

Abstract
Many cytokines, hormones and growth factors use the JAK (Janus kinase)/STAT (signal transducer and activator of transcription) pathway for cell signalling and specific gene activation. In the classical model, ligand is said to interact solely with the receptor extracellular domain, which triggers JAK activation of STATs at the receptor cytoplasmic domain. Activated STATs are then said to carry out nuclear events of specific gene activation. Given the limited number of STATs (seven) and the activation of the same STATs by cytokines with different functions, the mechanism of the specificity of their signalling is not obvious. Focusing on IFNγ (interferon γ), we have shown that ligand, receptor and activated JAKs are involved in nuclear events that are associated with specific gene activation, where the receptor subunit IFNGR1 (IFNγ receptor 1) functions as a transcription/co-transcription factor and the JAKs are involved in key epigenetic events. RTKs (receptor tyrosine kinases) such as EGFR [EGF (epidermal growth factor) receptor] and FGFR [FGF (fibroblast growth factor) receptor] also undergo nuclear translocation in association with their respective ligands. EGFR and FGFR, like IFNGR1, have been shown to function as transcription/co-transcription factors. The RTKs also regulate other kinases that have epigenetic effects. Our IFNγ model, as well as the RTKs EGFR and FGFR, have similarities to that of steroid receptor signalling. These systems consist of ligand–receptor–co-activator complexes at the genes that they activate. The co-activators consist of transcription factors and kinases, of which the latter play an important role in the associated epigenetics. It is our view that signalling by cytokines such as IFNγ is but a variation of specific gene activation by steroid hormones.
Keywords: cytokine, epigenetic change, interferon, Janus kinase (JAK), signal transducer and activator of transcription (STAT), steroid, steroid-like signalling
INTRODUCTION
The dominant role that has been placed upon STAT (signal transducer and activator of transcription) transcription factors in gene activation by cytokines may have obscured other important aspects of the complex events that must be associated with such activation. STATs have been shown to be essential for signalling by a host of proteins, including IFNs (interferons), most of the ILs (interleukins), growth factors such as PDGF (platelet-derived growth factor) and hormones such as growth hormone [1]. The prevailing view is that the ligand activates the cell solely via interactions with the extracellular domain of the receptor complex [1]. This in turn results in the activation of receptor-associated tyrosine kinases of the Janus or JAK (Janus kinase) family, leading to phosphorylation and dimerization of the STAT transcription factors, which dissociate from the receptor cytoplasmic domain and translocate to the nucleus. This view ascribes no further role to the ligand, JAKs or the receptor in the signalling process. It is not clear as to how specific gene activation, including the associated epigenetic events, fit into a model that focuses primarily or solely on the STATs. As indicated, the STATs form dimers when activated, but there are only seven different types of STATs, and the dimers are predominantly homomeric in nature. Given that there are functionally over 60 different types of ligands that use STATs, it is difficult to decipher the mechanism of their different specificities solely in the context of the particular STATs involved [2–4]. Furthermore, some ligands such as IFNγ and IL-10 use the same STATs, but have quite different effects on the same cells [2–4]. It is our view that resistance to ascribing any role of ligand, receptor and JAKs beyond STAT activation in cytokine signalling has seriously limited the value of the classical model of JAK/STAT signalling in providing a mechanism for specific gene activation, including the associated epigenetic events.
IFNγ signalling in the context of the classical model illustrates some of the problems with the model. The IFNγ receptor on cells consists of two chains, IFNGR1 (IFNγ receptor 1) and IFNGR2 (IFNγ receptor 2), that are non-covalently associated [5]. IFNγ in an asymmetric dimeric form binds predominantly to two IFNGR1 chains. The model contends that this cross-linking is responsible for the intracellular events that occur on the cytoplasmic domains of the receptor chains. The tyrosine kinase JAK1 is associated with IFNGR1, whereas JAK2 is associated with IFNGR2. IFNγ binding results in JAK2 moving from IFNGR2 to IFNGR1, where a sequence of events causes autophosphorylation of the JAKs and tyrosine phosphorylation of IFNGR1, followed by the recruitment of STAT1α and its subsequent tyrosine phosphorylation. Here, pSTAT1α (phosphorylated STAT1α) forms a dimer, dissociates from the receptor complex and goes to the nucleus, presumably via an intrinsic NLS (nuclear localization sequence). Structure studies have shown that dimeric STAT1α binds to the GAS element of the IFNG promoter [6], and this finding has been interpreted as validation of the model. Studies have shown, however, that contrary to the original assumptions, monomeric IFNγ can also stimulate the activation of STAT1α [7,8]. This raises the question of whether cross-linking of IFNGR1 is the determining event in subsequent signal transduction of IFNγ. Furthermore, there are several reports that STAT1α contains a novel intrinsic NLS [9–11], but there is disagreement concerning its properties, and nothing is presented as to how it functions in the complex low/high-affinity binding nature of the nuclear import apparatus (reviewed in [2]). The goal of the present review is to show that ligand signalling via JAK/STAT is but a variation of SR (steroid receptor) signalling, where the mechanism of specific gene activation and associated epigenetic events are better understood.
THE CLASSICAL MODEL OF JAK/STAT SIGNALLING WITH MODIFICATIONS
It has previously been acknowledged that the classical model of JAK/STAT signalling was oversimplified in its original form (Figure 1A) [12]. In the case of IFNγ, complexity beyond simple JAK/STAT activation in signal transduction is indicated in the demonstration that other pathways, including MAPK (mitogen-activated protein kinase), PI3K (phosphoinositide 3-kinase), CaMKII (Ca2+/calmodulin-dependent kinase II), NF-κB (nuclear factor κB) and others co-operate with or act in parallel with JAK/STAT signalling to regulate effects of IFNγ at the level of gene activation and cell phenotypes (Figure 1B) [12]. All of these pathways are generic in the sense that a plethora of cytokines with functions different from those of IFNγ also activate them. Also, the classical model lacks an orchestrating or co-ordinating centre. For IFNγ and other cytokines, uniqueness of function would seem to depend on cytokine control of complex and unique qualitative, quantitative and kinetic aspects of the activation of these pathways. We are not aware that any co-ordinator has been demonstrated for any cytokine in the context of the classical model of JAK/STAT signalling. The STATs are basically left to their own devices as though they know in isolation what to do to impart specificity.
Figure 1. Classical model of IFN signaling.

Left-hand panel: signalling through the transcription factors STATs. Binding of the cytokine to its cognate receptor begins a series of interactions that, through the participitation of the tyrosine kinases JAK1 and JAK2, result in the phosphorylation of STATs. STATs are then translocated to the nucleus to activate specific genes. Right-hand panel: alternative IFNγ signalling pathways. Multiple pathways through MAPK and IKK [IκB (inhibitor of NF-κB) kinase]/NF-κB signalling can activate the genes involved. CaMKII, Ca2 +/calmodulin-dependent protein kinase II; ERK, extracellular-signal-regulated kinase; MEKK, MEK (MAPK/ERK kinase) kinase; MKK, MAPK kinase; PKC, protein kinase C; Pyk2, proline-rich tyrosine kinase 2. See [12] for details.
A previous study introduced the technique of ChIP (chromatin immunoprecipitation) sequencing in assessing genome-wide profiles of STAT1 DNA association in HeLa cells treated with IFNγ [13]. The authors identified 41 582 and 11 004 putative STAT1-binding regions in IFNγ-stimulated and unstimulated cells respectively. Of 34 known loci that contained STAT1 IFNγ-responsive elements, ChIP sequencing identified 24. It is of note that approximately 85 % of the unstimulated STAT1 peaks overlapped those of IFNγ-stimulated cells. We find this to be a potentially interesting finding as it would suggest that IFNγ treatment of the cells caused a rearrangement of the STAT1 proteins. Related to this, the question arises as to the phosphorylated (activated) state of STAT1 in the genome of unstimulated compared with IFNγ-stimulated cells. ChIP was performed with antibodies against STAT1 protein, but selective ChIP with a phosphotyrosine-specific anti-STAT1 antibody could have provided some insight into the role of activated compared with non-activated STAT1 in the nucleus, particularly as it pertains to heterochromatin destabilization [14].
In another study using the same approach as above, the authors determined the genome-wide profile between H3K4 (Lys4 of histone H3) mono- and tri-methylation in HeLa cells treated with IFNγ [15]. Increased trimethylation of H3K4 from approximately 54 000 to approximately 76 100 was observed after IFNγ treatment. Increased trimethylation of H3K4 is associated with gene activation. But the proximal association of activation of STAT1 with such trimethylation was not clearly established. These bioinformatic-type studies, although of interest, provide little insight into the mechanism of specific gene activation by IFNγ via a strict focus on STAT1. In terms of epigenetics, one could come to similar conclusions concerning H3K4 trimethylation. There is no hint of a co-ordinator of the genomic events as they would pertain to specific gene activation. Approximately 70 000 STAT1-binding sites were observed in IFNγ-treated cells in the study by Robertson et al. [15], which is significantly greater than the 41 582 of the other study described above [13]. The authors did not address these differences. Thus variability of STAT1 binding from experiment to experiment is a possible concern.
