an increase in channel numbers. In the left panels of Figure 1D (top is cell-attached, bottom is inside-out) we overlay voltage-dependent activation currents for select voltage steps to demonstrate the increases in peak currents and kinetics of activation and inactivation for -30 mmHg (solid) vs. 0 mmHg (dot). In the right panels we show the resulting difference current, which is excess Na+ current when the patch is stretched (Idiff = I30mmHg – I0mmHg). Again, no qualitative difference between cell-attached and inside-out patches was noted for difference currents. In both preparations there was an increase (downward) in difference current for all time points at more hyperpolarized activating voltages (black, red and green traces). For the more depolarized voltages (blue traces) there was a biphasic response—the large negative early peaks in the difference current demonstrated the accelerated kinetics of activation, while faster inactivation resulted in upward deflection at later time points.
Pressure hyperpolarizes NaV1.5 half-point of voltage-dependence of activation and inactivation in inside-out patches
Previous work in cell-attached patches showed pressure-induced changes in NaV1.5 voltage-sensitivity.10,11 In excised inside-out patches at rest (0 mmHg) voltage-dependent peak currents were fit to a two-state Boltzmann function with half-point of activation V1/2a of -58.7 ± 5.4 mV, slope dVa of 8.8 ± 1.6 mV and Gmax of 1.7 ± 1.8 nS (range 0.27 to 5.85) (n = 9) (Fig. 2, black squares). When these patches were stimulated by a -30 mmHg pulse V1/2a was -68.8 ± 6.2 mV, dVa was 8.4 ± 1.7 mV and Gmax was 1.6 ± 1.8 nS (range 0.3 to 5.8 nS) (n = 7) (Fig. 2, red squares). There was a significant difference between -30 mmHg and 0 mmHg in half-points ΔV1/2a = -10.0 ± 1.5 mV (p < 0.01 by paired two-tailed t-test), while the changes in dVa and Gmax were not significant (n = 7).
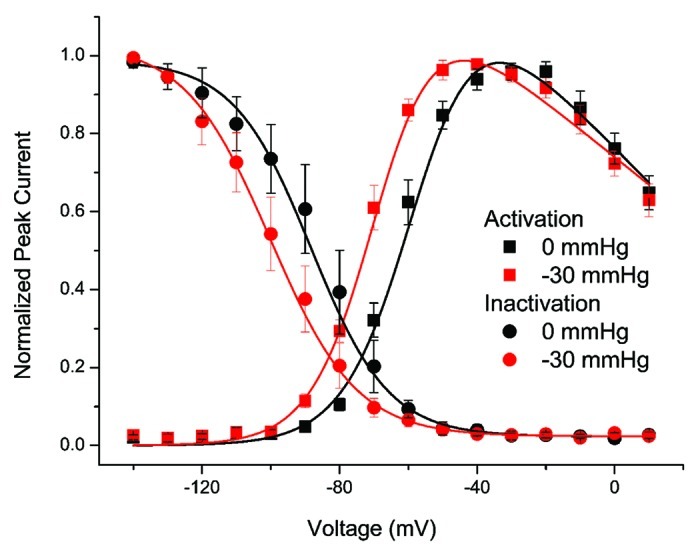
Figure 2. Negative pressure hyperpolarizes NaV1.5 voltage-dependence of activation (V1/2a) and inactivation (V1/2i). Shown here are voltage-dependent current vs. voltage (IV) of activation (squares) and inactivation (circles) from inside-out patches at resting pressure (0 mmHg, black) and with applied pressure (-30 mmHg, red). Data were fit to two-state Boltzmann functions (solid lines) and shows a hyperpolarizing shift in the half-points of voltage dependence of activation (ΔV1/2a = -10 mV) and inactivation (ΔV1/2i = -12 mV) (n = 5, p < 0.01).
In our protocol each 12 msec activation ladder voltage step was followed by a step to -10 mV for 4 msec to test voltage-dependence of fast inactivation (Fig. 1A). Voltage-dependent peak currents were then fit with two-state Boltzmann function. In inside-out patches at rest (0 mmHg) the half-point of voltage-dependence of inactivation (V1/2i) was -88.5 ± 12.0 mV and slope (dVi) was 10.5 ± 2.5 (n = 5) (Fig. 2, black circles). Pressure (-30 mmHg) in these patches resulted in V1/2i of -100.3 ± 10.3 mV and dVi of 11.5 ± 1.1 mV (n = 5) (Fig. 2, red circles). Therefore, pressure hyperpolarized the half-point of inactivation, with ΔV1/2i = -11.7 ± 2.8 mV (p < 0.01 by paired t-test) but there was no significant change in dVi.
These data show that in inside-out patches -30 mmHg pressure steps hyperpolarized the NaV1.5 half-points of voltage-dependence of activation (V1/2a) and inactivation (V1/2i) by -10 and -12 mV, respectively (Fig. 2).
Lidocaine decreases pressure-sensitivity of NaV1.5 half-point of voltage-dependence of activation (V1/2a) but not voltage-dependence of fast inactivation (V1/2i)
At rest and with 50µM lidocaine in the bath, average V1/2a was -56.7 ± 7.8 mV, dVa was -10.3 ± 2.1 mV and Gmax was 0.87 ± 0.75 nS (range 0.10 to 1.92 nS) (n = 7) (Fig. 3, black squares). With -30 mmHg pressure, V1/2a was -60.7 ± 5.1 mV, dVa was -10.6 ± 1.9 mV, Gmax was 0.87 ± 0.75 nS (range 0.075 to 1.92 nS) (n = 7) (Fig. 3, red circles). The resulting difference between 0 and -30 mmHg was a ΔV1/2a of -3.9 ± 4.3 mV (p < 0.05 by paired t-test) without significant differences noted for ΔdVa and ΔGmax (n = 7). However, compared with controls lidocaine resulted in a 6.1 mV smaller ΔV1/2a (p < 0.05 for control vs. lidocaine by two-sample t-test).