Other studies have shown a functional interaction between different STATs in gene activation/suppression, which provides more insight into STAT mediation of cytokine signalling. The induction of IL-17 by activated STAT3, for example, was countered by IL-2 activation of STAT5 [16]. It was demonstrated by ChIP sequencing that STAT3 and STAT5 bound to multiple common sites across the IL-17 gene locus, including non-coding sequences. Nothing was presented as to the phosphorylation state of these STATs. Activation of STAT5 by IL-2 resulted in more binding of STAT5 and less binding of STAT3 at these sites, whereas activation of STAT3 by IL-6 induced the opposite; the combination of the two STATs resulted in dynamic regulation of the IL-17 gene locus by the opposing effects of IL-2 (STAT5) and IL-6 (STAT3) [16]. A similar complementarity was observed with STAT4 and STAT6 with respect to Th1 and Th2 cell development, but with much less competition for binding sites at coding and non-coding regions of the gene [17]. These Yin–Yang interactions of STAT transcription factors are referred to as specification with respect to lymphocyte phenotypes. Important questions, however, are not addressed with respect to claims of specification and signalling specificity. For example, IL-6, IL-23 and IL-27 all activate STAT3 and are all involved in Th17 induction/differentiation and function [18–20]. Additionally, it has been shown that the IL-23 receptor is required for terminal differentiation of IL-17-producing effector Th cells [21]. Thus STAT3 does not seem to be the only factor required for activation and generation of Th17 cells. Rather, the requirements of IL-6 and IL-23 for Th17 induction/differentiation and IL-27 for suppression all involve activated STAT3 mediation through multiple unique ligand–receptor interactions. Interestingly, and contrary to the study described above [16,17], it has been demonstrated that IL-2 participates in expansion of Th17 cells in uveitis and scleritis [22].
The retinoic acid orphan nuclear receptor RORγt (thymus-type retinoic acid orphan receptor γ) is critical for T-cell development, and mice with RORγt deficiency have reduced Th17 cell differentiation [23,24]. RORγt in conjunction with STAT3 has been proposed as a central player in Th17 cell differentiation [25]. If activation of STAT3 is the key function of IL-23, then why can’t IL-6, IL-21 or even IL-12 replace or compensate for the IL-23 receptor requirement? Thus the connection between IL-23 and the events of specific gene activation remain to be determined and are not addressed currently with this cytokine, not withstanding recent results on ChIP sequencing in terms of STAT3 in Th17 cell specification [16]. One could come to a similar dichotomy with respect to IL-12 and Th1 cells.
Because of the involvement of JAKs, there is particular focus on tyrosine phosphorylation of the STATs. In the case of STAT1α, activated JAK1 and JAK2 are involved in phosphorylation of Tyr701 for IFNγ as well as for numerous other cytokines [26,27]. Ser727 phosphorylation is a critical non-tyrosine phosphorylation in the activation of STAT1α, but, again, this is not unique to a particular cytokine [26,27]. There is also evidence of pSTAT1α induction of the expression of unphosphorylated STAT1α [28]. The unphosphorylated STAT1α was shown to induce expression of several genes that function, at least in part, by association with unphosphorylated NF-κB. The unphosphorylated STAT1α has been shown to be involved in antiviral and immune responses, as well as in facilitating tumour growth. There are also reports of lysine acetylation of STAT1α in the nucleus via histone acetyltransferase [27]. This results in recruitment of T-cell tyrosine phosphatase TCP45 dephosphorylation of STAT1α, and its exit from the nucleus in a latent state. None of these modifications of STAT1α is unique to a specific cytokine, and thus these alterations do not convey a cytokine-specific function to the STAT.
A MORE COMPLEX MODEL OF IFNγ SIGNALLING
IFNγ has been known for some time to translocate to the nucleus of receptor-expressing cells with kinetics as rapid as those for the activation and nuclear translocation of the inducible transcription factor STAT1α that it activates [29,30]. More recently, IFNγ nuclear translocation has been shown to be driven by an NLS (R126KRKRSR) at its C-terminus which is similar to the prototypical SV40 TAg (simian virus 40 large T antigen) NLS (PKKKRKV) [31]. Mutations of the IFNγ NLS destroy its biological activity [32], which can be restored by reconstitution with the SV40 TAg NLS [33]. The SV40 TAg NLS is known to localize in the nucleus in an IMP (importin) α/β1/Ran-dependent fashion (reviewed in [34]); excess SV40 TAg NLS peptide inhibits IFNγ NLS-dependent nuclear import, suggesting that the IFNγ NLS mediates nuclear import through the same pathway [31]. Results from immunoprecipitation experiments, which detected endocytosed IFNγ bound to IMPα5 (NPI-1) in cells actively transporting IFNγ to the nucleus, are consistent with this conclusion [35,36]. Subsequent experiments showed that the receptor α-subunit, IFNGR1, of the heterodimeric IFNγ receptor also translocates to the nucleus in IFNγ-treated cells, in contrast with the β-subunit (IFNGR2), which remains in the plasma membrane [32,35,37]. Uptake of IFNγ is a receptor-mediated endocytic process, with previous studies indicating that plasma membrane lipid microdomains are primary sites for the endocytic events leading to nuclear translocation of IFNγ–IFNGR1, as well as of STAT1α [38]. The trafficking of IFNγ, the role of its NLS and how this relates to signal transduction/function have been the subject of previous studies [31–33,35,37–39]. The IFNγ NLS is known to contribute minimally to extracellular high-affinity receptor–ligand binding, but the C-terminal domain (including the NLS) appears to be able to interact with the intracellular cytoplasmic domain of IFNGR1 (residues 253–287) of the IFNγ –receptor complex [35]. This binding, which requires the NLS, also increases the affinity of the Janus family kinase JAK2 for IFNGR1 [39].
The IFNγ NLS has also been found to be required for internalization of IFNγ into the cell, even though, as indicated above, it contributes minimally to high-affinity receptor binding. Intracellular expression of a full-length non-secreted form of IFNγ can also affect IFNGR1 nuclear translocation, and activation and nuclear translocation of STAT1α, as well as induction of biological activities normally elicited by addition of extracellular IFNγ [32]. Intracellular expression of an IFNγ mutant where the basic residues of the NLS were replaced with alanine residues failed to induce nuclear translocation of IFNGR1 or STAT1α and led to a loss of IFNγ activities [32]. This suggests that the IFNγ NLS functions intracellularly, mediating interaction with specific intracellular components critical for IFNγ activity [32]. Unlike the strict species specificity displayed by extracellular IFNγ, where mouse IFNγ will not act extracellularly on human cells and vice versa, there appears to be no species specificity in terms of the response to intracellularly expressed IFNγ [32,35]. In this regard, previous studies have shown that the cytoplasmic domains of human and mouse IFNGR1 are interchangeable with respect to extracellular IFNγ function [5]. This cross-species functionality of intracellular IFNγ further highlights the fact that the cytoplasmic domain of IFNGR1 is the key target of intracellular interaction in the cytosol.
An intracellular excess of a peptide representing the cytoplasmic binding site on IFNGR1 for the C-terminus of IFNγ, IFNGR1 (residues 253–287), prevented the complexation of internalized IFNγ with the cytoplasmic domain of cell-surface IFNGR1 in cells that were actively internalizing IFNγ [35]. Moreover, such cells were also blocked with respect to the tyrosine phosphorylation of STAT1α. Thus internalized IFNγ appears to be able to interact with the cytoplasmic domain of IFNGR1 in intact cells as part of the signal transduction events leading to STAT1α tyrosine phosphorylation.
The IFNGR1 cytoplasmic domain would be on the outer surface of the endocytic vesicle following endocytosis, which suggests that IFNγ can traverse the membrane of the endocytic vesicle during internalization to contact the cytoplasmic domain of IFNGR1. Cytosolic injection of antibodies against IFNγ (residues 95–132) blocks STAT1α nuclear translocation in response to extracellular IFNγ [35], consistent with these observations. This further supports the idea that the C-terminus of endocytosed IFNγ accesses the cytosol, although the mechanism is as yet undetermined. The requirement of the IFNγ NLS for internalization, binding to the cytoplasmic domain of IFNGR1, activation of JAK2 and STAT1α, and nuclear translocation of activated STAT1α and IFNGR1 suggest that some or all of these processes may be coupled, presumably through the NLS. Consistent with this, it has been observed that, after internalization, extracellular IFNγ could be recovered directly associated with IMPα5 in a cytosolic complex of IFNγ –IFNGR1–pSTAT1α [32]. The formation of the complex was dependent on the IFNγ NLS. Similar results have been obtained with intracellular expression of non-secreted full-length IFNγ, which, as outlined above, induces nuclear translocation of IFNGR1 and STAT1α [32]. Intracellular expression of a non-secreted NLS-mutated IFNγ fails to induce complexation of IFNγ, IFNGR1 or STAT1α with IMPα5, and nuclear transport of STAT1α and IFNGR1 [32].
Others have reported the direct association of phosphorylated STAT1α in complexes with IMPα5 [9–11]. All of these NLSs were non-classical and there was no agreement with respect to the fact that they were different for all three reports. These differences have not been addressed, nor has the low binding affinity when compared with the classical polycationic NLS [2].
The clear implication of what is described above is that the IFNγ NLS plays a direct role in STAT1α nuclear transport at least in the context of specific gene activation. Thus a complex of IFNγ –IFNGR1–STAT1α with IMPα5, mediated by the IFNγ NLS, provides the link between the nuclear translocation of IFNGR1 and STAT1α and that of IFNγ, implying that one important function of nuclear transport of IFNγ may be to chaperone the nuclear transport of activated STAT1α to specific genes. A model representing the events in direct involvement of IFNγ and IFNGR1 in signal transduction is presented in Figure 2. Epigenetic aspects of this model are discussed in the following section.
Figure 2. Proposed model of IFNγ signaling.
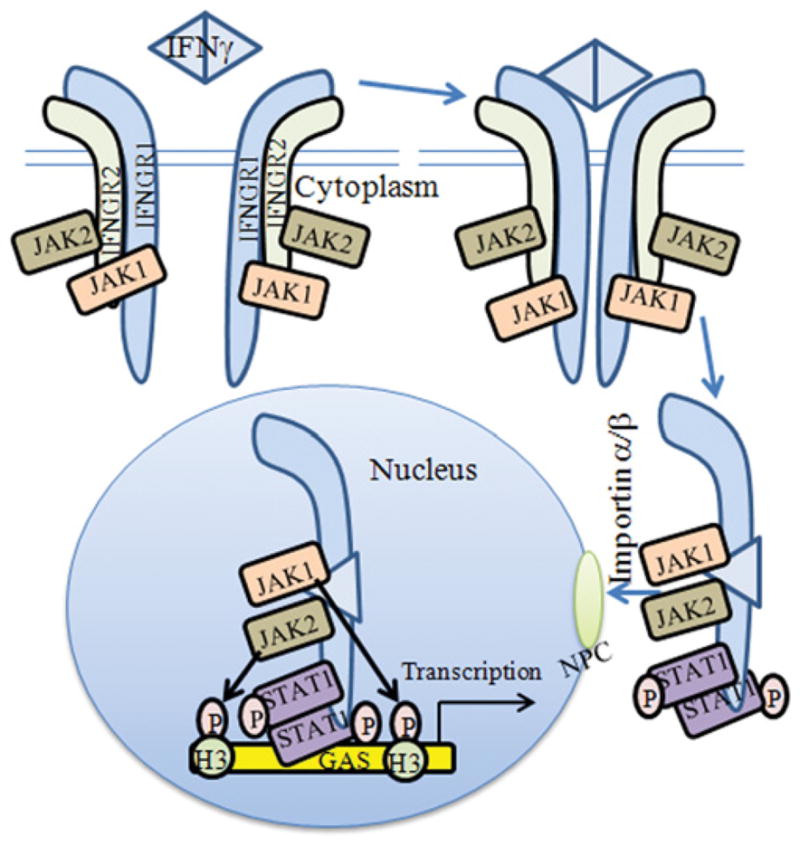
The following steps are involved. (i) Ligand binds to the receptor extracellularly. (ii) The receptor functions as a transcription/co-transcription factor. (iii) JAKs 1 and 2 are associated with the ligand–receptor complex, which translocates to the nucleus. (iv) The complex binds to response elements of specific genes. (v) Some of the cofactors of the complex, such as JAKs, are involved in the specific epigenetic events that cause heterochromatin destabilization and promoter activation. Histone H3 phosphorylation at Tyr41 by JAK1 and JAK2 (see arrows) illustrate a key epigenetic event in IFNγ/IFNGR1 gene activation. An animated version of this Figure is available at http://www.BiochemJ.org/bj/443/0329/bj4430329add.htm.
GENE ACTIVATION AND ASSOCIATED EPIGENETIC EVENTS IN THE CONTEXT OF THE MORE COMPLEX MODEL: IDENTIFICATION OF A NEW ROLE FOR JAKS
By ChIP followed by PCR, IFNγ, its receptor subunit IFNGR1 and STAT1α were found to be associated with the IFNγ-activated sequence (GAS) in the promoter of two of the genes stimulated by IFNγ [40]. Immunoprecipitated chromatin also showed the association of the IFNγ, IFNGR1 and STAT1α on the same DNA sequence. Examination of nuclear extracts from WISH cells treated with IFNγ revealed the specific binding of IFNγ, IFNGR1 and STAT1α to the biotinylated GAS nucleotide sequence. Association of IFNγ, IFNGR1 and STAT1α with the GAS promoter was also demonstrated by EMSA (electrophoretic mobility-shift assay). Transfection with a GAS–luciferase gene, together with the IFNGR1 and non-secreted IFNγ resulted in enhanced reporter activity. In addition, IFNGR1 fused to the yeast GAL4 DNA-binding domain resulted in enhanced transcription from a GAL4-response element, suggesting the presence of a transactivation domain in IFNGR1. These observations put IFNγ and its receptor subunit, IFNGR1, in direct contact with the promoter region of IFNγ-activated genes with associated increased activity, thus suggesting a transcriptional/co-transcriptional role for IFNγ–IFNGR1, as well as a possible role in determining the specificity of IFNγ action [40].
To address the possible epigenetic role of the JAKs that are activated, ChIP followed by PCR in IFNγ-treated WISH cells was carried out and showed association of pJAK1 (phosphorylated JAK1), pJAK2 (phosphorylated JAK2), IFNGR1 and STAT1 on the same DNA sequence of the IRF1 (IFN regulatory factor 1) gene promoter [41]. The ACTB (β-actin) gene, which is not activated by IFNγ, did not show this association. The movement of activated JAK to the nucleus and the IRF1 promoter was also confirmed by the combination of nuclear fractionation, confocal microscopy and DNA precipitation analysis using the biotinylated GAS promoter and is reflected in the model in Figure 2. Activated JAKs in the nucleus were associated with phosphorylated Tyr41 on histone H3 in the region of the GAS promoter. This is consistent with previous studies showing phosphorylation of Tyr41 on histone H3 in leukaemic cells expressing mutated JAK2, JAK2V617F and wild-type JAK2 in cells treated with the cytokine/growth factors PDGF, LIF (leukaemia-inhibitory factor) or IL-3 [42]. Unphosphorylated JAK2 was found to be constitutively present in the nucleus and was capable of undergoing activation in IFNγ-treated cells, most probably via nuclear IFNGR1. Association of pJAK2 and IFNGR1 with histone H3 in IFNγ-treated cells was demonstrated by histone H3 immunoprecipitation. Unphosphorylated STAT1 protein was associated with histone H3 of untreated cells. IFNγ treatment resulted in its disassociation and then reassociation as pSTAT1. It has been reported that unphosphorylated STAT in the nucleus was associated with heterochromatin stabilization and gene silencing in Drosophila, whereas pSTAT was associated with destabilization [14]. The results suggest a novel role for activated JAKs in epigenetic events for specific gene activation [41].