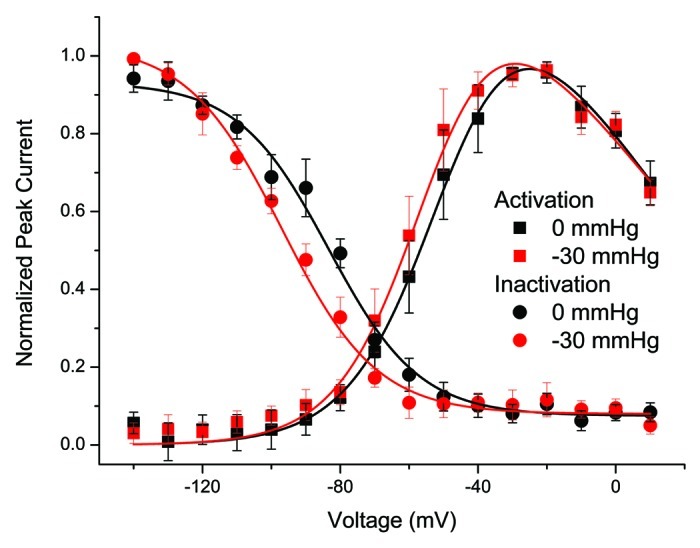
Figure 3. Lidocaine inhibits pressure-induced hyperpolarization of NaV1.5 voltage-dependence of activation (V1/2a) and but not inactivation (V1/2i). Shown here are voltage-dependent current vs. voltage (IV) of activation (squares) and inactivation (circles) from five inside-out patches at resting pressure (0 mmHg, black) and with applied pressure (-30 mmHg, red). Data was fit to two-state Boltzmann functions (solid lines) and shows a a -4 mV shift in the half-point of voltage dependence of activation (ΔV1/2a) (n = 7, p < 0.05), and a -10 mV shift in the half-point of voltage dependence of inactivation (n = 7, p < 0.05).
We also tested the effects of lidocaine on the pressure-induced shift in voltage-dependence of fast inactivation. Lidocaine is known to hyperpolarize the voltage-dependence of fast Nav1.5 inactivation.24 Indeed, at rest using our protocol with 50 µM lidocaine there was a shift in the voltage-dependence of inactivation (Fig. S3). In inside-out patches in the presence of 50 µM lidocaine in the bath at 0 mmHg the average voltage-dependence of half-point of inactivation (V1/2i) was -90.5 ± 3.8 mV, and slope dVi was 14.1 ± 3.8 mV (n = 5, p > 0.05 compared with no lidocaine controls by two-sample t-test). With -30 mmHg stimulation, V1/2i shifted to –101.0 ± 4.4 mV and dVi to 13.0 ± 2.3 mV (n = 5). The shift of V1/2i with stretch with lidocaine was significant for 0 mmHg vs. -30 mmHg ΔV1/2i of -10.5 ± 6.0 mV (p < 0.05 by paired t-test) and similar ΔV1/2i to controls of -11.7 ± 2.8 mV (p > 0.05 between lidocaine and control by two-sample t-test).
The data above show that lidocaine inhibits the pressure-induced hyperpolarizing shift of NaV1.5 half-point of voltage-dependence of activation (V1/2a) but does not inhibit the shift in the half-point of voltage-dependence of fast-inactivation (V1/2i) (Fig. 3). The effect of lidocaine on the mechanosensitivity of V1/2i is likely complicated by lidocaine’s hyperpolarizing effect on half-point of V1/2i at rest.24 Therefore, in the subsequent experiments we used voltage-dependence of activation to assess the effects of local anesthetics on NaV1.5 mechanosensitivity.
Lidocaine inhibits the pressure-induced increase in peak NaV1.5 current but pressure does not alter use-dependent block by lidocaine
In the absence of mechanical stimulation, lidocaine block of NaV1.5 channels is known to involve multiple mechanisms.25 The mechanism with the highest affinity is a use-dependent block by lidocaine.26 We examined the interaction between stretch and use-dependent block by lidocaine in inside-out patches. In controls, a 10 Hz use-dependence protocol resulted in a 10.8 ± 3.3% loss of current between the 1st and 20th steps (n = 3, p < 0.05, Fig. 4). At -30 mmHg, the same protocol resulted in a mean increase in peak current by 13.5 ± 2.6% at each step and a mean loss of current of 9.8 ± 9.7% between 1st and 20th steps (n = 3, p < 0.05, Fig. 4). When lidocaine (50 µM) was added to the bath, at rest (0 mmHg) this protocol produced a significant use-dependent decrease in mean current of 51.4 ± 6.0% between the 1st and 20th steps (n = 3, p < 0.05, Fig. 4). In the presence of lidocaine, pressure failed to increase peak currents at each step as in controls but did not otherwise alter use-dependent block by lidocaine. The average difference between peaks for all steps at 0 mmHg and -30 mmHg pressure was 0.3 ± 3% (p > 0.05 by paired t-test). These results suggest that the mechanisms of use-dependence and mechanosensitivity block of NaV1.5 by lidocaine may not overlap. Next, we explore whether lidocaine use-dependence and mechanosensitivity block mechanisms are related.
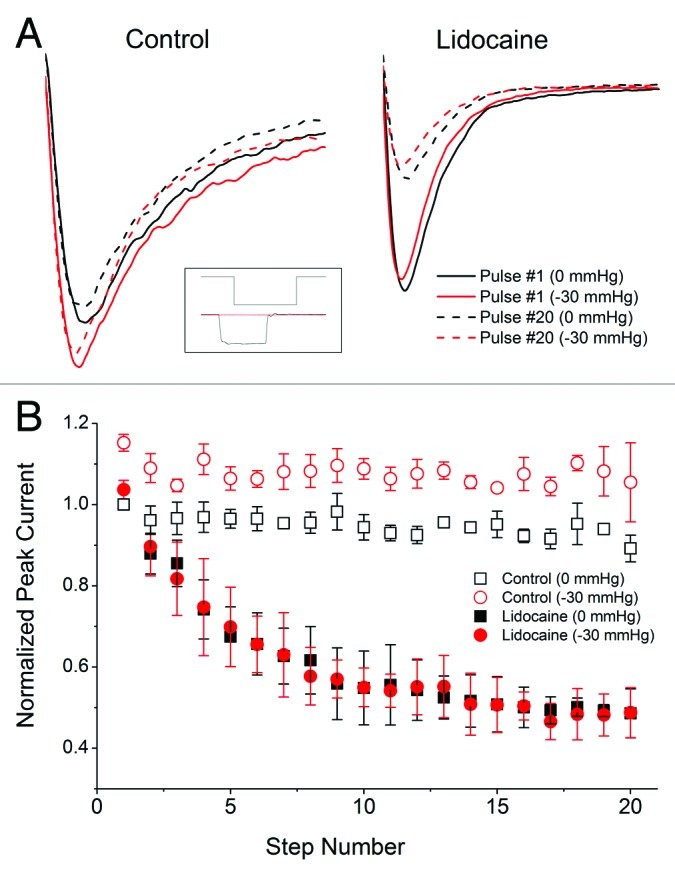
Figure 4. Lidocaine eliminates pressure-induced increase in NaV1.5 step peak current. Inset shows a voltage step (upper trace) of the 10 Hz use-dependence protocol with (lower black trace) and without (lower red trace) -30 mmHg pressure. (A) On the left, in a typical control inside-out patch at resting tension (0 mmHg) there was no significant difference between 1st (solid black) and 20th (dash black) steps. In the same patch, -30 mmHg pressure produced a tonic increase in peak current both 1st (solid red) and 20th (dash red) steps but no significant difference between steps. On the right, in a typical inside-out patch at rest lidocaine (50 uM) resulted in a significant use-dependent block as shown by a large difference between 1st (solid black) and 20th (dash black) steps. In the same patch, -30 mmHg pressure did not affect the relative morphology of the 1st (solid red) and 20th (dash red) steps compared with at rest controls. (B) Peak currents normalized to 1st step at 0 mmHg for all 20 steps in the 10 Hz use dependence protocol. Peak currents for all 20 steps are increased with pressure in the controls by 14% (n = 3, p < 0.05). In the presence of lidocaine there is a substantial use dependent block (51%, n = 3, p < 0.05) but no tonic increase in peak current with pressure at each step (0.3%, n = 3, p > 0.05).
Benzocaine inhibits pressure-induced shift in NaV1.5 half point of voltage-dependence of activation (V1/2a)
If the mechanism of NaV1.5 mechanosensitivity block by lidocaine is separate from the mechanism of use-dependent block, then other local anesthetics that are known to have no use dependent block may be effective blockers of NaV1.5 mechanosensitivity. We tested this hypothesis with benzocaine which is a neutral local anesthetic known to lack the ability of use-dependent block.27 Benzocaine also inhibited mechanosensitivity. In inside-out patches, 50 µM benzocaine added to the bath solution resulted in a pressure-induced shift in the voltage-sensitivity of activation ΔV1/2a of only -4.8 ± 3.3 mV (n = 4, p > 0.05 for 0 mmHg compared with -30 mmHg but p < 0.05 for ΔV1/2a for 0 mmHg vs. -30 mmHg for control vs. benzocaine). These data show that local anesthetics other than lidocaine modulate NaV1.5 mechanosensitivity and suggest that use-dependent block mechanism is distinct from blockade of mechanosensitivity.
Lidocaine inhibition of NaV1.5 mechanosensitivity does not require the F1760 binding site
We next examined whether the inhibition of NaV1.5 mechanosensitivity by lidocaine requires the established local anesthetic binding site. While multiple residues are known to affect local anesthetic binding within the pore of NaV1.5, F1760 is of most importance,28,29 and previously shown to be vital for use-dependent block.30 We introduced a point mutation in this residue (F1760A) and determined whether the inhibition of NaV1.5 mechanosensitivity was affected. In excised inside-out patches F1760A NaV1.5 showed voltage dependent activation properties similar to the wild-type NaV1.5 channels (V1/2a = -54.0 ± 2.9 mV, dVa = -8.5 ± 1.7 mV, Gmax = 1.5 ± 1.7 nS (range 0.65 to 4.0 nS), n = 4). At -30 mmHg, V1/2a was -64.6 ± 3.3, dVa was -9.8 ± 1.9 mV, Gmax was 1.5 ± 1.5 nS (range 0.42 – 3.78 nS) (n = 4) (Fig. 5A). Thus, negative pressure (-30 mmHg) produced a significant shift in the half-point of the voltage-dependence of activation (ΔV1/2a = -10.6 ± 3.6 mV, p < 0.05 by paired t-test) (Fig. 5, gray area in all panels) with no changes to other parameters (n = 4). The hyperpolarizing shift in V1/2a was not different from the wild-type channels (ΔV1/2a = -10.1 ± 1.5 mV) (p > 0.05 by two-sample t-test).
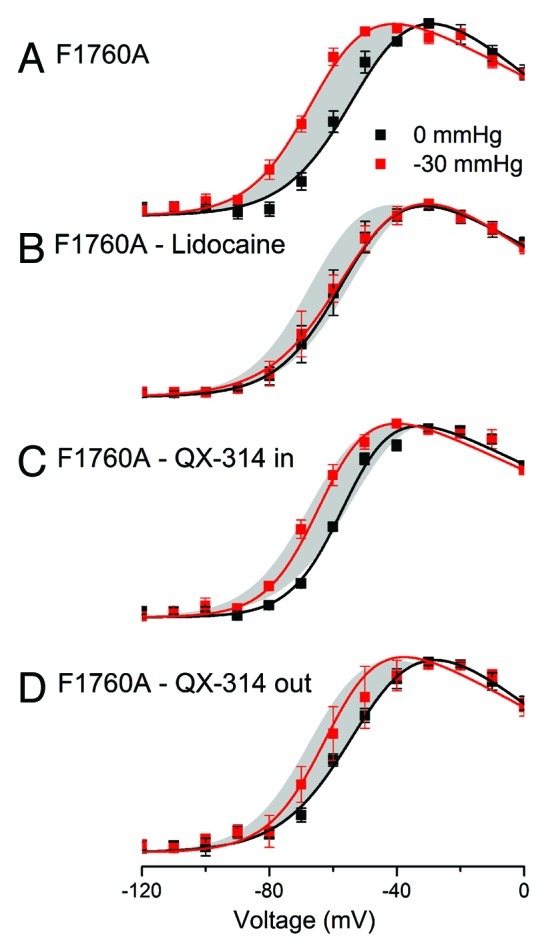
Figure 5. Lidocaine effect on NaV1.5 mechanosensitivity does not involve F1760 but requires a charge neutral form. Voltage-dependence of activation was tested at rest (0 mmHg, black) and with pressure (-30 mmHg, red curves) in the absence and presence of lidocaine and QX-314. (A) Voltage-dependent activation of F1760A at rest (0 mmHg, black) and in the presence of pressure (-30 mmHg, red), showing a -11 mV shift in half-point of activation with stretch (ΔV1/2a) (n = 4, p < 0.05). (B) With the addition of lidocaine (50 µM) inhibits the shift of the voltage-dependence of activation curve (ΔV1/2a = 1.4 mV) (n = , p > 0.05) (gray area is the shift by F1760A control). (C) With QX-314 (50 µM) applied to the inside the shift in the half-point of voltage-sensitivity with pressure is -9 mV (n = 4, p < 0.05). (D) With QX-314 (50 µM) applied to the outside the shift in the half-point of voltage-sensitivity with pressure is -8 mV (n = 4, p < 0.05).