INSIGHT INTO IFNγ MIMETIC DEVELOPMENT
Small peptide mimetics of IFNγ were developed not on the basis of the classical model of IFNγ-initiated signalling by extracellular interaction, but rather direct intracellular signalling by IFNγ. As indicated, IFNγ, its receptor subunit IFNGR1 and transcription factor STAT1α are transported to the nucleus of cells as a complex where IFNγ provides a classical polycationic NLS for such transport [32]. The C-terminus of IFNγ, represented by the mouse IFNγ peptide IFNγ-(95–132) was capable of also forming a complex with IFNGR1 and STAT1α when introduced intracellularly with an attached cell-penetrating group, and provided the NLS signalling for nuclear transport [32]. Importantly, mouse IFNγ-(95–132) and human IFNγ-(95–134) mimetics both induced an antiviral state and up-regulation of MHC class I molecules in cells similar to that of full-length IFNγ in a species non-specific manner [43,44]. This is consistent with the demonstration that the cytoplasmic domain of IFNGR1, unlike the extracellular domain, is not species-specific [5]. Both IFNγ and its peptide mimetics bind to an intracellular site, IFNGR1 (residues 253–287), on the cytoplasmic domain of receptor subunit IFNGR1. This binding plays a role in tyrosine phosphorylation events, catalysed by JAK1 and JAK2 at both the cytoplasmic and nuclear levels which result in the phosphorylation and binding of STAT1α to the cytoplasmic domain of IFNGR1. Important structural requirements for IFNγ mimetic activity are a polycationic NLS and an α-helix in the mimetics [40]. ChIP and reporter gene studies of IFNγ and IFNγ mimetic-treated cells indicate that they, along with IFNGR1 and STAT1α, bind to the IFNγ activation site element of IFNγ-activated genes and participate in STAT1α-mediated transcription [45]. IFNγ intracellular events played the key role in development of IFNγ mimetics [43,44]. In contrast with intact IFNγ the mimetics therefore do not bind to poxvirus IFNγ decoy receptor B8R protein and can thus initiate an antiviral response in the presence of B8R protein in cell culture and a mouse model of vaccinia virus infection [44]. The development of an IFNγ mimetic on the basis of our model of IFNγ signalling illustrates the dynamics of the model.
THE EGF (EPIDERMAL GROWTH FACTOR)/EGFR (EGF RECEPTOR) SYSTEM
RTKs (receptor tyrosine kinases), such as EGFR, also undergo nuclear translocation in association with their respective ligands. There are four isoforms of the EGFR family: EGFR, ErbB2 (HER-2), ErbB3 and ErbB4 [46,47]. The EGF family of peptides that bind the ErbB receptors are classified into three groups. The most recognized of these is the first group, which consists of EGF, TGFα (transforming growth factor α), amphiregulin and epigen, all of which bind to EGFR. Interestingly, ErbB2 does not bind any of the ligands, but interacts with other members of the EGFR family to form heterodimers. Two decades of research has provided unequivocal evidence that the ErbB-1 (EGFR) isoform in association with EGF ligand translocates to the nucleus [48]. The EGF–EGFR complex has been shown to bind to the CCND1 (cyclin D1) gene promoter in a sequence-specific fashion and to modulate CCND1 gene transcription [49]. Thus EGFR possesses transcription/co-transcription function. Furthermore, EGFR interacts with STAT3, STAT5 and E2F1 within the NOS2 (inducible NO synthase), PTGS2 (cyclo-oxygenase 2), MYBL2 (B-Myb) and Aurka (Aurora-A) promoter regions [50]. These interactions appear to be critical for EGFR transactivational activity at these different genes, and it is of interest as to whether the interactions occurred prior to binding to the particular promoters. It thus seems that these protein–protein interactions play an important role in the specificity of the gene activation.
EGF and TGFα play a critical role in EGFR movement from the plasma membrane to the appropriate promoters in the nucleus. Receptor endocytosis is initiated by interaction with EGF, which undergoes nuclear translocation in association with EGFR [49]. It has been shown, for example, that EGF binds tightly to chromatin and that cells lacking EGFR fail to accumulate EGF in the nucleus [49,50]. The presence of a well-defined polycationic NLS in the cytoplasmic domain of EGFR has been described, whereas EGF does not possess an NLS [51]. This provides further evidence that EGF and EGFR undergo nuclear translocation as a complex.
The EGFR system has been particularly useful in providing insight into how a plasma membrane protein with a hydrophobic transmembrane sequence migrates through the nuclear pore complex and functions as a transcription/co-transcription factor at promoters of activated genes. Upon treatment of MDA-MB-468 breast cancer cells with EGF, confocal immunofluorescence revealed that EGFR underwent retrograde movement to the Golgi and the ER (endoplasmic reticulum) where the N-terminus was within the lumen of the Golgi/ER and the C-terminus was exposed to the cytoplasm [48]. Retrograde trafficking was blocked by brefeldin A or dominant mutants of the small GTPase ARF (ADP-ribosylation factor), both of which resulted in disassembly of the COPI (coat protein complex 1) to the Golgi. The study concluded that the COPI regulated retrograde vesicular trafficking of EGFR from the Golgi to the ER. A related study showed that treatment of MDA-MB-468 cells with EGF resulted in trafficking of biotinylated cell-surface EGFR to the INM (inner nuclear membrane) through the nuclear pore complexes, while maintaining its membrane-bound state [52,53]. It was confirmed that IMPβ regulated EGFR nuclear transport to the INM as well as to the nucleus/nucleoplasm. EGF was associated with EGFR. Perhaps the most novel aspect of this study was the demonstration that Sec61β was found to be present in the INM and to associate with EGFR. Sec61β is a well-known ER-associated translocon [52]. Translocon is the term that refers to the conserved protein-conducting channel referred to as the Sec61 channel in eukaryotes. Knockdown of Sec61β expression reduced the level of EGFR in the nucleoplasm portion with concomitant accumulation in the INM. Thus the Sec61β translocon played an unexpected critical role in the release of membrane-anchored EGFR from the lipid bilayer of the INM to the nucleus. These findings provide insight into the mechanism of nuclear transport of a membrane-bound full-length protein that functions as a transcription/co-transcription factor.
Similar to IFNγ in Figure 1(B), EGFR signalling involves commonly shared cytoplasmic pathways such as MAPK and PI3K/Akt, and much of the specificity of normal signalling is ascribed to these pathways. EGFR family members are major players in many of the difficult, hard to treat cancers and in most cases aberrations in the MAPK, PI3K/Akt and other cytoplasmic signalling pathways are thought to be a causative factor and are the focus of some approaches to treat cancer [50]. To reiterate, these pathways are in a sense generic and have not been shown to be the mechanism of specific gene activation by EGFR or the ligands that activate it and the other related ErbB receptors.
In contrast with the fact that EGFR has been shown to undergo nuclear translocation as indicated above, most of the focus on EGFR endocytosis places particular emphasis on endosomal trafficking [52,53]. Upon ligand binding, EGFR is activated, ubiquitylated and recruited into clathrin-coated pits [54]. Blockage of ubiquitylation of EGFR results in inhibition of recruitment into coated pits. From the endosomal sorting complex, EGFR is either degraded in lysosomes or bypasses the lysosome stage and is recycled to the plasma membrane. Adherents of this basic model basically ignore events that result in nuclear transport of EGFR.
As indicated above, nuclear EGFR provides insight into specific gene activation. It also has a prognostic value in cancers that are thought to involve aberrant signalling by members of the EGFR family. The clinical studies of nuclear EGFR in several cancers warrants special mention. In one study, it was shown for the first time in a cohort of 130 breast carcinomas that, by immunohistochemical analysis, 37.7 % of the cohort was positive for nuclear EGFR, 6.9 % had high levels of expression of nuclear EGFR, and that a significant inverse correlation existed between high nuclear EGFR expression and overall survival [48]. In another cohort of 37 oral squamous cell carcinomas, it was shown that 24.3 % of the cases contained moderate/high levels of nuclear EGFR and that those with high EGFR had a tendency to poor survival [48]. Similar results were obtained in a study of 95 oropharyngeal carcinomas [46,49,55], as well as in a study of 221 cases of ovarian cancer [48,50,56]. Thus the presence of EGF/EGFR family members in the nucleus of cancer patients suggests not only a physiological function, but also most probably a considerable importance in cancer. Studies directed towards development of inhibitors of EGFR family member kinase activity may particularly be directed towards their nuclear function in gene activation.
THE FGF (FIBROBLAST GROWTH FACTOR)/FGFR (FGF RECEPTOR) SYSTEM
FGFs are a family of approximately 20 different growth factors and/or isoforms (reviewed in [57,58]). They interact at the cell surface with FGFRs, of which there are at least four different genes. Our focus in the present review is on the nuclear presence of FGF/FGFR in cells treated with FGF. The first demonstration of a nuclear function of FGF1 was the observation that it possessed a polycationic putative NLS at the N-terminus of the mature protein [59]. FGF1 lacking the NLS did not undergo nuclear translocation. Both FGF1 and FGF2 have been shown to undergo nuclear translocation in a FGFR-dependent fashion [58], where intact full-length FGF and FGFR have been recovered from the nucleus [60].