We next tested whether lidocaine blocked mechanosensitivity in F1760A NaV1.5. In inside-out patches the addition of 50 µM lidocaine to the bath solution of inside-out patches with F17060A NaV1.5 resulted in no changes to peak current or voltage sensitivity at 0 mmHg (Gmax = 1.5 ± 1.5 nS, V1/2a = -53.4 ± 4.2 mV, both p > 0.05 compared with wt NaV1.5) (Fig. 5B). In the presence of lidocaine, -30 mmHg stretch resulted in a shift of V1/2a to -54.8 ± 2.4 mV or a ΔV1/2a of -1.4 ± 2.0 mV for change from 0 mmHg to -30 mmHg (n = 4, p > 0.05 for 0 mmHg vs. -30 mmHg and p < 0.05 for control vs. 50 µM lidocaine) (Fig. 5B, gray area is ΔV1/2a for controls). The other parameters (dVa, Gmax) did not change significantly for 0 mmHg vs. -30 mmHg, consistent with controls.
These results show that in inside-out patches F1760A NaV1.5 are mechanosensitive to a similar extent as the wildtype NaV1.5 channels and that lidocaine inhibits mechanosensitivity of F1760A NaV1.5. This suggests that the mechanism of NaV1.5 mechanosensitivity block by lidocaine does not involve the principal component of the local anesthetic binding site.
QX-314 does not alter pressure sensitivity of F1760A NaV1.5
QX-314 is a permanently charged quaternary ammonium analog of lidocaine. It does not partition into the hydrophobic core of the lipid membrane. QX-314 efficiently and use-dependently blocks NaV1.5 from the intracellular side with slow off-rate and from the extracellular side as a slow block that requires permeation through the pore.31 We tested QX-314 on either side of the membrane to assess aqueous accessibility of binding sites for mechanosensitivity block and the need for membrane partitioning. In order to eliminate the use-dependent and slow off-rate block of NaV1.5 by QX-314 we used F1760A NaV1.5.
First, 50 µM QX-314 was added to the bath to assess the intracellular side of the inside-out patches. At rest and with 50 µM QX-314 in the bath, average V1/2a was -55.5 ± 1.5 mV, dVa was 8.7 ± 1.4 mV, Gmax was 2.0 ± 3.1 nS (range 0.4 to 6.6 nS) (n = 4) (Fig. 5C). With the application of -30 mmHg pressure to these patches, average V1/2a was -64.3 ± 3.8 mV, dVa was 9.0 ± 1.0 mV, Gmax was 2.2 ± 3.5 nS (range 0.4 – 7.5 nS) (n = 4). The result was a significant difference between 0 and -30 mmHg pressure for ΔV1/2a of -8.8 ± 2.6 mV (n = 4, p < 0.05 by paired t-test) with no differences in dVa and Gmax. Further, pressure sensitivity of ΔV1/2a was not different between F1760A NaV1.5 in the presence and absence of intracellular QX-314 (n = 4, p > 0.05 by two-sample t-test) (Fig. 5C, gray area is ΔV1/2a for controls).
Second, 50µM QX-314 was added to the patch pipette to assess the extracellular side of the inside-out patches. At rest and with 50µM QX-314 in the pipette, average V1/2a was -52.8 ± 5.8 mV, dVa was -8.1 ± 1.3 mV, Gmax was 1.3 ± 1.6 nS (range 0.4 to 3.7 nS) (Fig. 5D). With the application of -30 mmHg pressure to these patches, average V1/2a was -61.1 ± 6.1 mV, dV was 9.1 ± 1.6 mV, Gmax was 2.2 ± 3.5 nS (range 0.4 – 7.5 nS) (n = 4). The resulting significant difference between 0 and -30 mmHg pressure was ΔV1/2a of -8.4 ± 1.5 mV (n = 4, p < 0.05 by paired t-test), but no significant differences were found for dVa and Gmax (n = 4, p > 0.05, Figure 5D, gray area is ΔV1/2a for controls).
These data show that QX-314 applied separately from both intracellular and extracellular sites of the channel fails to block NaV1.5 mechanosensitivity, suggesting that mechanosensitivity block does not involve a site with solely aqueous access.
Discussion
NaV1.5 is robustly mechanosensitive in excised patches
Inside-out patches have an advantage over cell attached patches in that they allow precise control of the transmembrane voltage, partial elimination of cytoskeleton and other cytoplasmic factors and drug application to the cytoplasmic side. All of the previously published studies on NaV1.5 mechanosensitivity in patches used cell-attached configuration.9-11,20 Previous work in pressurized patches showed a fast capacitance rise (milliseconds), followed by a prolonged rise to steady-state (tens to hundreds of milliseconds).22 We assessed the fast and reversible response using a short 25 msec stretch stimulus and paired voltage ladder protocol, so patches were only stimulated by pressure for ~20% of the protocol duration (Fig. 1A and B). Given strong voltage-dependence of NaV1.5 we were able to test NaV1.5 voltage-dependent pressure sensitivity within 10 msec of pressure pulse initiation. In addition, these short pulses allowed testing without addition of lanthanides which block endogenous mechanosensitive channels,32 but are also known to affect NaV1.5 function.33 Reversible mechanosensitive response at the millisecond scale in excised patches further reinforces stand-alone mechanosensitivity of the NaV1.5 α-subunit.
Compared with the typical voltage-dependent behavior in whole-cells, both inside-out and cell-attached patches showed larger variability in the half-points of voltage-dependence (V1/2) and shallower slopes (dV). This may be due to non-zero resting tension34 and channel location in the rim of the seal.35 Nevertheless, our current protocol allowed adequate averaging in space and time and patches provide a robust platform for studying mechanosensitivity.
Membrane-permeable local anesthetics modulate NaV1.5 mechanosensitivity by a mechanism different than use-dependent block
NaV1.5 channels are mechanosensitive in excised inside-out patches, and the response type and amplitude of pressure sensitivity are similar to previously published data in cell-attached patches.10,11,20 Specifically, in response to -30 mmHg pressure applied to an inside-out patch we saw a hyperpolarizing shift in V1/2a and V1/2i of about -10 mV without changes in dVa, dVi or Gmax (Fig. 2). Lidocaine (50 µM) altered NaV1.5 mechanosensitivity as demonstrated by a decrease in shift of V1/2a with -30 mmHg pressure by 67% (ΔV1/2a from -12 mV for control to -4 mV for lidocaine), similar to previous finding in cell-attached patches.20 On the other hand, lidocaine did not alter the pressure-induced shift in V1/2i (Fig. 3). Since lidocaine stabilizes inactivation at rest (Fig. S3), resulting in a hyperpolarizing shift of V1/2i,24 the lack of pressure-induced effect on V1/2i in the presence of lidocaine likely relates to the drug’s effects at rest.
Block by lidocaine of NaV1.5 currents in the absence of pressure is known to be use-dependent and we next tested whether there were any changes to the use-dependence of lidocaine block of NaV1.5. Using a 10 Hz use-dependence protocol with and without -30 mmHg we show in control NaV1.5 (0 µM lidocaine) an increase in peak current at each step without use-dependent loss of current (Fig. 4A and C). Lidocaine blocked the pressure-induced increase in the peak current, but otherwise use-dependence did not change with application of pressure. We provide two pieces of evidence that suggest that mechanisms of inhibition of NaV1.5 mechanosensitivity and use-dependent block by lidocaine are separate mechanisms. First, we used another local anesthetic, benzocaine, which unlike lidocaine is permanently neutral and does not show use-dependent block of NaV1.5 likely due to escape via the hydrophobic pathway.27,36 Yet, benzocaine efficiently blocked NaV1.5 mechanosensitivity as shown by a decrease in shift of V1/2a by 59%. The magnitude of the NaV1.5 mechanosensitivity block by benzocaine was similar to lidocaine. Second, we mutated a critical residue for use-dependent block (F1760A).30 F1760A NaV1.5 were mechanosensitive to the same extent as the wild-type NaV1.5 (Fig. 5A). Lidocaine continued to block mechanosensitivity F1760A NaV1.5 at the level similar to the control wild-type channels (Fig. 5B). These data suggest that the mechanisms of lidocaine block of NaV1.5 mechanosensitivity and use-dependence are separate.
NaV1.5 mechanosensitivity block by local anesthetics may require membrane partitioning
Both lidocaine and benzocaine are membrane permeable and there are sufficient data that suggest that the mechanism of voltage-gated ion channel mechanosensitivity may be mediated by bilayer-protein interactions.17,18,37,38 We examined whether membrane partitioning by local anesthetics is critical for block of NaV1.5 mechanosensitivity. For this we used a membrane-impermeable permanently charged lidocaine homolog QX-314. QX-314 is structurally very similar to lidocaine, with the only difference being lidocaine’s secondary amine group being replaced by a charged quaternary ammonium. Thus, QX-314 and lidocaine access the same aqueous sites, but only lidocaine is able to access hydrophobic pathway.31 Unlike lidocaine and benzocaine, the application of QX-314 to either side of the membrane did not inhibit mechanosensitivity of F1760A NaV1.5 (Fig. 5C and D). First, these data suggest that not all local anesthetics modify mechanosensitivity, as lidocaine and benzocaine were effective inhibitors and QX-314 was not. Second, differential effects of NaV1.5 mechanosensitivity by QX-314 and lidocaine/benzocaine suggest that the mechanism of NaV1.5 mechanosensitivity involves the lipid bilayer and potentially the lipid-protein interface.
The potential mechanisms of NaV1.5 mechanosensitivity modulation by membrane permeable local anesthetics likely involve protein-bilayer interface
Membrane partitioning of local anesthetics may play a role in their inhibition of NaV1.5 mechanosensitivity. Several possibilities exist with respect to the mechanism, broadly divided into non-specific and specific effects. First, local anesthetics may non-specifically alter bulk mechanical or electrical membrane properties. Second, bilayer stretch may alter interactions between the membrane permeable local anesthetics and the bilayer. Third, local anesthetics may non-specifically alter lipid-protein interactions in vital lipid exposed channel domains (e.g., voltage-sensors, S4–5 linkers, pore). Fourth, specific binding of local anesthetics membrane accessible protein site(s) may inhibit NaV1.5 mechanosensitivity.
Bulk membrane mechanical (e.g., elasticity17 and thickness39) and electrical (e.g., electrostatic potential40 and capacitance41) properties are known to affect voltage-gated channel function. Previous work showed that increasing lipid thickness hyperpolarized NaV voltage-dependence of activation and inactivation,39 and disruption of the bilayer order by cholesterol depletion or amphiphiles hyperpolarized NaV1.4 voltage-dependence of inactivation.17 Further, local anesthetics partitioning into bilayers alter membrane structure42 and thus the mechanical43 and electrical44 functional properties. Interestingly, both charged and uncharged forms of local anesthetics interact with lipid bilayers, albeit the neutral forms show deeper penetration and larger effects.43 Yet, it is unlikely that the effects we have observed are due to alteration of bulk membrane mechanical properties at rest. First, at the 50 µM doses of local anesthetics used in this study the drug density is much less than 1% of the lipid, and unlikely to have a significant effect on bulk membrane properties.45 Second, the addition of lidocaine and benzocaine to NaV1.5 and QX-314 to F1760A NaV1.5 did not change the voltage-dependence properties (V1/2a and dVa) at rest (p > 0.05 for all compared with no drug NaV1.5 control).
Another possibility is that membrane stretch alters the interaction properties of the bilayer with the local anesthetics. It is known that the partition coefficient of the local anesthetics depends on the state of the bilayer, preferring to partition into the liquid-crystal over solid-gel membranes.45 This suggests that lidocaine partitioning likely increases with stretch, such that the drug’s effects on NaV1.5 may appear stretch dependent. In the use-dependence protocol lidocaine inhibited pressure-dependent increase in peak current, but otherwise did not show additional use-dependent block as may be expected if lidocaine availability increased with stretch. Nevertheless, this possibility exists and requires further exploration.