As is the case for EGFR and other transmembrane receptors that have been shown to undergo nuclear translocation, the question arises as to the nature of the FGFR mechanism(s) involved. As we indicated above, the Secβ61 translocon was shown to play a critical role in the release of EGFR from the INM to the nucleus. Early studies and modelling pointed to a role for Secβ61 in retrograde trafficking of receptors, such as FGFR from the transmembrane to the cytoplasm, but not with the specificity shown for EGFR release into the nucleus [61]. It has been shown that plasma membrane FGFR internalization was dependent on dynamin and ARF6 [62]. Dynamin has been used as a marker of clathrin-coated pit involvement in endocytosis, but its involvement with endocytic events can also involve non-clathrin pathways. ARF6 is a member of the small G-protein family [62]. Of the six small GTPases, only ARF6 has been shown to be present at the plasma membrane and is thought to play a role in non-clathrin and non-caveolar-type endocytosis [62]. There are other endocytic players, such as Rab5, but there is no definitive picture on the mechanism of FGFR endocytosis.
The translocation of full-length FGFRs to the nucleus upon stimulation with ligands such as FGF2 requires IMPβ1, but is independent of IMPα [63]. Interaction with cytoplasmic IMPβ1 means that the endocytosis is followed by exposure of FGFR to the cytoplasm. Interaction between FGFR1 and IMPβ is probably not direct since it does not contain a known NLS motif. It is possible that the NLS is provided by FGF. It was shown, for example, that the N-terminus of FGF1 contains the sequence N21YKKPKL, which was required for mutagenic activity of added FGF1, but not for stimulation of phosphorylation and transcription of c-Fos [59]. FGF1 lacking the sequence did not enter the nuclear fraction. Substitution of an NLS sequence, G30KKRKSK, from yeast histone H2A restored FGF1 nuclear translocation.
Assuming that IMPβ interacts with the NLS of FGF1 for active transport of the FGF1–FGFR1 complex into the nucleus via the nuclear pore complex, there is the question of how the FGF1 NLS becomes available. Upon interaction of FGF1 with the extracellular domain of FGFR1 and the subsequent endocytosis and endosome formation, the cytoplasmic domain of FGFR1 will be exposed to the cytoplasm, whereas FGF1 will be contained in the lumen of the endosome and thus not accessible to IMPβ. This is not a problem for those receptors that contain an NLS in their cytoplasmic domain, such as EGFR [52]. We addressed this issue with IFNγ receptor subunit IFNGR1, as the NLS was present at the C-terminus of IFNγ and not in the receptor (see the discussion on IFNγ/IFNGR1 above).
Both FGF1 and FGF2 have been shown to translocate across vesicular membranes by a common mechanism [64]. The authors of that study [64] used a clever approach to differentiate between exogenous and endogenous growth factor. FGF was modified to contain a farnesylation signal, a CaaX box (where a is an aliphatic amino acid). The logic was that farnesylation occurs only in the cytosol and nucleoplasm, and farnesylation of exogenous FGF2–CaaX is taken as evidence that FGF translocated across cellular membranes. Farnesylation of FGF2–CaaX was observed, thus demonstrating that FGF crosses from the lumen of the endosome and is thus exposed to the cytoplasm along with the cytoplasmic domain of FGFR1. Cytosolic HSP (heat-shock protein) 90 was required for the translocation as well as dissipation of the vesicular membrane potential. Since FGF and FGFR1 are associated in the IMPβ link for nuclear translocation, one could infer that FGFR1 binds FGF at a receptor cytoplasmic domain site. This inference must, however, be tested by an actual demonstration of such an interaction, as was done for IFNγ and IFNGR1 above.
There is evidence that FGFR1 possesses transcription-factor-like activity in the nucleus [57,65]. Nuclear FGFR1 induces c-Jun transcription and potentiates cyclin D1 expression, which is dependent on functional kinase activity as kinase-deficient point mutation results in loss of transcription activity. There are several other examples of FGFR1 transcriptional activity, an interesting example of which is the co-operation with CBP [CREB (cAMP-response-element-binding protein)-binding protein] [57]. Transcription of full-length FGFR1 fused to the GAL4 DNA-binding domain failed to stimulate luciferase activity from the GAL4 DNA-binding element, suggesting that the receptor lacks autonomous transactivation function [65]. It could, however, cooperate with co-transfected CBP. The N-terminal domain of the receptor was essential for the co-transcription activity. Thus FGFR1 appears to possess transcription and/or co-transcription function at genes that are induced in cells treated with FGF.
LESSONS FROM STEROID SIGNALLING
In a search for a precedent, it seems that our IFNγ, and probably EGF and FGF, have similarities to that of SR signalling. We now provide an overview of steroid/SR signalling to point out these similarities. SRs are a major subset of nuclear receptors. Basically, synthesis of SHs (steroid hormones) occurs in the adrenal cortex and in gonads [66]. SHs are derivatives of cholesterol that are biosynthesized through various biochemical pathways. This involves the conversion of cholesterol into pregnenolone, which is subsequently converted into 17-hydroxypregnenolone and progesterone. 17-Hydroxypregnenolone gives rise to testosterone, which can be converted into oestradiol via reduction. By a series of specific hydroxylations, progesterone gives rise to cortisol and aldosterone.
Broadly, the current model of SH signalling is as follows and is summarized in Figure 3. SH binds to SRs located in the cytoplasm or nucleus of the cell. In the absence of hormone, SR monomers are associated with HSPs and usually possess some basal level of phosphorylation. Upon binding of hormone, SRs dissociate from HSPs, dimerize and translocate to the nucleus where they bind to HREs (hormone-response elements) at genes that are activated by SHs. The complex of SH–SR recruits a series of co-activator complexes to both regulate target gene transcription as well as the associated epigenetic events that accompany such activation. Site-specific phosphorylation of receptors occurs subsequently to hormone binding with varied kinetics, depending on the kinase and the target in the receptor complex.
Figure 3. Current model of steroid signaling.
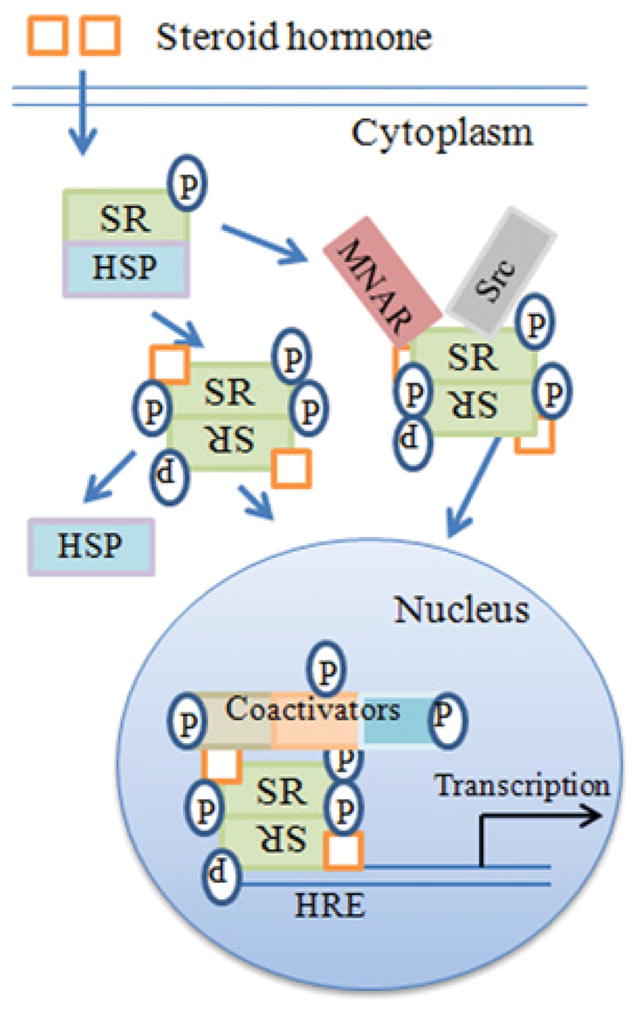
The following steps are involved. (i) Ligand binds to the receptor intracellularly. (ii) The receptor functions as a transcription/co-transcription factor. (iii) Co-activators such as kinases are associated with the ligand–receptor complex, which translocates to the nucleus. (iv) The complex binds to response elements of specific genes. (v) Some of the cofactors of the complex, such as the kinases, are involved in the specific epigenetic events that accompany specific gene activation. HRE, hormone-response element; MNAR, modulator of non-genomic action of oestrogen receptor; P, phosphotyrosine and non-phosphotyrosine kinase activity by co-factors and kinases. Adapted from [67] under the terms of the of the Creative Commons Non-Commercial Attribution License.
The kinases, although not the only components of the receptor associated co-activator complexes, are important for their action on members of the complex, as well as for specific epigenetic events of gene activation and thus act on histones as well as on members of the receptor complex. Many of the SH phosphorylation sites contain serine/threonine/proline motifs involving proline-specific kinases, such as the cyclin-dependent kinases and MAPKs [67,68]. Tyrosine kinases such as Src have also been shown to participate in SR signalling in the nucleus. SRs similarly cross-talk with RTKs, such as EGFR. EGFR family members are an important target in some of the most prevalent and difficult cancers, such as non-small cell lung carcinoma [46].
In addition to their presence in the cytoplasm, a subset of SRs are also membrane-associated through an S-palmitoylation linkage to the inner side of the plasma membrane [67]. It is generally thought that membrane-associated SR is the same as cytoplasmic SR, but this is not universally agreed upon. Membrane SR is involved in activation of MAPK and PI3K/Akt kinases.
In addition to the kinase-type activators described above, there are also so-called primary SRCs (SR co-activators), of which three are the most prominent [66]. SRC proteins are recruited to hormone-bound SRs and bind through their LXXL motifs. SRCs recruit secondary co-activators, such as the histone acetyltransferase p300/CBP, the histone methyltransferases PRMT1 (protein arginine N-methyltransferase 1) and CARM1 (co-activator-associated arginine methyltransferase 1), and the chromatin remodelling complex SWI/SNF. These secondary co-activators modify the chromatin and bridge the SR complex with the general transcription machinery. Although the various kinases are present, just how they associate with SR and SRCs is not precisely known. One does come up with, however, the general picture of SH–SR–co-activator complexes where the co-activators may be grouped as primary as the case for the SRCs or secondary as the case for the histone transferases. If one were to restrict primary co-activators to the SRCs, then the kinases could also possibly be called secondary co-activators.
A comparison of IFNγ signaling in Figure 2 and SH signalling in Figure 3 suggests the following similar features. Ligand associates with the receptor intracellularly. In the case of IFNγ, first there is extracellular binding to IFNGR1 and then intracellular binding in conjunction with the endocytosis. SH penetrates the plasma membrane and binds the cytoplasmic SR. In both cases the receptors function as transcription/co-transcription factors. Co-activators are associated with the ligand–receptor complex. An overview of similarities between IFNGR1, EGFR, FGFR and SR systems is presented in Table 1. Currently, much more is known concerning the SH–SR complex than the IFNγ–IFNGR1 complex, but STAT1α and the kinases JAK1 and JAK2 are associated in the cytoplasm and the nucleus. In both cases, the ligand–receptor–co-activator complex binds to response elements of genes that are specifically activated. Some of the co-factors, such as the kinases, are involved in specific epigenetic events for both systems. We do not feel that IFNs are a special case with respect to protein ligands with associated tyrosine kinase activity or with RTKs, as EGFR and FGFR have similarities to the IFNs in receptor involvement in nuclear aspects of gene activation. We further feel that all of the cytokines, hormones and growth factors that use the JAK/STAT pathway are likely to also share these similarities. In our view the template for all of this resides in the SH–SR system of specific gene activation.
Table 1. Receptors as co-ordinators of complex formation, and function in genetic and epigenetic changes in gene activation.
In IFNγ, EGF, FGF and SR systems, similarities in terms of the steps involved in specific gene activation are indicated. References are provided, and detailed references for the other molecules involved are indicated in the text. ERK, extracellular-signal-regulated kinase; Msk1, mitogen- and stress-activated kinase 1; Rsk1, ribosomal S6 kinase 1; Src, cytoplasmic tyrosine kinase.
| Component | Signalling systems
|
|||
|---|---|---|---|---|
| IFNγ | EGF | FGF | SR | |
| Ligand | IFNγ: (i) activates receptor; (ii) provides NLS [31,32] | EGF, TGFα: (i) activate receptor | FGF: (i) activates receptor; (ii) provides NLS | SH: (i) activates receptor |
| Receptor | IFNGR1: (i) transcription factor/co-transcription factor [40] | EGFR: (i) transcription factor/co-transcription factor; (ii) provides NLS; (iii) RTK and other kinase activity for epigenetic modification | FGFR1, FGFR2: (i) transcription factor/co-transcription factor; (ii) RTK activity for epigenetic modification | SR: (i) transcription factor/co-transcription factor; (ii) platform for co-activators |
| JAKs | IFNGR2: (i) moves JAK2 to IFNGR1 [39] (i) STAT activation; (ii) epigenetic modification [41] |
(i) STAT activation; (ii) epigenetic modification | (i) STAT activation; (ii) epigenetic modification | |
| STATs | STAT1α: (i) transcription factor in activated state [41]; (ii) heterochromatin stabilizer in non-phosphorylated state [41] | STATs 1, 3, 5: (i) transcription factors | STAT5: (i) transcription factors | STAT5: (i) transcription factor for progesterone receptor [70] |
| Other associated nuclear kinases and co-factors | MAPK ERK1/2 NF-κB [12] |
MAPK Src [69] |
MAPK Rsk1 [70] |
MAPK SRC 1, 2, 3 [71] Msk1 and ERK [71] |
CONCLUSIONS
Steroid signalling provides invaluable insight into the specificity of gene activation. Steroid hormone receptors are intracellular proteins that are activated by internalized SHs. The essence of specific gene activation is the formation of a complex of steroid–SR–co-activator. The steroid ligand causes allosteric changes in the receptor. This results in dissociation of HSPs and the attraction of co-activators, some of which serve as a platform for other co-activators such as serine/threonine/proline and tyrosine kinases. The complex translocates to the nucleus via a receptor NLS. Receptor functions as a transcription/co-transcription factor and the kinases participate in the epigenetic events associated with specific gene activation. Our studies with IFN in JAK/STAT signalling as well as those of others with RTKs, such as EGFRs and FGFRs, suggest that similar mechanisms of specific gene activation are involved. Although it is difficult to approach the mechanisms of specific gene activation by JAK/STAT via the classical model, our model readily provides insight into such mechanisms. It is but a variation of specific gene activation by SHs.
Acknowledgments
FUNDING
Work of the authors is supported by the National Institutes of Health [grant number R01 056152 (to H.M.J.).
Abbreviations used
- ARF
ADP-ribosylation factor
- ChIP
chromatin immunoprecipitation
- CBP
CREB (cAMP-response-element-binding protein)-binding protein
- COPI
coat protein complex 1
- EGF
epidermal growth factor
- EGFR
EGF receptor
- ER
endoplasmic reticulum
- FGF
fibroblast growth factor
- FGFR
FGF receptor
- H3K4
Lys4 of histone H3
- HSP
heat-shock protein
- IFN
interferon
- IFNGR1
IFNγ receptor 1
- IFNGR2
IFNγ receptor 2
- IL
interleukin
- IMP
importin
- INM
inner nuclear membrane
- IRF1
IFN regulatory factor 1
- JAK
Janus kinase
- MAPK
mitogen-activated protein kinase
- NF-κB
nuclear factor κB
- NLS
nuclear localization sequence
- PDGF
platelet-derived growth factor
- PI3K
phosphoinositide 3-kinase
- pJAK2
phosphorylated JAK2
- RORγt
thymus-type retinoic acid orphan receptor γ
- RTK
receptor tyrosine kinase
- SH
steroid hormone
- SR
steroid receptor
- SRC
SR co-activator
- STAT
signal transducer and activator of transcription
- pSTAT
phosphorylated STAT
- SV40 TAg
simian virus 40 large T antigen
- TGFα
transforming growth factor α
References
- 1.Brivanlou AH, Darnell JE., Jr Signal transduction and the control of gene expression. Science. 2002;295:813–818. doi: 10.1126/science.1066355. [DOI] [PubMed] [Google Scholar]
- 2.Johnson HM, Subramaniam PS, Olsnes S, Jans DA. Trafficking and signaling pathways of nuclear localizing protein ligands and their receptors. BioEssays. 2004;26:993–1004. doi: 10.1002/bies.20086. [DOI] [PubMed] [Google Scholar]