It is clear that the lipid bilayer serves as an important pathway for the function of local anesthetics,24 and the neutral forms of these drugs pack readily within the lipids. Voltage-gated ion channel function depends on lipids46 and the recent crystal structure of NaVAb shows lipids as important structural elements, intruding through side entries into the pore of the channel.47 The findings of this study do not allow for specific localization of the target of the membrane permeable anesthetics with respect to the mechanism of NaV1.5 mechanosensitivity modulation. However, potential targets are hydrophobic protein domains that may either rely on lipids for appropriate function46 or identified binding sites for local anesthetics.48
In conclusion, in this study we demonstrate fast and reversible NaV1.5 mechanosensitivity in excised inside-out patches using a protocol with short pressure steps. Patch pressure hyperpolarizes NaV1.5 the half-points of voltage-dependence of activation and inactivation by about -10 mV. Lidocaine inhibits the pressure-induced shift in the voltage-dependence of activation but not inactivation. The uncoupling by lidocaine of the pressure-induced left-shifts in the half-points of voltage dependence of activation and inactivation may have therapeutic potential by decreasing pressure-induced excitability.49 The mechanism of modulation of NaV1.5 mechanosensitivity by local anesthetics appears to require membrane partitioning and likely protein-lipid interaction.
Methods
Cell culture
Human embryonic kidney 293 (HEK) cells were cultured and transfected as previously described.10 Cells were grown in T25 flasks in Minimum Essential Medium, 10% horse serum, non-essential amino acids, sodium pyruvate and penicillin-streptomycin (Invitrogen, Carlsbad, CA). Cells were grown to about 75% confluency. One day prior to the experiments, flasks were transfected with SCN5A encoding hH1c3 or hH1c1 F1760A (kind gift of Dr. Jonathan Makielski), with green fluorescent protein (GFP) pEGFP-C1 (Clontech, Palo Alto, CA), using Lipofectamine 2000 reagent and OPTI-MEM I reduced serum medium (Invitrogen, Carlsbad, CA). On the morning of experiments, cells were trypsinized, plated onto poly-l-lysine glass coverslips and used after about 5 h.
Recording solutions and pharmacology
For cell-attached patches, bath solution (extracellular) contained (in mM): 149 KCl, 2.45 CaCl2, 5.0 HEPES, 5.5 mM glucose. For excised inside-out patches, bath solution (intracellular) contained (in mM): 120 CsCl, 40 CsF, 1 NaCl, 2 MgCl2, 0.68 CaCl2, 10 HEPES, 2 EGTA. Free Ca2+ was calculated to be 100 nM (http://www.stanford.edu/~cpatton/CaEGTA-TS.htm). The patch pipette solution (extracellular) contained (in mM): 160 NaCl, 1CsCl, 1 MgCl2, 2 CaCl2 and 10 HEPES. For all solutions osmolality was 300 ± 10 mmol/kg and pH 7.2. Local anesthetic solutions (lidocaine, benzocaine and QX-314) were prepared daily from 100 mM ethanol stocks.
Electrophysiology
Electrodes were pulled using Sutter Instruments P-97 puller and coated with heat cured Dow Corning R6101 compound from Garner 8250 glass. Electrodes were fire polished specifically for patch mechano-activation as previously published.10 Axon 200A amplifier, Digidata 1322A, and Clampex 9 software were used for voltage-clamp and data acquisition. For voltage-ladder protocols, a 1.7 Hz stimulation frequency was used with no inter-pulse delay. Patches were held at -100 mV, and preceding each test step were pre-pulses of 10 msec to -204 mV and 4 msec to -140 mV which accelerated recovery from inactivation. Following the pre-pulse steps were 16 test voltage steps in 10 mV increments from -140 mV to 10 mV for 12 msec per step to test activation and finally a step to -10 mV for 4 msec to test availability. At each tested voltage a pair of pulses were obtained for control (0 mmHg) and pressure (-30 mmHg), which were separated by 230 msec. Currents were corrected with a P/6 protocol (hyperpolarizing steps from HP -120 mV) and a 5x average was obtained for each voltage step.
Mechanical activation of voltage-clamped currents
A specialized rapid pressure clamp was used to apply pressure (courtesy of Dr. Fred Sachs laboratory).50 Negative pressure was monitored during patch formation and pressure delivery for all protocols.32 Due to the mechanical remodeling of patches with prolonged stretch pulses32 we used a rapid pressure clamp (rise time to -30 mmHg in 5 msec). Stretch steps were limited to 25 msec obtained during activation/inactivation steps only. Seal history is important for such experiments,32 therefore we recorded the pressures required for patch formation. Patches were typically formed at < 5 mmHg. Patches with seals that were formed at > 10 mmHg were discarded.
Data analysis
Voltage dependence of activation for each experiment was fit individually and averages provided in results. The figures show averages and fits of raw data. For voltage-dependence of activation we used a two-state Boltzmann model {I = [(V-Vrev)Gmax/(1+e(V-V1/2a)/dVa)]}, where peak current (I) is a function of voltage (V), V1/2a was half-point, dVa was the slope, Vrev was reversal potential, and Gmax was maximal conductance. We fit voltage dependence of inactivation using a two-state Boltzmann model [I = (A1-A2)/(1+e(V-V1/2i)/dVi)+A2], where A1 and A2 were constants, V1/2i was half-point, dVi was the slope. Voltage dependence and dose-response curves were fit in pClamp9 and Origin 8.61.
Supplementary Material
Acknowledgments
The authors would like to thank Drs. David Linden and Arpad Mike for their critical review of the manuscript. This work was supported by NIDDK 52766 and P30DK84567.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental materials may be found here: www.landesbioscience.com/journals/channels/article/21202
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/21202
References
- 1.Gellens ME, George AL, Jr., Chen LQ, Chahine M, Horn R, Barchi RL, et al. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci U S A. 1992;89:554–8. doi: 10.1073/pnas.89.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ou Y, Gibbons SJ, Miller SM, Strege PR, Rich A, Distad MA, et al. SCN5A is expressed in human jejunal circular smooth muscle cells. Neurogastroenterol Motil. 2002;14:477–86. doi: 10.1046/j.1365-2982.2002.00348.x. [DOI] [PubMed] [Google Scholar]
- 3.Bennett PB, Yazawa K, Makita N, George AL., Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–5. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- 4.Farrugia G. Ionic conductances in gastrointestinal smooth muscles and interstitial cells of Cajal. Annu Rev Physiol. 1999;61:45–84. doi: 10.1146/annurev.physiol.61.1.45. [DOI] [PubMed] [Google Scholar]
- 5.Strege PR, Mazzone A, Kraichely RE, Sha L, Holm AN, Ou Y, et al. Species dependent expression of intestinal smooth muscle mechanosensitive sodium channels. Neurogastroenterol Motil. 2007;19:135–43. doi: 10.1111/j.1365-2982.2006.00844.x. [DOI] [PubMed] [Google Scholar]
- 6.Strege PR, Ou Y, Sha L, Rich A, Gibbons SJ, Szurszewski JH, et al. Sodium current in human intestinal interstitial cells of Cajal. Am J Physiol Gastrointest Liver Physiol. 2003;285:G1111–21. doi: 10.1152/ajpgi.00152.2003. [DOI] [PubMed] [Google Scholar]
- 7.Locke GR, 3rd, Ackerman MJ, Zinsmeister AR, Thapa P, Farrugia G. Gastrointestinal symptoms in families of patients with an SCN5A-encoded cardiac channelopathy: evidence of an intestinal channelopathy. Am J Gastroenterol. 2006;101:1299–304. doi: 10.1111/j.1572-0241.2006.00507.x. [DOI] [PubMed] [Google Scholar]
- 8.Saito YA, Strege PR, Tester DJ, Locke GR, 3rd, Talley NJ, Bernard CE, et al. Sodium channel mutation in irritable bowel syndrome: evidence for an ion channelopathy. Am J Physiol Gastrointest Liver Physiol. 2009;296:G211–8. doi: 10.1152/ajpgi.90571.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morris CE, Juranka PF. Nav channel mechanosensitivity: activation and inactivation accelerate reversibly with stretch. Biophys J. 2007;93:822–33. doi: 10.1529/biophysj.106.101246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beyder A, Rae JL, Bernard C, Strege PR, Sachs F, Farrugia G. Mechanosensitivity of Nav1.5, a voltage-sensitive sodium channel. J Physiol. 2010;588:4969–85. doi: 10.1113/jphysiol.2010.199034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banderali U, Juranka PF, Clark RB, Giles WR, Morris CE. Impaired stretch modulation in potentially lethal cardiac sodium channel mutants. Channels (Austin) 2010;4:12–21. doi: 10.4161/chan.4.1.10260. [DOI] [PubMed] [Google Scholar]
- 12.Ou Y, Strege P, Miller SM, Makielski J, Ackerman M, Gibbons SJ, et al. Syntrophin gamma 2 regulates SCN5A gating by a PDZ domain-mediated interaction. J Biol Chem. 2003;278:1915–23. doi: 10.1074/jbc.M209938200. [DOI] [PubMed] [Google Scholar]
- 13.Strege PR, Holm AN, Rich A, Miller SM, Ou Y, Sarr MG, et al. Cytoskeletal modulation of sodium current in human jejunal circular smooth muscle cells. Am J Physiol Cell Physiol. 2003;284:C60–6. doi: 10.1152/ajpcell.00532.2001. [DOI] [PubMed] [Google Scholar]
- 14.Shcherbatko A, Ono F, Mandel G, Brehm P. Voltage-dependent sodium channel function is regulated through membrane mechanics. Biophys J. 1999;77:1945–59. doi: 10.1016/S0006-3495(99)77036-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang JA, Lin W, Morris T, Banderali U, Juranka PF, Morris CE. Membrane trauma and Na+ leak from Nav1.6 channels. Am J Physiol Cell Physiol. 2009;297:C823–34. doi: 10.1152/ajpcell.00505.2008. [DOI] [PubMed] [Google Scholar]
- 16.Tabarean IV, Juranka P, Morris CE. Membrane stretch affects gating modes of a skeletal muscle sodium channel. Biophys J. 1999;77:758–74. doi: 10.1016/S0006-3495(99)76930-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lundbaek JA, Birn P, Hansen AJ, Søgaard R, Nielsen C, Girshman J, et al. Regulation of sodium channel function by bilayer elasticity: the importance of hydrophobic coupling. Effects of Micelle-forming amphiphiles and cholesterol. J Gen Physiol. 2004;123:599–621. doi: 10.1085/jgp.200308996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lundbaek JA, Koeppe RE, 2nd, Andersen OS. Amphiphile regulation of ion channel function by changes in the bilayer spring constant. Proc Natl Acad Sci U S A. 2010;107:15427–30. doi: 10.1073/pnas.1007455107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fredj S, Sampson KJ, Liu H, Kass RS. Molecular basis of ranolazine block of LQT-3 mutant sodium channels: evidence for site of action. Br J Pharmacol. 2006;148:16–24. doi: 10.1038/sj.bjp.0706709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beyder A, Strege PR, Reyes S, Bernard CE, Terzic A, Makielski J, et al. Ranolazine decreases mechanosensitivity of the voltage-gated sodium ion channel NaV1.5: a novel mechanism of drug action. Circulation. 2012;125:2698–706. doi: 10.1161/CIRCULATIONAHA.112.094714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arnadóttir J, Chalfie M. Eukaryotic mechanosensitive channels. Annu Rev Biophys. 2010;39:111–37. doi: 10.1146/annurev.biophys.37.032807.125836. [DOI] [PubMed] [Google Scholar]
- 22.Suchyna TM, Besch SR, Sachs F. Dynamic regulation of mechanosensitive channels: capacitance used to monitor patch tension in real time. Phys Biol. 2004;1:1–18. doi: 10.1088/1478-3967/1/1/001. [DOI] [PubMed] [Google Scholar]