- 3.Subramaniam PS, Torres BA, Johnson HM. So many ligands, so few transcription factors: a new paradigm for signaling through the STAT transcription factors. Cytokine. 2001;15:175–187. doi: 10.1006/cyto.2001.0905. [DOI] [PubMed] [Google Scholar]
- 4.Johnson HM, Ahmed CM. Gamma interferon signaling: insights to development of interferon mimetics. Cell Mol Biol. 2006;52:71–76. [PubMed] [Google Scholar]
- 5.Muthukumaran G, Donnelly RJ, Ebensperger C, Mariano MT, Garotta G, Pestka S. The intracellular domain of the second chain of the interferon γ receptor is interchangeable between species. J Interferon Cytokine Res. 1996;16:1039–1045. doi: 10.1089/jir.1996.16.1039. [DOI] [PubMed] [Google Scholar]
- 6.Chen X, Vinkemeier U, Zhao Y, Jeruzalmi D, Darnell JE, Kuriyan J. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998;93:827–839. doi: 10.1016/s0092-8674(00)81443-9. [DOI] [PubMed] [Google Scholar]
- 7.Krause CD, Lunn CA, Izotova LS, Mirochnitchenko O, Kotenko SV, Lundell DJ, Narula SK, Pestka S. Signaling by covalent heterodimer of interferon γ Evidence for one-sided signaling in the active tetrameric receptor complex. J Biol Chem. 2000;275:2995–3004. doi: 10.1074/jbc.M909607199. [DOI] [PubMed] [Google Scholar]
- 8.Landar A, Curry B, Parker MH, DiGiacomo R, Indelicato SR, Nagabhushan TL, Rizzi G, Walter MR. Design, characterization, and structure of a biologically active single-chain mutant of human IFNγ. J Mol Biol. 2000;299:169–179. doi: 10.1006/jmbi.2000.3734. [DOI] [PubMed] [Google Scholar]
- 9.McBride KM, McDonald C, Reich NC. Nuclear export signal located within the DNA-binding domain of the STAT1 transcription factor. EMBO J. 2000;19:6196–6206. doi: 10.1093/emboj/19.22.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melen K, Kinnunen L, Julkunen I. Arginine/lysine-rich structural element is involved in interferon-induced nuclear transport. J Biol Chem. 2001;276:16447–16455. doi: 10.1074/jbc.M008821200. [DOI] [PubMed] [Google Scholar]
- 11.Begitt A, Meyer T, van Rossum M, Vinkemeier U. Nucleocytoplasmic translocation of STAT1 is regulated by a leucine-rich export signal in the coiled-coil domain. Proc Natl Acad Sci USA. 2000;97:10418–10423. doi: 10.1073/pnas.190318397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gough DJ, Levy DE, Johnsotone RW, Clarke CJ. IFNγ signaling: does it mean JAK-STAT? Cytokine Growth Factor Rev. 2008;19:383–394. doi: 10.1016/j.cytogfr.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 13.Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- 14.Yan SJ, Lim SJ, Shi S, Dutta P, Li WX. Unphosphorylated STAT and heterochromatin protect genome stability. FASEB J. 2011;25:232–241. doi: 10.1096/fj.10-169367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robertson AG, Bilenky M, Tam A, Zhao Y, Zeng T, Thiessen N, Cezard T, Fejes AP, Wederell ED, Cullum R, et al. Genome-wide relationship between histone H3 lysine 4 mono- and tri-methylation and transcription factor binding. Genome Res. 2008;18:1906–1917. doi: 10.1101/gr.078519.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Grainger JR, Hirahara K, Sun HW, Wei L, Vahedi G, Kanno Y, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei L, Vahedi G, Sun HW, Watford WT, Takatori H, Ramos HL, Takahashi H, Liang J, Gutierrez-Cruz G, Zang C, et al. Discrete roles of STAT4 and STAT6 transcription factors in tuning epigenetic modifications and transcription during T helper cell differentiation. Immunity. 2010;32:840–851. doi: 10.1016/j.immuni.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, Lee J, de Sauvage FJ, Ghilardi N. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. 2006;7:929–936. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- 19.Stumhofer JS, Laurence A, Wilson EH, Huang E, Tato CM, Johnson LM, Villarino AV, Huang Q, Yoshimura A, Sehy D, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7:937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 20.Colgan J, Rothman P. All in the family: IL-27 suppression of Th17 cells. Nat Immunol. 2006;7:899–901. doi: 10.1038/ni0906-899. [DOI] [PubMed] [Google Scholar]
- 21.McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, McClanahan TK, O’Shea JJ, Cua DJ. The interleukin 23 receptor is essential for the terminal differentiation of interleukin-17 producing effector cells. Nat Immunol. 2009;3:314–324. doi: 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, Gery I, Lee YS, Egwuagu CE. Th17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat Med. 2007;6:711–718. doi: 10.1038/nm1585. [DOI] [PubMed] [Google Scholar]
- 23.Sun Z, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, Mebius RE, Littman DR. Requirement for RORγ in thymocyte survival and lymphoid organ development. Science. 2000;288:2369–2373. doi: 10.1126/science.288.5475.2369. [DOI] [PubMed] [Google Scholar]
- 24.Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirahara K, Ghoreschi K, Laurence A, Yang XP, Kanno Y, O’Shea JJ. Signal transduction pathways and transcriptional regulation in Th17 cell differentiation. Cytokine Growth Factor Rev. 2010;21:425–434. doi: 10.1016/j.cytogfr.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 27.Kramer OH, Heinzel T. Phosphorylation-acetylation switch in the regulation of STAT1 signaling. Mol Cell Endocrinol. 2010;315:40–48. doi: 10.1016/j.mce.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 28.Cheon H, Yang J, Stark GR. The functions of signal transducers and activators of transcriptions 1 and 3 as cytokine-inducible proteins. J Interferon Cytokine Res. 2011;31:33–40. doi: 10.1089/jir.2010.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bader T, Weitzerbin J. Nuclear accumulation of interferon γ. Proc Natl Acad Sci USA. 1994;91:11831–11835. doi: 10.1073/pnas.91.25.11831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacDonald HS, Kushnaryov VM, Sedmak JJ, Grossberg SE. Transport of γ-interferon into the cell nucleus may be mediated by nuclear membrane receptors. Biochem Biophys Res Commun. 1986;138:254–260. doi: 10.1016/0006-291x(86)90273-1. [DOI] [PubMed] [Google Scholar]
- 31.Subramaniam PS, Mujtaba MG, Paddy MR, Johnson HM. The carboxyl terminus of interferon-γ contains a functional polybasic nuclear localization sequence. J Biol Chem. 1999;274:403–407. doi: 10.1074/jbc.274.1.403. [DOI] [PubMed] [Google Scholar]
- 32.Ahmed CM, Burkhart MA, Mujtaba MG, Subramaniam PS, Johnson HM. The role of IFNgamma nuclear localization sequence in intracellular function. J Cell Sci. 2003;116:3089–3098. doi: 10.1242/jcs.00528. [DOI] [PubMed] [Google Scholar]
- 33.Subramaniam PS, Green MM, Larkin J, Torres BA, Johnson HM. Nuclear translocation of IFNγ is an intrinsic requirement for its biologic activity and can be driven by a heterologous nuclear localization sequence. J Interferon Cytokine Res. 2001;21:951–959. doi: 10.1089/107999001753289569. [DOI] [PubMed] [Google Scholar]
- 34.Jans DA, Xiao CY, Lam MN. Nuclear targeting signal recognition: a key control point in nuclear transport? BioEssays. 2000;22:532–544. doi: 10.1002/(SICI)1521-1878(200006)22:6<532::AID-BIES6>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 35.Subramaniam PS, Larkin J, Mujtaba MG, Walter MR, Johnson HM. The COOH-terminal nuclear localization sequence of interferon gamma regulates STAT1α nuclear translocation at an intracellular site. J Cell Sci. 2000;113:2771–2781. doi: 10.1242/jcs.113.15.2771. [DOI] [PubMed] [Google Scholar]
- 36.Sekimoto T, Imamoto N, Nakajima K, Hirano T, Yoneda Y. Extracellular signal-dependent nuclear import of Stat1 is mediated by nuclear pore-targeting complex formation with NPI-1, but not Rch1. EMBO J. 1997;16:7067–7077. doi: 10.1093/emboj/16.23.7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Larkin J, III, Johnson HM, Subramaniam PS. Differential nuclear localization of the IFNGR-1 and IFNGR-2 subunits of the IFNγ receptor complex following activation by IFNγ. J Interferon Cytokine Res. 2000;20:565–576. doi: 10.1089/10799900050044769. [DOI] [PubMed] [Google Scholar]