- 23.Gu CX, Juranka PF, Morris CE. Stretch-activation and stretch-inactivation of Shaker-IR, a voltage-gated K+ channel. Biophys J. 2001;80:2678–93. doi: 10.1016/S0006-3495(01)76237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nau C, Wang GK. Interactions of local anesthetics with voltage-gated Na+ channels. J Membr Biol. 2004;201:1–8. doi: 10.1007/s00232-004-0702-y. [DOI] [PubMed] [Google Scholar]
- 26.Sheets MF, Fozzard HA, Lipkind GM, Hanck DA. Sodium channel molecular conformations and antiarrhythmic drug affinity. Trends Cardiovasc Med. 2010;20:16–21. doi: 10.1016/j.tcm.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanck DA, Nikitina E, McNulty MM, Fozzard HA, Lipkind GM, Sheets MF. Using lidocaine and benzocaine to link sodium channel molecular conformations to state-dependent antiarrhythmic drug affinity. Circ Res. 2009;105:492–9. doi: 10.1161/CIRCRESAHA.109.198572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pless SA, Galpin JD, Frankel A, Ahern CA. Molecular basis for class Ib anti-arrhythmic inhibition of cardiac sodium channels. Nat Commun. 2011;2:351. doi: 10.1038/ncomms1351. [DOI] [PubMed] [Google Scholar]
- 29.Mike A, Lukacs P. The enigmatic drug binding site for sodium channel inhibitors. Curr Mol Pharmacol. 2010;3:129–44. doi: 10.2174/1874467211003030129. [DOI] [PubMed] [Google Scholar]
- 30.Carboni M, Zhang ZS, Neplioueva V, Starmer CF, Grant AO. Slow sodium channel inactivation and use-dependent block modulated by the same domain IV S6 residue. J Membr Biol. 2005;207:107–17. doi: 10.1007/s00232-005-0805-0. [DOI] [PubMed] [Google Scholar]
- 31.Qu Y, Rogers J, Tanada T, Scheuer T, Catterall WA. Molecular determinants of drug access to the receptor site for antiarrhythmic drugs in the cardiac Na+ channel. Proc Natl Acad Sci U S A. 1995;92:11839–43. doi: 10.1073/pnas.92.25.11839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris CE, Juranka PF, Lin W, Morris TJ, Laitko U. Studying the mechanosensitivity of voltage-gated channels using oocyte patches. Methods Mol Biol. 2006;322:315–29. doi: 10.1007/978-1-59745-000-3_22. [DOI] [PubMed] [Google Scholar]
- 33.Li GR, Baumgarten CM. Modulation of cardiac Na(+) current by gadolinium, a blocker of stretch-induced arrhythmias. Am J Physiol Heart Circ Physiol. 2001;280:H272–9. doi: 10.1152/ajpheart.2001.280.1.H272. [DOI] [PubMed] [Google Scholar]
- 34.Ursell T, Agrawal A, Phillips R. Lipid bilayer mechanics in a pipette with glass-bilayer adhesion. Biophys J. 2011;101:1913–20. doi: 10.1016/j.bpj.2011.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bae C, Markin V, Suchyna T, Sachs F. Modeling ion channels in the gigaseal. Biophys J. 2011;101:2645–51. doi: 10.1016/j.bpj.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quan C, Mok WM, Wang GK. Use-dependent inhibition of Na+ currents by benzocaine homologs. Biophys J. 1996;70:194–201. doi: 10.1016/S0006-3495(96)79563-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmidt D, MacKinnon R. Voltage-dependent K+ channel gating and voltage sensor toxin sensitivity depend on the mechanical state of the lipid membrane. Proc Natl Acad Sci U S A. 2008;105:19276–81. doi: 10.1073/pnas.0810187105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kraichely RE, Strege PR, Sarr MG, Kendrick ML, Farrugia G. Lysophosphatidyl choline modulates mechanosensitive L-type Ca2+ current in circular smooth muscle cells from human jejunum. Am J Physiol Gastrointest Liver Physiol. 2009;296:G833–9. doi: 10.1152/ajpgi.90610.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hendry BM, Elliott JR, Haydon DA. Further evidence that membrane thickness influences voltage-gated sodium channels. Biophys J. 1985;47:841–5. doi: 10.1016/S0006-3495(85)83988-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jogini V, Roux B. Dynamics of the Kv1.2 voltage-gated K+ channel in a membrane environment. Biophys J. 2007;93:3070–82. doi: 10.1529/biophysj.107.112540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conti F, Fioravanti R, Segal JR, Stühmer W. Pressure dependence of the sodium currents of squid giant axon. J Membr Biol. 1982;69:23–34. doi: 10.1007/BF01871238. [DOI] [PubMed] [Google Scholar]
- 42.Pinto LM, Yokaichiya DK, Fraceto LF, de Paula E. Interaction of benzocaine with model membranes. Biophys Chem. 2000;87:213–23. doi: 10.1016/S0301-4622(00)00196-4. [DOI] [PubMed] [Google Scholar]
- 43.Högberg CJ, Maliniak A, Lyubartsev AP. Dynamical and structural properties of charged and uncharged lidocaine in a lipid bilayer. Biophys Chem. 2007;125:416–24. doi: 10.1016/j.bpc.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 44.Högberg CJ, Lyubartsev AP. Effect of local anesthetic lidocaine on electrostatic properties of a lipid bilayer. Biophys J. 2008;94:525–31. doi: 10.1529/biophysj.107.104208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang J, Hadlock T, Gent A, Strichartz GR. Tetracaine-membrane interactions: effects of lipid composition and phase on drug partitioning, location, and ionization. Biophys J. 2007;92:3988–4001. doi: 10.1529/biophysj.106.102434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu Y, Ramu Y, Lu Z. Removal of phospho-head groups of membrane lipids immobilizes voltage sensors of K+ channels. Nature. 2008;451:826–9. doi: 10.1038/nature06618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature. 2011;475:353–8. doi: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fraceto LF, Oyama S, Jr., Nakaie CR, Spisni A, de Paula E, Pertinhez TA. Interaction of local anesthetics with a peptide encompassing the IV/S4-S5 linker of the Na+ channel. Biophys Chem. 2006;123:29–39. doi: 10.1016/j.bpc.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 49.Morris CE. Voltage-gated channel mechanosensitivity: fact or friction? Front Physiol. 2011;2:25. doi: 10.3389/fphys.2011.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Besch SR, Suchyna T, Sachs F. High-speed pressure clamp. Pflugers Arch. 2002;445:161–6. doi: 10.1007/s00424-002-0903-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.