- 38.Subramaniam PS, Johnson HM. Lipid microdomains are required sites for the selective endocytosis and nuclear translocation of IFNγ, its receptor chain IFNγ receptor-1, and the phosphorylation and nuclear translocation of STAT1α. J Immunol. 2002;169:1959–1969. doi: 10.4049/jimmunol.169.4.1959. [DOI] [PubMed] [Google Scholar]
- 39.Szente BE, Subramaniam PS, Johnson HM. Identification of IFNγ receptor binding sites for JAK2 and enhancement of binding by IFNγ and its C-terminal peptide IFNγ (95–133) J Immunol. 1995;155:5617–5622. [PubMed] [Google Scholar]
- 40.Ahmed CM, Johnson HM. IFNγ and its receptor subunit IFNGR1 are recruited to the IFNγ-activated sequence element at the promoter site of IFNγ-activated genes: evidence of transactivational activity in IFNGR1. J Immunol. 2006;177:315–321. doi: 10.4049/jimmunol.177.1.315. [DOI] [PubMed] [Google Scholar]
- 41.Noon-Song EN, Ahmed CM, Dabelic R, Canton J, Johnson HM. Controlling nuclear JAKs and STATs for specific gene activation by IFNγ. Biochem Biophys Res Commun. 2011;410:648–653. doi: 10.1016/j.bbrc.2011.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dawson MA, Bannister AJ, Gottgens B, Foster SD, Bartke T, Green AR, Kouzarides T. JAK2 phosphorylates histone H3Y41 and excludes HP1α from chromatin. Nature. 2009;461:819–822. doi: 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szente BE, Johnson HM. Binding of IFNγ and its C-terminal peptide to a cytoplasmic domain of its receptor that is essential for function. Biochem Biophys Res Commun. 1994;201:215–221. doi: 10.1006/bbrc.1994.1691. [DOI] [PubMed] [Google Scholar]
- 44.Ahmed CM, Burkhart MA, Subramaniam PS, Mujtaba MG, Johnson HM. Peptide mimetics of γ-interferon possess antiviral properties against vaccinia virus and other viruses in the presence of poxvirus B8R protein. J Virol. 2005;79:5632–5639. doi: 10.1128/JVI.79.9.5632-5639.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szente BE, Weiner IJ, Jablonsky MJ, Krishna NR, Torres BA, Johnson HM. Structural requirements for agonist activity of a murine interferon-γ as assessed by monoclonal antibodies. J Interferon Cytokine Res. 1996;16:813–817. doi: 10.1089/jir.1996.16.813. [DOI] [PubMed] [Google Scholar]
- 46.Lo HW, Hung MC. Nuclear EGFR signaling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br J Cancer. 2006;94:184–188. doi: 10.1038/sj.bjc.6602941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177–184. doi: 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 48.Wang YN, Yamaguchi H, Hung MC. Nuclear trafficking of the epidermal growth factor receptor family membrane proteins. Oncogene. 2010;29:3997–4006. doi: 10.1038/onc.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang YN, Wang H, Yamaguchi H, Lee HJ, Lee HH, Hung MC. COPI-mediated retrograde trafficking from the Golgi to the ER regulates EGFR nuclear transport. Biochem Biophys Res Commun. 2010;399:498–504. doi: 10.1016/j.bbrc.2010.07.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lo HW. Nuclear mode of EGFR signaling network: biology, prognostic value, and therapeutic implications. Discovery Med. 2010;10:44–51. [PMC free article] [PubMed] [Google Scholar]
- 51.Wang YN, Yamaguchi H, Huo L, Du Y, Lee HJ, Lee HH, Wang H, Hsu JM, Hung MC. The translocon sec61β localized in the inner nuclear membrane transports membrane-embedded EGF receptor to the nucleus. J Biol Chem. 2010;285:38720–38729. doi: 10.1074/jbc.M110.158659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lo HW, Ali-Seyed M, Wu Y, Bartholomeusz G, Hsu SC, Hung MC. Nuclear-cytoplasmic transport of EGFR involves receptor endocytosis, importin β1 and CRM1. J Cell Biochem. 2006;98:1570–1583. doi: 10.1002/jcb.20876. [DOI] [PubMed] [Google Scholar]
- 53.Sorkin A, Goh KL. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res. 2009;315:683–696. doi: 10.1016/j.yexcr.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 54.Lo HW, Xia W, Wei Y, Ali-Seyed M, Huang SF, Hung MC. Novel prognostic value of nuclear epidermal growth factor receptor in breast cancer. Cancer Res. 2005;65:338–348. [PubMed] [Google Scholar]
- 55.Psyrri A, Yu Z, Weinberger PM, Sasaki C, Haffty B, Camp R, Rimm D, Burtness BA. Quantitative determination of nuclear and cytoplasmic epidermal growth factor receptor expression in oropharyngeal squamous cell cancer by using automated quantitative analysis. Clin Cancer Res. 2005;11:5856–5862. doi: 10.1158/1078-0432.CCR-05-0420. [DOI] [PubMed] [Google Scholar]
- 56.Xia W, Wei Y, Du Y, Liu J, Chang B, Yu YL, Huo LF, Miller S, Hung MC. Nuclear expression of epidermal growth factor receptor is a novel prognostic value in patients with ovarian cancer. Mol Carcinog. 2009;48:610–616. doi: 10.1002/mc.20504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bryant DM, Stow JL. Nuclear translocation of cell surface receptors: lessons from fibroblast growth factor. Traffic. 2005;6:947–954. doi: 10.1111/j.1600-0854.2005.00332.x. [DOI] [PubMed] [Google Scholar]
- 58.Wesche J, Malecki J, Wieldocha A, Skjerpen CS, Claus P, Olsnes S. FGF-1 and FGF-2 require the cytosolic chaperone Hsp70 for translocation into the cytosol and the cell nucleus. J Biol Chem. 2006;281:11405–11412. doi: 10.1074/jbc.M600477200. [DOI] [PubMed] [Google Scholar]
- 59.Imamura T, Engleka K, Zhan X, Tokita Y, Forough R, Roeder D, Jackson A, Maier JA, Hla T, Maciag T. Recovery of mitogenic activity of a growth factor mutant with a nuclear translocation. Science. 1990;249:1567–1570. doi: 10.1126/science.1699274. [DOI] [PubMed] [Google Scholar]
- 60.Wiedlocha A, Nilsen T, Wesche J, Sorensen V, Malecki J, Marcinkowska E, Olsnes S. Phosphorylation-regulated nucleo-cytoplasmic trafficking of internalized fibroblast growth factor-1. Mol Biol Cell. 2005;16:794–810. doi: 10.1091/mbc.E04-05-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Romisch K. Surfing the Sec61 channel: bidirectional protein translocation across the ER membrane. J Cell Sci. 1999;112:4185–4191. doi: 10.1242/jcs.112.23.4185. [DOI] [PubMed] [Google Scholar]
- 62.Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 63.Reilly JF, Maher PA. Importin β-mediated nuclear import of fibroblast growth factor receptor: role in cell proliferation. J Cell Biol. 2001;152:1307–1312. doi: 10.1083/jcb.152.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Malecki J, Wesche J, Skjerpen CS, Wiedlocha A, Olsnes S. Translocation of FGF-1 and FGF2 across vesicular membranes occurs during G1-phase by a common mechanism. Mol Biol Cell. 2004;15:801–814. doi: 10.1091/mbc.E03-08-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stachowiak MK, Maher PA, Stachowiak EK. Integrative nuclear signaling in cell development: a role for FGF receptor-1. DNA Cell Biol. 2007;26:811–826. doi: 10.1089/dna.2007.0664. [DOI] [PubMed] [Google Scholar]
- 66.Stanisic V, Lonard DM, O’Malley BW. Modulation of steroid hormone receptor activity. Prog Brain Res. 2010;181:153–176. doi: 10.1016/S0079-6123(08)81009-6. [DOI] [PubMed] [Google Scholar]
- 67.Weigel NL, Moore NL. Kinases and protein phosphorylation as regulators of steroid hormone action. Nucl Recept Signaling. 2007;5:e005. doi: 10.1621/nrs.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vicent GP, Nacht AS, Zaurin R, Ballare C, Clausell J, Beato M. Role of kinases and chromatin remodeling in progesterone signaling to chromatin. Mol Endocrinol. 2010;24:2088–2098. doi: 10.1210/me.2010-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jaganathan S, Yue P, Paladino DC, Bogdanovic J, Huo Q, Turkson J. A functional nuclear epidermal growth factor receptor, Src and STAT3 heteromeric complex in pancreatic cancer cells. PLoS ONE. 2011;6:e19605. doi: 10.1371/journal.pone.0019605. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 70.Cerliani JP, Guillardoy T, Giuanelli S, Vaque JP, Gutkind JS, Vanzulli SI, Martins R, Zeitlin E, Lamb CA, Lanari C. Interaction between FGFR-2, STAT5, and progesterone receptors in breast cancer. Cancer Res. 2011;71:3720–3731. doi: 10.1158/0008-5472.CAN-10-3074. [DOI] [PubMed] [Google Scholar]
- 71.Vicent GP, Ballare C, Nacht AS, Clausell J, Subtil-Rodriguez A, Quiles I, Jordan A, Beato M. Induction of progesterone target gene requires activation of Erk and Msk kinases and phosphorylation of histone H3. Mol Cell. 2006;24:367–381. doi: 10.1016/j.molcel.2006.10.011. [DOI] [PubMed] [Google Scholar]